0022-538X/05/$08.00⫹0 doi:10.1128/JVI.79.2.955–965.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Novel Adeno-Associated Virus Vector Vaccine Restricts Replication
of Simian Immunodeficiency Virus in Macaques
Philip R. Johnson,
1* Bruce C. Schnepp,
1Mary J. Connell,
1Daniela Rohne,
2Suzanne Robinson,
2Georgia R. Krivulka,
2Carol I. Lord,
2Rebekah Zinn,
3David C. Montefiori,
3Norman L. Letvin,
2and K. Reed Clark
1Center for Gene Therapy, Columbus Children’s Research Institute, Columbus Children’s Hospital, and Department of Pediatrics, College of Medicine and Public Health, Ohio State University, Columbus, Ohio1; Division of Viral
Pathogenesis, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, Massachusetts2; and
Department of Surgery, Duke University Medical Center, Durham, North Carolina3
Received 6 July 2004/Accepted 3 September 2004
Gene transfer vectors based on recombinant adeno-associated virus (rAAV) are simple, versatile, and safe. While the conventional applications for rAAV vectors have focused on delivery of therapeutic genes, we have developed the system for delivery of vaccine antigens. In particular, we are interested in generating rAAV vectors for use as a prophylactic human immunodeficiency virus type 1 (HIV-1) vaccine. To that end, we constructed vaccine vectors that expressed genes from the simian immunodeficiency virus (SIV) for evaluation in the monkey SIV model. After a single intramuscular dose, rAAV/SIV vaccines elicited SIV-specific T cells and antibodies in macaques. Furthermore, immunized animals were able to significantly restrict replication of a live, virulent SIV challenge. These data suggest that rAAV vaccine vectors induced biologically relevant immune responses, and thus, warrant continued development as a viable HIV-1 vaccine candidate.
Vectors based on recombinant adeno-associated virus (rAAV) are unique among available gene transfer vehicles. Depending on the desired application, vectors can be designed for sustained expression of self-genes (so-called gene therapy) or for expression of nonself genes (vaccines). The former ap-plication is widely appreciated, with 18 protocols listed in the National Institutes of Health (NIH) Office of Biotechnology Activities Genetic Modification Clinical Research Information System (GeMCRIS) database of human gene transfer proto-cols (www.gemcris.od.nih.gov). However, the potential of rAAV vectors as vaccine vehicles has only recently been rec-ognized (17, 19, 28, 53, 66, 68, 71, 100, 111, 112).
Several important features make rAAV attractive as a vac-cine vector. First, the vector system is simple. Vector genomes do not contain endogenous wild-type AAV genes (see Fig. 1), and systems based on stable cell lines or large-scale transient transfection have been developed for commercial scale pro-duction (16). Second, rAAV vectors can be easily administered into muscle, an accessible and practical site for vaccine admin-istration. Importantly, muscle has been shown to be a highly susceptible target for rAAV transduction (1, 8, 19, 26, 27, 32, 34, 49, 50, 56, 90, 96, 98, 105, 110, 113). Finally, rAAV vectors have an outstanding safety profile in more than 8 years of evaluation in preclinical models and human clinical trials (22, 32, 35, 72, 77, 99, 102).
In the past several years, we have been interested in devel-oping the rAAV vector system as a vaccine for human immu-nodeficiency virus type 1 (HIV-1). In previous work, we showed that rAAV vectors efficiently expressed genes from
simian immunodeficiency virus (SIV) and that rAAV vectors engendered immune responses to a nonself transgene in ani-mals (19, 20). In the work reported herein, we present the results of immunogenicity and challenge trials in rhesus ma-caques. We have constructed a series of rAAV vectors that express SIV genes and used these vectors to immunize cohorts of macaques in three distinct studies. After immunization, an-imals were challenged with live, virulent SIV. Our results showed that rAAV/SIV vaccines were immunogenic after a single dose in macaques and that vaccinated animals signifi-cantly restricted replication of SIV after challenge.
MATERIALS AND METHODS
Animals.Rhesus macaques (Indian origin) were housed in the vivarium at the Columbus Children’s Research Institute in accordance with standards set forth by the American Association for Accreditation of Laboratory Animal Care. All animals were negative for antibodies to SIV, simian type D retrovirus, and simian T-cell lymphotropic virus type 1.
