Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Mutations within the
Autographa californica
Nucleopolyhedrovirus
FP25K
Gene Decrease the Accumulation of ODV-E66
and Alter Its Intranuclear Transport
SHARON C. BRAUNAGEL,1JARED K. BURKS,2GERMAN ROSAS-ACOSTA,2
ROBERT L. HARRISON,2,3H. MA,4ANDM. D. SUMMERS1,2,4*
Texas Agricultural Experiment Station,1Department of Entomology,2and Department of Biochemistry and Biophysics,4
Texas A&M University, College Station, Texas 77843-2475, and Department of Entomology, Iowa State University, Ames, Iowa 500113
Received 27 January 1999/Accepted 23 June 1999
Previous reports indicate that mutations within theAutographa californicanucleopolyhedrosis virusFP25K
gene (open reading frame 61) significantly reduce incorporation of enveloped nucleocapsids into viral occlu-sions. We report that FP25K is a nucleocapsid protein of both the budded virus (BV) and occluded virus (ODV), and we describe the effects of twoFP25Kmutations (480-1 [N-terminal truncation] and FP-gal [C-terminal fu-sion]) on the expression and cellular localization of ODV-E66 and ODV-E25. Significantly decreased amounts of ODV-E66 are detected in cells infected with 480-1 or FP-gal viral mutants, even though during FP-gal infection, steady-state levels of ODV-E66 transcripts remain unchanged. While ODV-E66 is normally detected in intranuclear microvesicles and ODV envelopes by 24 h postinfection (p.i.), ODV-E66 remains cytosolic throughout infection in cells infected with 480-1 virus (up to 96 h p.i.), and its intranuclear localization is not detected until 96 h p.i. in cells infected with the FP-gal mutant virus. The nuclear localization of ODV-E25 is not affected during infection by the FP-gal mutant; however, its trafficking is significantly delayed during infection by the 480-1 mutant. Temporal Western blot analyses of cell lysates show that both 480-1 and FP-gal mutant virus infections result in altered accumulation patterns of several structural proteins, including gp67, BV/ODV-E26, and the major capsid protein p39. In addition to BV/ODV-E26, ODV-E66 and gp67 may interact with FP25K, and ODV-E25 and p39 may also be components of a protein complex containing ODV-E66 and FP25K. Together, these data suggest that FP25K and its associated protein complex(es) may play an important role in the targeting and intracellular transport of viral proteins during infection.
Autographa californica nucleopolyhedrovirus (AcMNPV) few polyhedra (FP) mutants were first observed by Hink and Vail in infectedTrichoplusia nicultures (16). By definition, FP mutants are viral isolates which produce few (⬍10) polyhedra compared to wild-type (WT) viral isolates, which produce 50 or more polyhedra per cell. Mutations in several regions of the AcMNPV genome can result in an FP phenotype. Examples include insertion of the copia-like transposable element (TE-D) in the HindIII-K region of the baculovirus genome (map units [m.u.] 85.1 to 87.2 [24]), deletions within thePstI-G fragment (m.u. 8.6 to 10.2 [21]), deletions within the PstI-I fragment (m.u. 14.3 to 17.9 [21]), and mutations within the
FP25Kgene. Using nine FP mutants and marker rescue, Fraser et al. (12) identified a region of the genome that was consis-tently altered in an FP phenotype (HindIII-I region, m.u. 33.8 to 37.7) and noted that the FP virus-infected cells were missing a 25-kDa protein. The coding region for this 25-kDa protein, called theFP25Kgene, was sequenced by Beames and Sum-mers (2), andFP25Kgene sequences are now available for five baculovirus species (Fig. 1). This study addresses only features of an AcMNPV FP phenotype due to mutations within the
FP25Kgene.
The plaque phenotype caused by mutations within the
FP25Kgene is a decreased number of nuclear occlusions; how-ever, upon more careful examination, the effects of FP25K
gene mutations are more complex. The number of occlusions
produced by an FP mutant can vary according to cell type. In vitro, FP mutant infections produce fewer occlusions in theT. nicell line (TN-368) than inSpodoptera frugiperdacells lines (Sf21 or Sf9) (reference 11 and unpublished observations), and when larvae are infected by hemocoelic injection with an FP mutant, the number of occlusions produced per cell varies according to the type of infected tissue (27). In addition, FP mutant occlusions contain fewer virions than WT occlusions, predominantly of the enveloped single nucleocapsid type, and the envelopment of the occluded baculovirus form (occluded derived virus [ODV]) is impaired (10, 23, 25, 27). The altered and decreased ODV envelopment may correlate with the ob-served altered morphology and electron density of virus-in-duced intranuclear membranes (13). Cells infected with FP mutants release more budded virus (BV) into the medium (10, 13, 26), and virus progeny are still observed budding at the plasma membrane at 72 h post infection (p.i.) (26), whereas in cells infected with WT virus, BV production has decreased to barely detectable levels by 72 h p.i. (33). The prolonged period of BV cell surface maturation is reflected in increased BV titers. Potter et al. (26) showed that FP mutants of T. ni
nucleopolyhedrovirus increased in frequency in vitro from un-detectable levels up to 93% of total plaques in 10 serial pas-sages, while Fraser and Hink (10) demonstrated that the FP phenotype inGalleria melonellaNPV increased from undetect-able levels up to nearly 100% of the cells showing an FP phenotype in five serial passages. TheLymantria dispar NPV produces an FP phenotype even more rapidly, with several isolates showing a 92% plaque FP phenotype in only one passage (30). Thus, the FP phenotype provides a selection
* Corresponding author. Mailing address: Texas A&M University, Department of Entomology, College Station, TX 77843-2475. Phone: (409) 847-9036. Fax: (409) 845-8934. E-mail: [email protected].
8559
on November 9, 2019 by guest
http://jvi.asm.org/
advantage for in vitro virus maturation. However, there is no observed selection for the FP phenotype when virus is pas-saged by per os feeding of insect larvae (10). Hence, the FP phenotype resulting in reduced ODV and enhanced BV mat-uration is not an advantage for horizontal transmission from insect to insect in the natural infection of insect populations.
In an FP mutant-infected cell the rate of polyhedrin mRNA expression is significantly reduced compared to that of WT infection (15), and polyhedrin localizes less efficiently to the nucleus during the early occlusion phase of infection (24 h p.i.) (20), yet polyhedrin mRNA stability is similar in WT- and FP mutant-infected cells (15). While polyhedrin gene expression is significantly altered, the steady-state mRNA levels of another very late gene, the p10 gene, remain unaltered (15).
In summary, while the FP plaque phenotype is the most easily identified effect of mutations within the FP25K gene, such mutations also result in (i) increased production of BV, (ii) decreased amounts of viral occlusions, (iii) decreased amounts of ODV production, (iv) altered morphology of in-tranuclear membranes, (v) decreased amounts of polyhedrin mRNA, and (vi) altered transport of polyhedrin protein into the nucleus. However, little is known about the function(s) of
the FP25K protein, even though immunoelectron microscopy detects FP25K protein in electron-dense regions in both the cytoplasm and nucleus (13). Considering the varied effects of mutations within theFP25Kgene, it is possible that FP25K has more than one function during the invasion and infection pro-cess.
In this study we show that FP25K is a structural protein of the nucleocapsid of AcMNPV and that it may interact with or influence the levels or rate of protein accumulation of several structural proteins of the virus. Infection by two FP mutants, 480-1 and FP-gal, result in significantly decreased amounts of ODV-E66 protein; however, steady-state levels ofODV-E66
[image:2.612.61.522.83.415.2]transcripts remain unchanged compared to the WT. Transport of ODV-E66 into the nucleus is impaired in both of these mutants, with the 480-1 mutant having the most significant effect. Transport of ODV-E25 (28) into the nucleus is different for the two mutants: ODV-E25 transport is delayed during infection by 480-1 but is unaffected when cells are infected with FP-gal. Studies designed to identify proteins interacting with FP25K suggest that ODV-E26 and ODV-E66 may interact directly with FP25K and that ODV-E25 and p39 may be com-ponents of a complex containing FP25K.