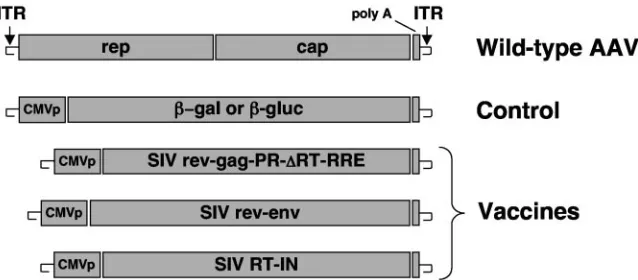
Vaccines.The genetic structures for the vaccines are depicted in Fig. 1 and are described further below. All SIV genes used in the vaccines were derived from an infectious molecular clone of SIVsm (SIVsm/H4) (51). rAAV vectors were constructed, produced, and quantified as previously described (16, 18). All rAAV vectors used in these studies were based on the AAV serotype 2 capsid and inverted terminal repeats (ITRs). Plasmid DNA (used in study C) was grown on a large scale in the Ohio State University Fermentor facility and purified using the Ultrapure 100 System (QIAGEN, Valencia, Calif.). Final preparations were tested for endotoxin levels using the Limulus Amebocyte Lysate assay (Cape
Cod, Falmouth, Mass.) and contained⬍0.7 endotoxin unit/g. All vaccines were
administered by deep intramuscular injection into the right quadriceps. For study A, the rAAV/SIV vaccine was a mixture of two vectors given as a single injection. One vector contained a subgenomic fragment of the SIVsm/H4 molecular clone (gag, protease, and part of the reverse transcriptase [RT]; nucleotides 1049 to 3053) flanked upstream by the first exon of rev and down-stream by the rev response element (RRE) and second exon of rev (rAAV/
SIVrev-gag-PR-⌬RT-RRE) (Fig. 1). Transcription was driven by the standard
human cytomegalovirus (CMV) immediate-early promoter. The polyadenylation signal was derived from simian virus 40. The second vector also contained a subgenomic fragment of the SIVsm/H4 molecular clone (nucleotides 6481 to
* Corresponding author. Mailing address: Columbus Children’s Hospital, Room WA3011, 700 Children’s Dr., Columbus, OH 43205. Phone: (614) 722-2735. Fax: (614) 722-3273. E-mail: johnsonp @pediatrics.ohio-state.edu.
955
on November 8, 2019 by guest
http://jvi.asm.org/
9208) that began with the first exon of rev and ended with the stop codon of gp160 (rAAV/SIVrev-env) (Fig. 1). Doses of the vaccine were based on quanti-fication of DNase-resistant particles (DRP), which are equivalent to
encapsi-dated vector genomes (18). For the first dose (time zero), animals received 1013
DRP of rAAV/SIVrev-gag-PR-⌬RT-RRE and 3.2 ⫻ 1012
DRP of rAAV/ SIVrev-env (given as a single injection). For the second dose (24 weeks), animals
received 1013
DRP of rAAV/SIVrev-gag-PR-⌬RT-RRE and 5⫻1012
DRP of rAAV/SIVrev-env (again as a single injection). Control animals were vaccinated
with an rAAV vector expressing theEscherichia coli-galactosidase gene (4⫻
1012DRP at time zero) followed by an rAAV vector expressing the murine
-glucuronidase gene (1013
DRP at 24 weeks). These vectors have been de-scribed elsewhere (19, 92).
For study B, the rAAV/SIV vaccine was a mixture of three vectors given as a single injection at time zero and again at 12 weeks. The same vaccines used in study A were used in study B. In addition, a third vector representing the remainder of RT and integrase (IN) (Fig. 1, SIV RT-IN; corresponds to nucle-otides 2839 to 5395 in SIVsm/H4) was constructed using synthetically derived codon-optimized sequences (Operon, Inc., Almeda, Calif.). SIV vaccine doses
for each immunization were as follows: rAAV/SIVrev-gag-PR-⌬RT-RRE, 1013
DRP; rAAV/RT-IN, 1013DRP; and rAAV/SIVrev-env, 5⫻1012DRP. The
control animals received rAAV/murine-glucuronidase (1013
DRP) at time zero and again at 12 weeks.
For study C, animals were immunized three times with plasmid DNA (either control or SIV DNA) and then given a booster dose of plasmid DNA (either control or SIV DNA) or a single dose of rAAV/SIV (see Table 1). SIV genes (as described above for studies A and B) were cloned into NotI restriction sites in
place of thelacZgene in pAAV/CMV/-gal. Thus, the plasmid DNAs
repre-sented exactly the same DNA sequences that were used to derive the rAAV vectors (including the ITRs). Each dose contained 9 mg of DNA representing a
mixture of three plasmid DNAs (gag-PR-⌬RT, RT-IN, and rev-env) at 3 mg
each. The DNA was dissolved in sterile phosphate-buffered saline. The control
DNA was pAAV/CMV/-gal given at a dose of 9 mg. The rAAV vaccines were
as described in study B.