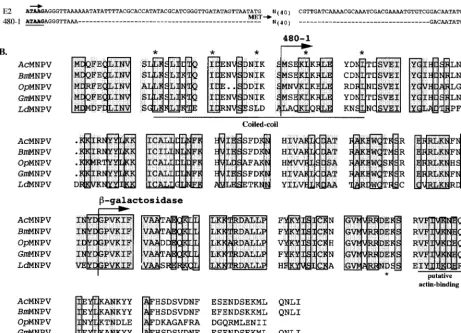
FIG. 1. Genomic constructs and sequence comparison of FP25K proteins. (A) The 5⬘untranslated region of FP25K (E2) is shown, along with the TAAG transcription initiation sequence (arrow). In the 480-1 mutant virus 120 nt have been removed, as indicated by dashes. (B) The amino acid sequences of five baculovirus FP25K protein sequences are compared. Identical amino acids are shaded and outlined, while conservative changes are shown in the shaded regions. The 480-1 N-terminal methionine is identified (arrow), as is the site of the-gal fusion (arrow). The N-terminal region of FP25K contains a conserved coiled-coil domain (underlined and marked with asterisks), and the C-terminal region contains a putative actin binding helix (underlined; the asterisk shows the requisite E or Q). Rules used to assign conservation are as follows: A⫽G⫽S⫽T, V⫽L⫽I⫽M⫽F⫽Y⫽W, N⫽Q⫽D⫽E, and R⫽K⫽H. Accession numbers: BmMNPV (B. morinucleopolyhedrovirus), L33180 (nt 43656 to 44298); GmMNPV (G. melonellanucleopolyhedrovirus), M29140; OpMNPV (Orgyia pseudosugata nucleopolyhedro-virus), U75930 (nt 50697 to 51323); AcMNPV, L22858 (nt 48513 to 49155); and LdMNPV (L. disparnucleopolyhedrovirus), U58676.
on November 9, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Insect cell lines and virus.S. frugiperdaIPLB-Sf21-AE clonal isolate 9 (Sf9) cells were cultured in suspension at 27°C in TNMFH medium (31) supplemented with 10% fetal bovine serum (complete medium). AcMNPV (strain E2) was used to infect cells at a known multiplicity of infection (MOI), with time zero set at the time of virus addition. After 1 h of adsorption, cells were washed and resus-pended in fresh complete medium. The FP mutant viruses 480-1 and FP-gal were described by Beames and Summers (1, 2) and are summarized in Fig. 1.
Western blot analysis of infected cells, virus, and virus fractionation.Sf9 cells were infected with either AcMNPV (WT) or 480-1 or FP-gal virus (MOI, 20), and at an appropriate time p.i., the cells were collected and washed once with phosphate-buffered saline (PBS). Cell pellets were resuspended in PBS contain-ing protease inhibitors (20g of leupeptin per ml, 20g of aprotinin per ml, 20 g of pepstatin A per ml, 0.5 mM phenylmethylsulfonyl fluoride, 1M E64). Cells were broken by sonication, and protein concentrations were determined by the method of Bradford (4).
BV was purified from the cell culture supernatant of infected cells (36 h p.i.), and ODV was purified from infected-cell lysates (72 h p.i.), by the technique described by Braunagel and Summers (7). Purified virus was further fractionated into envelope and nucleocapsid fractions (7). The purified virus and respective envelope and nucleocapsid fractions were analyzed by using the ODV envelope marker proteins ODV-E66, ODV-E56, ODV-E18, ODV-E25, BV/ODV-E26, and ODV-EC27, and the BV was characterized by using the envelope markers gp64 and BV/ODV-E26. ODV, BV, and viral fractions were analyzed by using the nucleocapsid marker p39. An example of such a characterization is shown in Fig. 3F.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed as described by Laemmli (22) (4% stacking gel, 12.5% separating gel). Samples were incubated in 1.5% SDS–0.5%-mercaptoethanol–25 mM Tris-HCl (pH 6.8)–7% glycerol for 15 min at 65°C. Test gels were run and stained with Coomassie blue to visually confirm that protein was loaded at equal concentra-tions per sample. Following electrophoresis, the gels were transferred onto Immobilon-P membranes (Millipore, Bedford, Mass.). The membranes were blocked with TTBS-BLOTTO (150 mM NaCl, 10 mM Tris, and 0.1% Tween 20 [pH 8.0] supplemented with 1% nonfat dry milk). Antibody was bound overnight (4°C), the blots were washed twice with TBS, and horseradish peroxidase-linked immunoglobulin G (1:5,000) was bound for 1 h at room temperature. The blots were washed three times with TTBS, reacted for 1 min with ECL (Amersham, Arlington Heights, Ill.) chemiluminescence reagent, and exposed to X-ray film. For each antibody determination the entire experiment was performed as a matched set; thus, a direct comparison of signal intensities reflects differing amounts of bound antibody.
The following antibodies and dilutions were used for Fig. 3: anti-FP25K, no. 2804 (1:1,000); anti-E66, no. 5297 (1:1,000); anti-E25 (provided by G. Rohr-mann, Oregon State University, Corvallis) (1:2,000); anti-E56, no. 6543 (1:1,000); anti-E18, no. 7350 (1:1,000); anti-EC27, no. 7351 (1:1,000); anti-gp67 B12D5
(provided by L. Volkman, University of California, Berkeley) (1:1,000); anti-E26, no. 7554 (1:1,000); anti-p78/83 (provided by C. Richardson, Amgen Institute, Toronto, Ontario, Canada) (1:2,000); anti-p39, p10C6(provided by L. Volkman)
(1:1,000); and anti-gp41, monoclonal antibody 3.10, 6.31 (provided by P. Faulk-ner, Queens University, Kingston, Ontario, Canada) (1:1,000).
Immunofluorescence microscopy.Cells were processed for light microscopy by using a modification of previously described procedures (8). Sf9 cells were in-fected (MOI, 20), and at the appropriate time p.i. cells were rinsed with Grace’s medium and fixed with 3.7% paraformaldehyde in PBS (20 mM phosphate, 140 mM NaCl, pH 7.2) for 10 min at room temperature. The fixative was removed, and cells were washed and permeabilized with methanol (10 min) and subse-quently treated with 0.5% Triton-X 100 (10 min), followed by two rinses with PBS. The cells were blocked for 1 h in 1% normal goat serum–3% bovine serum albumin and then incubated with primary antibody (anti-FP25K, no. 2804, 1:1,000; anti-E66, no. 5297, 1:1,000; or anti-E25, 1:2,000 in 1% normal goat serum in PBS) overnight at 4°C. Cells were rinsed three times, and secondary antibody (fluorescein isothiocyanate [FITC] (Sigma, St. Louis, Mo.; 1:100 in PBS) was added and incubated for 1 h. The cells were washed, and the nucleus was visualized by staining with DAPI (4⬘,6-diamidino-2-phenylindole) (0.1g/ml in PBS). Cells were viewed and photographed with a Zeiss Axiovert 135 pho-tomicroscope. Each experiment was performed three to five times, and obser-vations were made by using both FITC- and tetramethyl rhodamine isothiocya-nate-labeled secondary antibodies. Thus, several thousand cells were viewed per experiment, and representative cells were chosen for data presentation.