Challenge virus.The SIV challenge virus used in these studies was SIVsm/ E660 (52, 55), which was kindly contributed by Vanessa Hirsch (National Insti-tute of Allergy and Infectious Diseases [NIAID], NIH). The titer of the challenge stock for infectivity in rhesus macaques had been previously determined. For
studies B and C, a standard intravenous inoculum of 50 MID50(50% infectious
dose in macaques) was used. For study A, a dose that represented approximately
2 MID50was given intravenously.
PCR assay for detection of SIV DNA sequences.Genomic DNA was isolated from peripheral blood mononuclear cells (PBMCs) or lymph node tissue using the QIAamp DNA mini kit (QIAGEN). Approximately 100 ng of DNA was used as the template in a nested PCR to detect sequences in the SIV RT gene or SIV long terminal repeat (LTR). The first round of PCR was performed using the Taq PCR core kit (QIAGEN) following the manufacturer’s protocol with the
SIV primer set (500 ng each) (SIVpolF [5⬘-AATGGCAGTTCATTGTATGAA
T-3⬘] and SIVpolR [5⬘-CTGCAAGTCCACCATGCCCATCC-3⬘) or the SIV
LTR primer set (SIVLTRF [5⬘-CTCTGCGGAGAGGCTGGC-3⬘] and SIVLTRR
[5⬘-GGGTCCTAACAGACCAGG-3⬘] ). Cycling conditions were as follows: (i) 3
min at 94°C; (ii) 30 cycles, with 1 cycle consisting of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C; and (iii) 10 min at 72°C. The second round of PCR was performed as
described above except that 1l of the first-round PCR product was used as the
template with the SIV RT nested primer set (500 ng each) (SIVpolNF [5⬘-GGGG
AGGAATAGGGGATATGAC-3⬘] and SIVpolNR [5⬘-TATGATGGGGCACAT
AACAAG-3⬘] ) or the SIV LTR nested primer set (SIVLTRNF [5⬘-GATTGAGC
CCTGGGAGGT-3⬘] and SIVLTRNR [5⬘-ACCAGGCGGCGACTAGGA-3⬘] ).
Tetramer and CTL assays.Gated CD3⫹CD8⫹T cells from anti-coagulated
whole blood were quantified for staining with the p11C, C-M (Gag), p54m (Env), or p68A (Pol) tetramers as previously described (31, 36, 65). Cytotoxic T
lym-phocytes (CTL) were functionally assessed by a standard51Cr release assay (31).
SIV neutralizing antibody assays.Serum neutralizing antibodies to SIVsm were determined as previously described (24, 80). For each determination, sera were tested against both SIVsm/H4 (the molecularly cloned virus that was the basis of the vaccine) and SIVsm/E660 (the challenge virus).
AAV neutralizing antibody assays.Serial dilutions of macaque sera were
incubated with rAAV2/-gal for 1 h at room temperature and were then added
to duplicate wells of C12 cells (20, 21) for another hour. After the inoculum was removed, adenovirus type 5 was added to each well, and the plates were
incu-bated for 18 h at 37o
C. Color was developed by adding the Pierce All-in-One Reagent. Plates were read with 405-nm-wavelength light.
Viral load assays.Quantification of SIV in plasma after challenge was deter-mined using the Bayer SIV branched DNA (bDNA) assay (97).
Statistical methods.The differences in viral loads between groups at various time points were determined by the Mann-Whitney test.
RESULTS
Study designs. Three immunization and challenge studies (studies A, B, and C) were performed in the SIV-macaque model. The design of each study is summarized in Table 1. In all three studies, rhesus macaques were immunized with rAAV2 vaccine vectors carrying either a control insert or SIV genes (Fig. 1). For study C, naked DNA as a priming vaccine was also evaluated. At various times after the last vaccine dose, each animal was challenged intravenously with SIVsm/E660, a well-characterized pathogenic SIV (52, 55). Immune responses and outcome after challenge for each study are described in the sections that follow.