Quantitative primer extension.Sf9 cells were infected with either AcMNPV or FP-gal (MOI, 10), and infected-cell RNA was isolated by the method of Chir-gwin et al. (9). Primer extensions were performed with 30g of RNA hybridized to specific oligonucleotide probes labeled with [␥-32P]ATP (29). The
oligonucleo-tide sequence of the ODV-E66 probe was 5⬘-GATAGGTACAAAAAACATAT TAAAAATATTA CAACTATGAC-3⬘, and that of the vp39 probe was 5⬘-CGC GAAAATGCAGCGATTAACTCTCATTTGTCGCGGCGCC-3⬘. RNA-prim-er hybrids wRNA-prim-ere precipitated with ethanol, washed with 70% ethanol, and resus-pended in 30l of reverse transcription reaction mix (50 mM Tris [pH 7.6], 60 mM KCl, 10 mM MgCl2, 0.66 mM deoxynucleoside triphosphates, 1 mM
dithio-threitol, 40l of RNasin, 50l of actinomycin D per ml, and 150 U of Moloney
murine leukemia virus reverse transcriptase [U.S. Biochemicals, Cleveland, Ohio]). Extension of the annealed primers was performed at 42°C for 2 h. The reaction products were ethanol precipitated and resuspended in 2l of 0.1 N NaOH. After a 30-min incubation to eliminate the RNA template, 4l of se-quencing stop buffer was added, and the samples were boiled for 3 min and analyzed by electrophoresis on a urea–6% polyacrylamide gel together with a sequencing ladder generated by using the same oligonucleotides. The gels were dried and subjected to autoradiography, and the primer extension products were quantitated with the FUJIX BAS2000 bioimaging analyzer system (Fuji Photo Film Co., Tokyo, Japan).
Immunoprecipitation.A total of 1.5⫻106infected cells (MOI, 20) were used
for each immunoprecipitation. At the appropriate time, cells were collected and resuspended in 500l of TBN buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 10g of leupeptin per ml, 10g of aprotinin per ml, 1g of pepstatin A per ml, 1 mM phenylmethylsulfonyl fluoride, 1 mM E64) supplemented with either 0.2% Tween 20 or 1% Nonidet P-40 (NP-40). Cells were incubated for 20 min at 4°C and then lysed by passage through a 25-gauge needle four times. The lysed extract was centrifuged (3,000 rpm, 10 min, 4°C, Microfuge), and the supernatant was preabsorbed for 1 h with 25l of preimmune serum at 4°C. Protein A-aga-rose (Sigma) (40l of a 50% slurry) was added to the extract and incubated for 1 h at 4°C. The immune complexes formed during preadsorption were pelleted at 3,000 rpm for 15 min in a Microfuge. The preadsorbed extract was then immunoprecipitated with the appropriate antibody overnight (25l, 4°C) (ODV-E66, no. 5297; FP25K, no. 2804; gp67, AcV1). Protein A-agarose (40l, 50%
slurry) was added and incubated for 2 h at 4°C. The agarose beads were washed three times in TBN and then once in TBS. The washed beads (20l) were prepared for SDS-PAGE (4% stacking gel, 12.5% separating gel).
Yeast two-hybrid library construction and screen.Sf9 cells were infected (MOI, 20), and after 1 h of adsorption, the virus inoculum was removed and the cells were resuspended in complete medium. At 18 or 24 h p.i., cells were collected and mRNA was isolated by using either the Poly A Tract System 1000 (Promega, Madison, Wis.) or the Poly(A) Pure mRNA Isolation Kit (Ambion, Austin, Tex.). The cDNA library was then constructed by using the Two Hybrid cDNA Construction Kit (Clontech, Palo Alto, Calif.). The libraries were ampli-fied, and the resulting titers of the amplified libraries were as follows: 18 h p.i., 1.31⫻1012; 24 h p.i., 3.25⫻1013. To harvest large quantities of DNA from each
library, a 1-ml aliquot of amplified library was diluted and grown on 200 Luria broth-ampicillin supplemented plates (150-mm diameter), bacteria were har-vested, and plasmid DNA was purified by using the Plasmid Giga Kit (Qiagen, Valencia, Calif.).
The appropriate genes were cloned into the yeast binding domain vector. ⌬23-E66 was constructed in the yeast vector pAS2-1 such that the N-terminal Met was followed by the FLAG epitope (D-Y-K-D-D-D-D-K) (Kodak, New Haven, Conn.) followed by amino acids 24 to 704 of ODV-E66 (16). Library screens were performed with the Matchmaker Two Hybrid System 2 (Clontech) and the complementation assay with blue selection for-galactosidase (-gal) activity. The colonies that showed a positive blue interacting color reaction within an 8-h period were confirmed by using a secondary reaction. The yeast activation domain plasmid was then purified and transformed intoEscherichia coliDH5␣, and DNA was amplified and sequenced.
RESULTS
FP25K homology comparison and selection of mutant vi-ruses. One goal of this study was to examine the effects of
FP25K deletions and potential interactions between FP25K and structural proteins of AcMNPV. To consider a design for
FP25Kgene mutations, we examined the predicted structural features of the FP25K protein. Computer-assisted analysis re-vealed a highly conserved coiled-coil domain (a structural mo-tif often involved in protein-protein interactions) at the N terminus and a putative actin binding helix (Fig. 1). Two pre-viously reported viral mutants (1, 2), each lacking one of these regions, were selected for these studies. The 480-1 mutant virus contains a 120-nucleotide (nt) deletion extending from posi-tion ⫺45 to ⫹77 (relative to the FP25K initiation codon), resulting in translation initiation at an internal Met (Fig. 1A). Thus, in this mutant the putative coiled-coil domain was de-leted, resulting in an FP25K protein with a 31-amino-acid N-terminal truncation (13) (Fig. 1). The FP-gal mutant lacks the C-terminal half of FP25K, including the putative acting binding helix, and instead contains the amino acids-gal. Note that although the FP25K protein is highly conserved through-out the putative actin binding region, the last 20 to 25 amino acid residues show little conservation (Fig. 1B).
on November 9, 2019 by guest
http://jvi.asm.org/
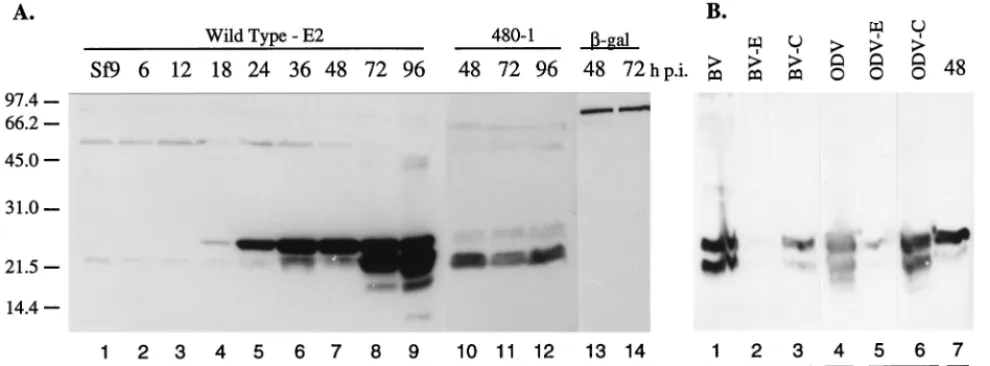
Two forms of FP25K are present during infection, and FP25K is a structural protein of AcMNPV. Previous work using Western blot analysis detected trace amounts of FP25K in purified BV, ODV, and viral occlusions; however, the levels were too low to be convincing (14). To clarify this, we used the more sensitive detection techniques involving horseradish per-oxidase-labeled secondary antibody and chemiluminescence. This confirmed that FP25K was a structural protein in the nucleocapsids of both BV and ODV and further identified two forms, of 25 and 23 kDa (Fig. 2B, lanes 1 to 6). We know from previous work that BV can be contaminated with nonoccluded ODV if purified late during infection (unpublished observa-tions). To decrease this potential contamination, we purified the BV from the supernatant of a 36-h-p.i. infected sample and tested the BV for the presence of ODV envelope proteins (see Fig. 3F). No ODV proteins were detected with the BV. A time course analysis of infected cells showed that the 25-kDa pro-tein was the first to accumulate to detectable levels, while the 23-kDa protein was detected, and accumulated, later during the infection (Fig. 2A, lanes 2 to 9). The molecular mass of the truncated protein produced from the 480-1 mutant virus was similar to that of the 23-kDa FP25K species observed in WT-infected cells (Fig. 2A, compare lanes 8 to 11). The molecular mass of the FP-gal fusion was high (⬎110 kDa), and only one major protein was detected throughout infection (Fig. 2A, lanes 13 and 14).
It is possible that the 23-kDa protein detected in WT infec-tion was a by-product of protein degradainfec-tion. Hom and Volk-man (17) showed that the viral cysteine protease, v-CATH, is activated by SDS-PAGE sample buffer, and the protease in-hibitor E64 must be included in the loading dye to prevent protease activity. Thus, even though a battery of protease in-hibitors were used in the preparation of the infected-cell ly-sates, it was possible that the FP25K protein was degraded while in SDS-PAGE sample buffer. When FP25K was analyzed by SDS-PAGE with E64 incorporated at all phases of sample handling, the 23-kDa form was still present and was at the same relative levels as observed in Fig. 2A (data not shown). Alternatively, it is possible that a second product is translated, starting from amino acid 32 of FP25K (internal Met); thus, the
resultant 23-kDa form may be functionally significant. While the FP-gal fusion also contains the internal Met due to the large size of the fusion protein (⬎110 kDa), it is unlikely that removal of a few kilodaltons would be detectable. We note that degradation of the-gal fusion protein is not detected, sug-gesting that protein degradation is not occurring at high levels. Mutations within the FP25Kgene alter structural protein profiles during infection. Harrison and Summers (13) ob-served aberrant envelope-nucleocapsid interactions within the nuclei of FP25K mutant-infected cells. Instead of circular, well-formed microvesicles, some intranuclear membranes are elongated, angular, and unusual in shape and electron density. Additionally, although nucleocapsid assembly is apparently normal, there are few ODVs (intranuclear enveloped nucleo-capsids) present in an FP25K mutant-infected cell (9, 23, 27). These observations show that intranuclear membrane forma-tion, ODV envelopment, and the process of viral occlusion formation are altered in an FP mutant infection and suggest that these maturation processes could be at least partially in-fluenced by FP25K. We decided to determine if mutations within theFP25Kgene resulted in detectable changes in the amount or localization of baculovirus structural proteins. In a matched experiment, Sf9 cells were infected with WT, 480-1, or FP-gal virus, and the cells were harvested at various times p.i., protein concentrations were determined, and test gels were stained with Coomassie blue to visually confirm equivalent protein amounts per lane (data not shown). Matched gels and Western blots were then prepared, bound to antibodies and exposed to X-ray film for chemiluminescent detection. The exposure time was set to be optimal for protein detection in WT infection. Thus, differences in blot intensities represent quantitative changes in protein amounts relative to those of WT virus (E2 strain). During WT infection, the FP25K protein (25 kDa) was first detected at 18 h p.i. (Fig. 3A). The second form of FP25K (23 kDa) was detected at 36 h p.i. and accu-mulated to high levels by 72 h p.i. (see Fig. 2 for the molecular masses of two forms of FP25K and the protein produced by 480-1 virus). The truncated protein produced by the 480-1 mutant virus (23 kDa) was not detected until 36 h p.i. and did not accumulate to significant levels until 48 h p.i. (note that this FIG. 2. FP25K is a structural protein of baculovirus. (A) Time course of FP25K accumulation in E2-infected cells (lanes 2 to 9) and size and late accumulation in cells infected with 480-1 mutant virus (lanes 10 to 12) and FP-gal recombinant virus (lanes 13 and 14). (B) BV and ODV were purified and separated into envelope (E) and nucleocapsid (C) fractions (lanes 1 to 6). WT AcMNPV-infected cell extract (48 h p.i.) were used a positive control (lane 7). Samples were separated by SDS-PAGE, Western blotted onto a polyvinylidene difluoride membrane, and reacted with antibody to FP25K. The amount of protein analyzed per lane is indicated. Numbers on the left are molecular masses in kilodaltons.
on November 9, 2019 by guest
http://jvi.asm.org/
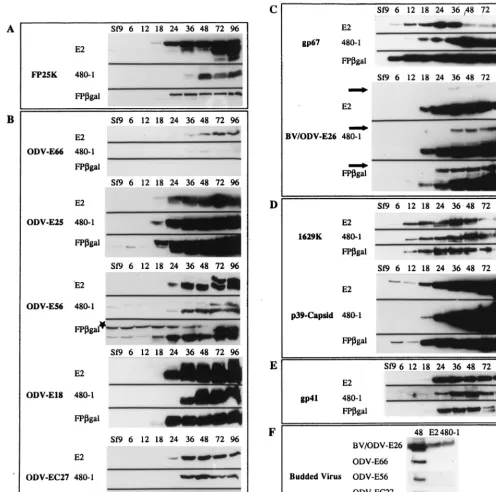
[image:4.612.57.552.77.260.2]pattern of protein accumulation was very similar to the appear-ance of the 23-kDa form during WT infection). The temporal detection of the FP-gal fusion was similar to that of the WT protein (Fig. 3A).
The amount of detectable ODV-E66 decreased significantly in cells infected with the FP mutant viruses (Fig. 3B). Indeed, ODV-E66 was barely detectable by Western blot analyses in either the 480-1- or FP-gal-infected cells. In contrast, ODV-E25 accumulated to higher levels in the FP-gal
mutant-in-fected cells than in the WT-inmutant-in-fected cells (Fig. 3B). While the results suggest that levels and temporal accumulation of ODV-E56 are mostly unchanged, the Western blot of the FP-gal infection time course shows a higher background against the 70-kDa cellular protein (Fig. 3B) (5). Levels of ODV-E18 and ODV-EC27 remained mostly unchanged (6); however, both proteins were detected slightly later in the 480-1-infected cells (Fig. 3B).
[image:5.612.53.549.76.568.2]A screen of BV envelope proteins revealed that both gp67 FIG. 3. Structural protein-temporal expression. (A to E) Matched sets of cell lysates (25g/lane) were collected at defined times p.i. and analyzed for protein content with the appropriate antibody. SDS-PAGE, Western blotting, and antibody reactions were performed for matched sets, and the optimal exposure was set for WT AcMNPV. (A) FP25K; (B) ODV envelope proteins; (C) BV envelope proteins; (D) capsid-associated proteins; (E) tegument-associated protein. (F) Purified BV from both WT AcMNPV- and 480-1-infected-cell supernatants. Virus was loaded at 10g/lane, and that for the 48-h-p.i. time point was loaded at 15g/lane. Antibodies to each of the structural proteins were used to determine protein composition. The star in panel B shows a higher background for ODV-56 against the 70-kDa cellular protein. The arrows in panel C show increased levels of the higher-molecular-mass form in FP mutant-infected cells.
on November 9, 2019 by guest
http://jvi.asm.org/
and BV/ODV-E26 accumulated to higher levels during FP mutant infection than during WT infection (Fig. 3C). Addi-tionally, the higher-molecular-mass, immunoreactive form of BV/ODV-E26 (3) was also detected at increased levels in the FP mutant-infected cells than in the WT-infected cells (Fig. 3C). The levels of the capsid protein p78/83 (1629K) was not altered compared those in WT infection. However, another nucleocapsid protein, p39, was detected in significantly higher quantities in 480-1 mutant-infected cells but not during FP-gal infection (Fig. 3D). We also observed that in both WT-and FP-gal-infected cells, p39 was detected at 6 h p.i. Earlier studies of p39 suggest that transcription of the p39 gene occurs late (32); however, an early transcription consensus motif (CAGT) is present at nt⫺317, just 4 nt away from a utilized late transcription initiation motif. Thus, it is possible thatp39
is transcribed at low levels early in infection. p39 was not detected at 2 or 4 h p.i. (data not shown). Effects on the only known tegument protein, gp41, were minimal; however, a de-creased amount of gp41 was observed very late in FP-gal infection (72 to 96 h p.i.) (Fig. 3E).
Decreased levels of ODV-E66 protein are not due to de-creased steady-state levels ofODV-E66transcripts.Mutations in theFP25K gene result in decreased steady-state levels of polyhedrin gene transcripts (15). To determine if reduced ODV-E66 transcription could also explain the significant de-crease in ODV-E66 protein, quantitative primer extension analysis ofODV-E66 was performed. As an internal control, primer extension was performed forvp39. Figure 4A shows the results for primer extension products ofvp39and ODV-E66. The placement of these products was determined by using a matched sequencing ladder (not shown). The extension prod-ucts for each primer extension were quantitated by using a Bio-Imaging Analyzer, and the ratio ofODV-E66tovp39was determined. Figure 4B shows that in two different experiments at 24 and 48 h p.i., the ODV-E66/vp39 ratio remained un-changed for WT- and FP-gal-infected cells. These data sug-gest that the decreased amount of detectable ODV-E66 pro-tein was not due to transcriptional down-regulation at the
ODV-E66locus. Quantitative transcription was not done for the 480-1 mutant, but we note that both mutants result in a significant decrease in ODV-E66 protein.
Mutations within the FP25K gene alter the intranuclear trafficking of ODV-E66.Since steady-state levels ofODV-E66
transcripts were not altered in FP-gal mutant infection, pos-sible mechanisms to explain the lack of detectable ODV-E66 protein include decreased ODV-E66 translation, altered ODV-E66 targeting or transport, and/or altered ODV-E66 protein stability. To test if either FP25K mutant virus resulted in abnormal transport of ODV-E66, matched infections of WT and FP mutant viruses were performed, and ODV-E66 local-ization was determined by immunofluorescence with rabbit antisera raised against ODV-E66 and FITC-conjugated anti-rabbit secondary antibodies. (Trying to show these data by using an overview of infected cells is difficult, so representative cells from these experiments were chosen for enlargement. An example of how we made this choice is shown in Fig. 7A and B. Since showing phase-contrast, FITC, DAPI, and dual expo-sures for every data set [which was performed for each exper-iment] would make the figures large, cumbersome, and difficult to present, only the FITC and FITC-DAPI dual exposures [with DAPI defining the area of the nucleus] are presented. One example of phase-contrast–FITC dual exposure is shown in Fig. 7g2 for reference. Additionally, control antibody reac-tions [uninfected and preimmune sera] were performed for every experiment; however, the background cross-reactivity was minimal and was reproduced as a black field. An example of such a control is shown in Fig. 7D.)
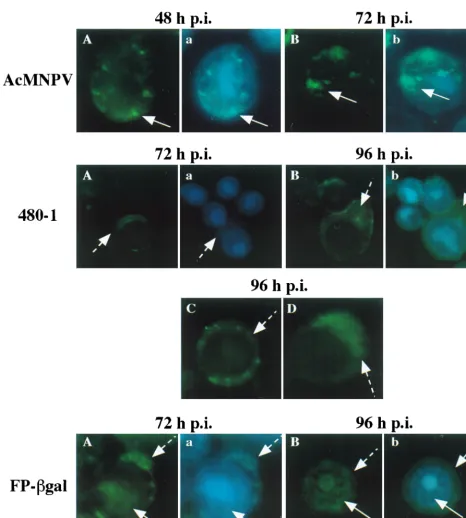
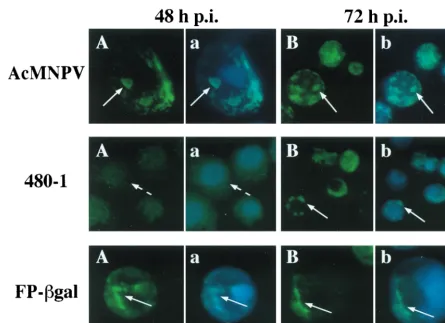
By 24 h p.i., in WT-infected cells, ODV-E66 was detected in the cytoplasm; however, labeling was enriched within the nu-cleus (data not shown). By 48 and 72 h p.i., ODV-E66 was easily visualized in discrete foci within the nucleus (Fig. 5, AcMNPV), confirming our previous electron microscopy and immunogold localization studies, which showed that ODV-E66 localizes to virus-induced intranuclear microvesicles and ODV envelope (18). Consistent with the intensity of the signal of ODV-E66 detected by Western blotting, ODV-E66 protein was detected at very low levels in 480-1- and FP-gal-infected cells. In 480-1-infected cells, -E66 was detected starting at 72 h p.i., and it was located predominantly in a diffuse pattern in the cytoplasm (Fig. 5A, 480-1). By 96 h p.i. there was an increase in detectable levels of ODV-E66 protein; however, it still ac-cumulated within the cytoplasm (Fig. 5B to D, 480-1). The pattern of ODV-E66 localization during FP-gal infection was intermediate to that observed during WT and 480-1 infections. ODV-E66 did not accumulate to detectable levels until late in infection (72 h p.i.), but by this time ODV-E66 was detected in both the cytoplasm and nucleus (Fig. 5A, FP-gal). By 96 h p.i. the accumulated amount of ODV-E66 increased slightly, and it was still detected in both the cytoplasm and nucleus (Fig. 5B, FP-gal).
Because infection by both 480-1 and FP-gal results in in-creased BV production (13) (as determined by inin-creased virus titers), we considered the possibility that in Sf9 cells infected with such mutants ODV-E66 was translated at WT levels but that instead of being transported to nuclear membranes, it was exported to the plasma membrane and potentially incorpo-rated in the envelope of BV. To test this, BV was purified from WT- and 480-1-infected cells, and Western blot analysis was performed to detect the presence of ODV-E66 and other ODV envelope proteins. A 48-h-p.i. cell lysate was used as a positive control. As shown in Fig. 3F, none of the ODV enve-lope proteins (E66, E56, EC27, or ODV-E25) were present in BV purified from WT- or 480-1-infected cells, while the BV envelope proteins, BV/ODV-E26 and gp67, were present (gp67 data not shown). These data show that in 480-1-infected cells, ODV-E66 and other ODV envelope pro-teins are not inappropriately incorporated into BV.
FIG. 4. Quantitative primer extension analysis. (A) Quantitative primer ex-tension ofp39and ODV-E66 from Sf9 cells infected with WT AcMNPV (E2) or FP-gal virus. Primer extensions were with a mock-infected RNA sample (lane M) and E2 and FP-gal RNA samples with primers for vp39 or ODV-E66. The locations of ODV-E66andvp39 were established by using DNA sequencing ladders (not shown) and are indicated to the right and left of the autoradiograph. (B) Calculated ratios ofODV-E66tovp39RNA for two replicate sets of exper-iments at 24 and 48 h p.i.
on November 9, 2019 by guest
http://jvi.asm.org/
480-1 mutant infection delays, but does not inhibit, trans-port of ODV-E25 into the nucleus.While the overall amino acid sequences of ODV-E66 and ODV-E25 do not show sig-nificant homology, the amino acids at the N termini of ODV-E66 and ODV-E25 (23 and 24 amino acids, respectively) are similar and sufficient to target fusion reporter proteins to in-tranuclear microvesicles and ODV envelope (19). If these N-terminal sequences interact with FP25K, then altered transport
of ODV-E25 into the nucleus during FP25K mutant infection might be expected. To test this, the nuclear localization of ODV-E25 in WT- and FP25K mutant-infected cells was deter-mined. By 24 h p.i. ODV-E25 localized very efficiently to dis-crete regions in the nuclei of WT-infected cells (data not shown), and the intensity of localization and intranuclear flu-orescence increased at 48 and 72 h p.i. (Fig. 6A and B, Ac
[image:7.612.71.537.71.589.2]M-NPV). In 480-1 mutant-infected cells, there was a significant FIG. 5. Cellular localization of ODV-E66. Sf9 cells were infected with WT AcMNPV, 480-1, or FP-gal virus. At the indicated times p.i., cells were collected, fixed, and reacted with antibody to ODV-E66 (no. 5297; 1:1,000) and FITC-conjugated secondary antibody. To visualize the nucleus, cells were also stained with DAPI, and a dual exposure of FITC and DAPI was performed. Dashed arrows show cytoplasmic labeling, and solid arrows show intranuclear labeling. (A, B, C, and D) FITC exposure; (a and b) dual exposure of DAPI and FITC labeling.
on November 9, 2019 by guest
http://jvi.asm.org/
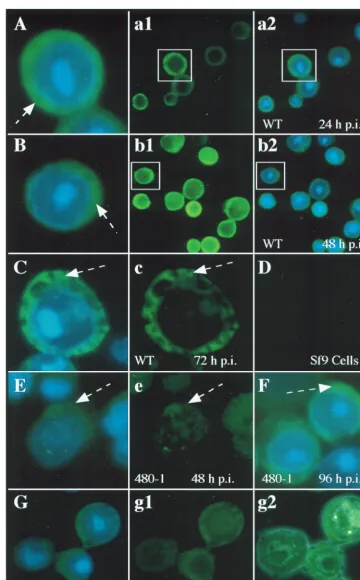
delay of localization of ODV-E25 in the nucleus. By 48 h p.i. ODV-E25 was located predominantly in the cytoplasm (Fig. 6A, 480-1); however, by 72 h p.i., ODV-E25 was detected in the nucleus in a pattern similar to that for WT infection (Fig. 6B, 480-1). Like for WT infection, immunoelectron microscopy showed that at 72 h p.i. ODV-E25 was present in intranuclear microvesicles and the ODV envelope in the 480-1 mutant-infected cells (data not shown). The nuclear localization of ODV-E25 in the FP-gal mutant-infected cells was indistin-guishable from that in WT infection: by 24 h p.i. ODV-E25 was detected predominantly within the nucleus (data not shown), and it remained intranuclear throughout infection (Fig. 6A and B, FP-gal).
Localization of FP25K in WT- and FP mutant-infected cells. To provide an overview of FP25K localization during infection, we used immunofluorescence microscopy of WT-, 480-1-, or FP-gal-infected cells (Fig. 7). Figure 7 also exemplifies how the representative cells shown throughout this work were se-lected. An example of these is shown in Fig. 7a1, a2, b1, and b2 and then enlarged in Fig. 7A and B. This analysis showed that significant amounts of FP25K were present in the cytoplasm throughout infection (Fig. 7A to C). It is known from previous work that FP25K protein is present in both cytoplasmic and nuclear fraction, and it accumulates in electron-dense regions (14). These discrete regions enriched in FP25K could be de-tected as the microscope operator “focused through” the cell; however, they were not easily discernible by using a single
focus, as shown in Fig. 7. The mutant 480-1 FP25K protein was not detected until 48 h p.i. (Fig. 7E), and even then the levels were too low to convincingly localize this protein; however, most of the truncated protein appeared to be cytoplasmic. By 72 and 96 h p.i., the localization of the truncated protein more closely resembled that seen in WT infection (Fig. 7F and data not shown). The FP-gal fusion protein showed a pattern of localization similar to that in WT infection (Fig. 7G, g1, and g2). Since the results for FP-gal were so similar to those for the WT, only one time point is shown (48 h p.i.), and an example of the matched phase-contrast–FITC dual exposure is also shown (Fig. 7g2). Like the results seen with antibodies to ODV-E66 and ODV-E25, background FITC levels of FP25K antibody tested against uninfected cells were reproduced as black field (Fig. 7D).
FP25K potentially interacts with other baculovirus struc-tural proteins.The Western blot analysis showed that com-pared to WT infection, infections by the FP25K mutant viruses resulted in increased protein levels of both BV envelope pro-teins, BV/ODV-E26 and gp67 (Fig. 3C). Previous data show that FP25K is capable of interacting with BV/ODV-E26 (3) (summarized in Table 1). Antibody to gp67 (AcV1)
[image:8.612.77.523.73.396.2]coprecipi-tated FP25K, and the total amount of precipicoprecipi-tated FP25K decreased as the amount of gp67 decreased (Fig. 8A, lanes 2 to 5). When the reciprocal experiment was performed with anti-body to FP25K, gp67 was not coprecipitated; however, the FIG. 6. Cellular localization of ODV-E25. Sf9 cells were infected with WT AcMNPV, 480-1, or FP-gal virus. At the indicated times p.i., cells were collected, fixed, and reacted with antibody to ODV-E25 (1:1,000) and FITC-conjugated secondary antibody. To visualize the nucleus, cells were also stained with DAPI, and a dual exposure of FITC and DAPI was performed. Dashed arrows show cytoplasmic labeling, and solid arrows show intranuclear labeling. (A and B) FITC exposure alone; (a and b) dual exposure of DAPI and FITC labeling.
on November 9, 2019 by guest
http://jvi.asm.org/
FIG. 7. Cellular localization of FP25K. (A to C) Cells were infected with WT AcMNPV. Panels a1, a2, b1, and b2 show an overview of the cell population (a1 and b1, FITC exposure; a2 and b2, FITC-DAPI dual exposure; the cells chosen as representative of the population are indicated (boxes) and then enlarged in panels A and B (FITC-DAPI dual exposure). (E and F) Cells infected with the 480-1 mutant virus (E and F, FITC-DAPI; e, FITC). (G) Cells infected with FP-gal recombinant virus (g1, FITC; G, FITC-DAPI; g2, FITC–phase-contrast dual exposure). The time p.i. for each set is indicated. (D) Background labeling of FP25K antibody against uninfected Sf9 cells. Dashed arrows show cytoplasmic labeling.
on November 9, 2019 by guest
http://jvi.asm.org/
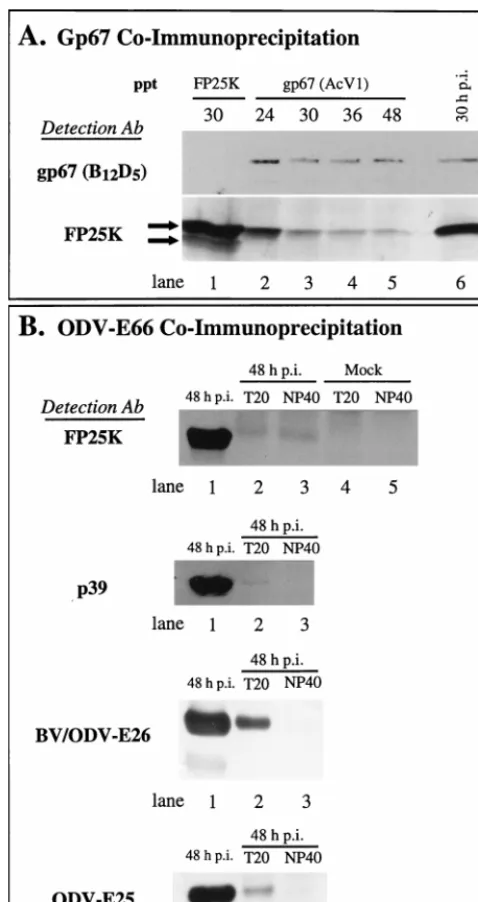
FP25K antibodies precipitated both the 23- and 25-kDa forms (Fig. 8A, lane 1).
We also tested FP25K antibody-immunoprecipitated com-plexes for the presence of ODV-E66 and ODV-E25, and we were unable to detect either protein. Indeed, FP25K antibody-immunoprecipitated samples also lack BV/ODV-E26 (data not shown). Because these results suggest that FP25K antibodies may be unable to recognize bound FP25K protein, antibodies to ODV-E66 were used to further characterize the possible
interaction between FP25K and ODV-E66. FP25K coprecipi-tated with ODV-E66 (Fig. 8B; FP25K, lanes 2 and 3). Because FP25K migrates at a molecular weight similar to that of im-munoglobulin light chain, control experiments using unin-fected cells were performed, and no background signal due to an interaction between the secondary antibody and the light chain of rabbit was detected (Fig. 8B, FP25K, lanes 4 and 5). In addition, antibody to E66 coprecipitated p39, ODV-E25, and BV/ODV-E26 at 48 h p.i. (Fig. 8B), and coimmuno-precipitation of these proteins was optimal with Tween 20 (0.2%). We note that each of the coprecipitated proteins is present in a different relative amount in the precipitated com-plex. This may reflect the conditions for coprecipitation, the nature of binding within the complex (affinity), or the stoichi-ometric ratio of binding.
Infected-cell cDNA libraries constructed at different times p.i. were screened for protein-protein interactions by using the yeast two-hybrid system. Because nuclear localization is a req-uisite event for successful detection of yeast two-hybrid pro-tein-protein interactions and hydrophobic regions are known to interfere with this transport, the library was screened with a construct which effectively deleted the hydrophobic N-terminal domain of ODV-E66 (amino acids 2 to 23). These screens detected positive interactions between E66 and ODV-E25 and between ODV-E66 and FP25K at 18 h p.i., while at 24 h p.i. a positive interaction between E66 and ODV-E25 was detected. Similar yeast two-hybrid analyses also sug-gested protein-protein interactions between ODV-E66 and p39 and between FP25K and BV/ODV-E26. A summary of yeast two-hybrid results and coprecipitation data is shown in Table 1.
DISCUSSION
The amino acid sequence of FP25K is highly conserved among baculoviruses. AcMNPV andBombyx mori nucleopoly-hedrovirus have 96.7% overall similarity, and a comparison of all the sequenced FP25K genes show that the only region lacking significant conservation is the last 19 to 26 C-terminal amino acids (Fig. 1). Immunoblot analyses performed with purified AcMNPV ODV and BV show that FP25K is a struc-tural protein in the nucleocapsids of both progeny viruses (Fig. 2); however, immunofluorescence microscopy indicates that a large fraction of FP25K remains cytoplasmic throughout infec-tion (Fig. 7). Two major forms of FP25K are observed during infection and in purified virus: 25 and 23 kDa. While the 23-kDa form may correspond to translation at an internal Met, further analyses will be required to characterize this second form and clarify its functional significance.
[image:10.612.52.291.71.522.2]Computer analyses of the primary structure of FP25K pre-dicted two regions of possible structural relevance: a coiled-coil region and a putative actin binding helix (Fig. 1). Viral mutants lacking either of these regions were chosen for further FIG. 8. Immunoprecipitation analyses. Sf9 cells were infected with WT
Ac-MNPV and at the indicated times p.i., and cell lysates were prepared in the presence of either Tween 20 or NP-40. (A) Protein complexes were immuno-precipitated with antibody (Ab) to gp67 (lanes 2 to 5) or FP25K (lane 1). Lane 6, 30-h-p.i. cell lysate sample analyzed as positive control. (B) Protein complexes were immunoprecipitated with antibody to ODV-E66 and two different deter-gent conditions (0.2% Tween 20 [T20] or 1% NP-40). The immunoprecipitated protein complex (20l) was analyzed by SDS-PAGE, Western blotted to a polyvinylidene difluoride membrane, and reacted with antibodies to FP25K, p39, BV/ODV-E26, or ODV-E25. A 48-h-p.i. cell lysate was used as positive control (lane 1 for each sample).
TABLE 1. Summary of protein-protein interactions Protein paira Immunoprecipitation Yeast two-hybrid analysis
*E66-FP25K ⫹ ⫹(library screen)
*E66-p39 ⫹ ⫹(direct cross)
*E66-E25 ⫹ ⫹(library screen)
*E66-E26 ⫹ ⫺
FP25K-E26* ⫹ ⫹(direct cross)
FP25K-gp67* ⫹ ⫺
aAsterisks indicate the antibody used for the immunoprecipitation
experi-ment.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:10.612.313.553.83.158.2]study, and infection by either FP25K mutant resulted in a decreased amount of detectable ODV-E66 (Fig. 3). Immuno-fluorescence microscopy showed that during infection by both mutants, ODV-E66 displayed an altered pattern of intranu-clear localization. This effect was more dramatic during infec-tion by the 480-1 mutant, with infecinfec-tion resulting in a cytoplas-mic location of ODV-E66. In FP-gal-infected cells, some ODV-E66 was detected within the nucleus; however, this traf-ficking was delayed and diminished compared to that with WT infection (Fig. 5). Since the two mutants produced equivalent amounts of ODV-E66, the effect of the 480-1 mutation on nuclear transport of ODV-E66 is likely not related to a mini-mal protein requirement for initiation of nuclear transport. Additionally, while the amounts of ODV-E25 which accumu-late during WT and 480-1 infection are similar, nuclear trans-port of ODV-E25 was delayed by approximately 48 h, while intranuclear transport was not affected, in cells infected with FP-gal. These results suggest that the N-terminal region of FP25K may be important for normal trafficking of ODV-E66 and ODV-E25. Lack of the N-terminal region does not result in detectable relocalization of ODV envelope proteins to the plasma membrane (data not shown) or incorporation into BV (ODV-E66, ODV-E56, ODV-EC27, and ODV-E25) (Fig. 3F). While the mutant FP-gal virus was generated from the pa-rental E2 virus, 480-1 is a spontaneous, naturally occurring
FP25Kmutant virus, and it is possible that additional muta-tions are present (1).
Western blot analysis of a time course of infected cells showed that in addition to that of ODV-E66, accumulation levels of several baculovirus structural proteins were altered. Compared to those in WT-infected cells, the amounts of BV envelope proteins gp67 and BV/ODV-E26 and the major cap-sid protein p39 increased in mutant-infected cells. We note that the proteins which exhibited an altered temporal accumu-lation pattern during FP mutant infection (Fig. 3) were the same proteins that protein-protein interaction studies sug-gested interact with FP25K (Fig. 8 and Table 1). Results from immunoprecipitation studies suggest that FP25K may be a member of protein complexes which include gp67, ODV-E66, and BV/ODV-E26. While FP25K coprecipitates with each of these proteins, these proteins are not precipitated in the re-ciprocal experiment (with antibody to FP25K). This suggests that FP25K may be an integral component of these complexes, and when bound, FP25K is masked from antibody recognition. The interactions of FP25K and ODV-E66 and of FP25K and BV/ODV-E26 have been confirmed by using the complemen-tary yeast two-hybrid technique (Table 1) (3). However, a yeast two-hybrid cross between FP25K and gp67 was negative, sug-gesting that this interaction may be mediated by other proteins. Only the 25-kDa form is detected coprecipitating with gp67, ODV-E66, or BV/ODV-E26 (Fig. 8) (3), while antibody to FP25K precipitates both the 25- and 23-kDa forms. These data suggest that these two forms may be functionally distinct.
The data presented here indicate that in addition to the other, better-characterized effects of mutations within the
FP25Kgene, the accumulation patterns of several structural proteins are altered, and intranuclear transport of both ODV-E66 and ODV-E25 is compromised. We note that in the ab-sence of a fully functional FP25K gene, intranuclear local-ization of polyhedrin is also compromised during the early occlusion phase of infection (20). FP25K could be affecting protein accumulation and trafficking by regulating transcrip-tion, mRNA stability, translatranscrip-tion, or altered protein stability of one or many viral proteins. While transcription of ODV-E66 was not altered during an infection with the FP-gal virus (Fig. 4), FP mutant virus infection can alter steady-state levels of
polyhedrin (15). The phenotypic hallmarks of the FP mutant, including apparently normal numbers of nucleocapsids but lack of ODV envelopment and a decreased amount of viral occlusions, may not be due to the mutant FP25K protein per se but may result from improper stoichiometry, localization, or protein complex formation of other viral proteins. Studies aimed to further characterize the composition of protein com-plexes containing FP25K and their role in the trafficking path-way(s) of ODV envelope proteins are under way.
ACKNOWLEDGMENTS
We thank Gabriella Marcano and Shawn Williamson for the devel-opment of infected-cell cDNA yeast two-hybrid libraries. We thank Tao Hong, Gabriella Marcano, and Shawn Williamson for cloning genes into yeast two-hybrid vectors and Erin Pinkerton and Bill Keyes for their excellent technical support. We thank the following people for providing antisera: Christopher Richardson (Amgen Institute, To-ronto, Ontario, Canada) (p78/83), George Rohrmann (Oregon State University, Corvallis) (ODV-E25, p78/83), Loy Volkman (University of California, Berkeley) (capsid, gp64), and Peter Faulkner (Queen’s University, Kingston, Ontario, Canada) (gp41).
This work was supported in part by National Institutes of Health grant 2RO1GM47552 (to M.D.S. and S.C.B.) and Texas Agricultural Experiment Station Project TEXO8078 (to M.D.S.).
REFERENCES
1.Beames, B., and M. D. Summers.1988. Comparisons of host cell DNA insertions and altered transcription at the site of insertions in few polyhedra baculovirus mutants. Virology162:206–220.
2.Beames, B., and M. D. Summers.1989. Location and nucleotide sequence of the 25K protein missing from baculovirus few polyhedra (FP) mutants. Virology168:344–353.
3.Beniya, H., S. C. Braunagel, and M. D. Summers.1998.Autographa califor-nicanuclear polyhedrosis virus: subcellular localization and protein traffick-ing of BV/ODV-E26 to intranuclear membranes and viral envelopes. Virol-ogy240:64–75.
4.Bradford, M. M.1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem.72:248–254.
5.Braunagel, S. C., D. M. Elton, H. Ma, and M. D. Summers.1996. Identifi-cation and analysis of anAutographa californicanuclear polyhedrosis virus structural protein of the occlusion-derived virus envelope: ODV-E56. Virol-ogy217:97–110.
6.Braunagel, S. C., H. He, P. Ramamurthy, and M. D. Summers.1996. Tran-scription, translation, and cellular localization of threeAutographa califor-nicanuclear polyhedrosis virus structural proteins: ODV-E18, ODV-E35 and ODV-EC27. Virology222:100–114.
7.Braunagel, S. C., and M. D. Summers.1994.Autographa californicanuclear polyhedrosis virus, PDV, and ECV viral envelopes and nucleocapsids: struc-tural proteins, antigens, lipid and fatty acid profiles. Virology202:315–328. 8.Charlton, C. A., and L. E. Volkman.1991. Sequential rearrangement and nuclear polymerization of actin in baculovirus-infectedSpodoptera frugiperda cells. J. Virol.65:1219–1227.
9.Chirgwin, J. M., A. E. Przbyla, R. J. MacDonald, and W. J. Rutter.1979. Isolation of biologically active ribonucleic acid from sources enriched in ribonuclease. Biochemistry18:5294–5299.
10. Fraser, M. J., and W. F. Hink.1982. The isolation and characterization of the MP and FP plaque variants ofGalleria mellonellanuclear polyhedrosis virus. Virology117:366–378.
11. Fraser, M. J., and W. F. Hink.1982. Comparative sensitivity of several plaque assay techniques employing TN-368 and IPLB-SF 21AE insect cell lines for plaque variants ofGalleria mellonellanuclear polyhedrosis virus. J. Invert. Pathol.40:89–97.
12. Fraser, M. J., G. E. Smith, and M. D. Summers.1983. Acquisition of host cell DNA sequences by baculoviruses: relationship between host DNA in-sertions and FP mutants ofAutographa californicaandGalleria mellonella nuclear polyhedrosis virus. J. Virol.47:287–300.
13. Harrison, R. L., and M. D. Summers.1995. Mutations in theAutographa californicamultinucleocapsid nuclear polyhedrosis virus 25 kDa protein gene result in reduced virion occlusion, altered intranuclear envelopment and enhanced virus production. J. Gen. Virol.76:1451–1459.
14. Harrison, R. L., and M. D. Summers.1995. Biosynthesis and localization of theAutographa californica nuclear polyhedrosis virus 25K gene product. Virology208:279–288.
15. Harrison, R. L., D. L. Jarvis, and M. D. Summers.1996. The role of the AcMNPV 25K gene, “FP25,” in baculoviruspolhandp10expression. Virol-ogy226:34–46.
on November 9, 2019 by guest
http://jvi.asm.org/
16.Hink, W. F., and P. V. Vail.1973. A plaque assay for titration of alfalfa looper nuclear polyhedrosis virus in a cabbage looper (TN-368) cell line. J. Invert. Pathol.22:168–174.
17. Hom, L. G., and L. E. Volkman.1998. Preventing proteolytic artifacts in the baculovirus expression system. BioTechniques25:18–20.
18. Hong, T., S. C. Braunagel, and M. D. Summers.1994. Transcription, trans-lation, and cellular localization of PDV-E66: a structural protein of the PDV envelope ofAutographa californicanuclear polyhedrosis virus. Virology204:
210–222.
19. Hong, T., M. D. Summers, and S. C. Braunagel.1997. N-terminal sequences fromAutographa californicanuclear polyhedrosis virus envelope proteins ODV-E66 and ODV-E25 are sufficient to direct reporter proteins to the nuclear envelope, intranuclear microvesicles and the envelope of the occlu-sion derived virus. Proc. Natl. Acad. Sci. USA94:4050–4055.
20. Jarvis, D. L., D. A. Bohlmeyer, and A. Garcia.1992. Enhancement of poly-hedrin nuclear localization during baculovirus infection. J. Virol.66:6903– 6911.
21. Kumar, S., and L. K. Miller.1987. Effects of serial passage ofAutographa californicanuclear polyhedrosis virus in cell culture. Virus Res.7:335–349. 22. Laemmli, U. K.1970. Cleavage of structural proteins during assembly of the
head of bacteriophage T4. Nature227:680–695.
23. MacKinnon, E. A., J. F. Henderson, D. B. Stoltz, and P. Faulkner.1974. Morphogenesis of nuclear polyhedrosis virus under conditions of prolonged passagein vitro. J. Ultrastruct. Res.49:419–435.
24. Miller, D. W., and L. K. Miller.1982. A virus mutant with an insertion of a copia-like transposable element. Nature299:562–564.
25. Potter, K. N., P. Faulkner, and E. A. MacKinnon.1976. Strain selection
during serial passage ofTrichoplusia ninuclear polyhedrosis virus. J. Virol.
18:1040–1050.
26. Potter, K. N., R. P. Jaques, and P. Faulkner.1978. Modification of Trichop-lusia ninuclear polyhedrosis virus passagedin vivo. Intervirology9:76–85. 27. Ramoska, W. A., and W. F. Hink.1974. Electron microscope examination of
two plaque variants from a nuclear polyhedrosis virus of the alfalfa looper, Autographa californica. J. Invert. Pathol.23:197–201.
28. Russell, L. Q., and G. F. Rohrmann.1993. A 25-kDa protein is associated with the envelopes of occluded baculovirus virions. Virology195:532–540. 29. Sambrook, J., E. F. Fritsch, and T. Maniatis.1989. Molecular cloning: a
laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
30. Slavicek, J. M., N. Hayes-Plazolles, and M. E. Kelly.1995. Rapid formation of few polyhedra mutants ofLymantria disparmultinucleocapsid nuclear polyhedrosis virus during serial passage in cell culture. Biol. Control5:251– 261.
31. Summers, M. D., and G. E. Smith.1987. A manual of methods for baculo-virus vectors and insect cell culture procedures. Texas Agric. Exp. Station Bull.1555:10–18; 38–48.
32. Thiem, S. M., and L. K. Miller.1989. Identification, sequence, and tran-scriptional mapping of the major capsid protein gene of the baculovirus Autographa californicanuclear polyhedrosis virus. J. Virol.63:2008–2018. 33. Volkman, L. E., M. D. Summers, and C.-H. Hsieh.1976. Occluded and
nonoccluded nuclear polyhedrosis virus grown inTrichoplusia ni: compara-tive neutralization, comparacompara-tive infectivity, and in vitro growth studies. J. Vi-rol.19:820–832.