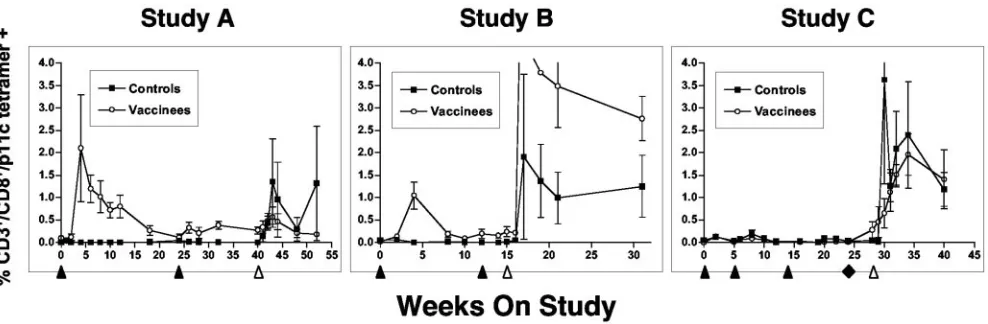
[image:2.585.134.453.69.209.2]Study A (low-dose challenge). Ten macaques were immu-nized as described in Table 1. In this study, 7 of 10 animals (all 5 SIV vaccinees and 2 of 5 controls) were selected for the major histocompatibility complex class I Mamu Aⴱ01 allele to facilitate analysis of antigen-specific T-cell responses using a tetramer based on the SIV gag epitope p11C, C-M (31). As
FIG. 1. Genetic composition of vaccines. All rAAV constructs were generated in the context of the AAV serotype 2 genome (shown as wild-type AAV). Transgenes were inserted as an expression cassette between the AAV ITRs as previously described (20). Transcription was driven by a standard human CMV promoter (CMVp) in each vaccine. Vector production, purification, and characterization were performed as previously described (18). The positions of the-galactosidase (-gal) and-glucuronidase (-gluc) genes are shown.
on November 8, 2019 by guest
http://jvi.asm.org/
shown in Fig. 2, in study A, no tetramer-positive cells were detected in the control group prior to challenge; after chal-lenge, both animals exhibited responses. All five vaccinees (all were Mamu Aⴱ01⫹) had measurable tetramer responses after the first rAAV/SIV immunization. However, a second vacci-nation 24 weeks later did not significantly increase the numbers of tetramer-positive cells. After challenge, none of the rAAV2/ SIV-vaccinated animals showed a significant increase in tet-ramer-positive cells.
To confirm that the tetramer-positive cells were functional, a portion of the fresh, unfractionated PBMCs from each time point were expanded in the presence of the p11C, C-M peptide and then divided into two pools. One pool was stained with tetramer, and the other pool was subjected to a standard51Cr
release assay using autologous B-lymphoblastoid cell lines (BLCL) derived from each individual animal prior to study enrollment. In each case, tetramer staining from fresh whole
blood correlated with significant expansion of tetramer-posi-tive CD8⫹ T cells after stimulation with peptide. Moreover, these stimulated CD8⫹T cells effectively killed target BLCL that had been pulsed with the p11C, C-M peptide.
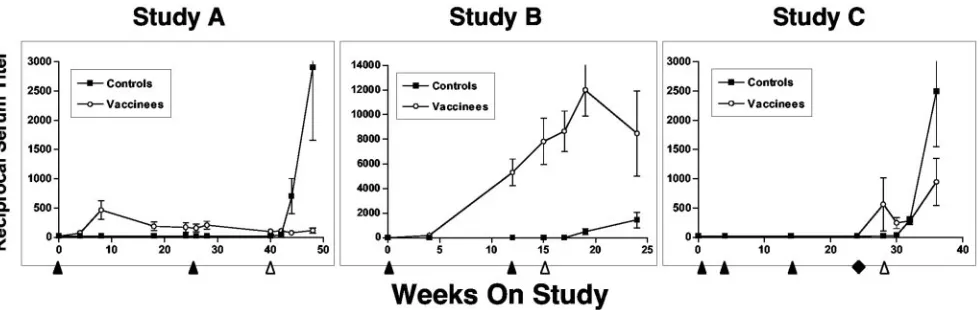
[image:3.585.42.551.81.225.2]Serum neutralizing antibodies against the vaccine virus (SIVsm/H4) were also measured and are shown in Fig. 3. None of the control animals responded before challenge infection, while the antibody titers for three of five animals were higher after challenge in study A. In contrast, all of the vaccinees had low but measurable titers after the first rAAV/SIV immuniza-tion that failed to increase after the second vaccinaimmuniza-tion 24 weeks later. Importantly, none of the vaccinated animals showed an increase in titer after challenge. None of the sera from vaccinated animals (or controls) neutralized the SIVsm/ E660 challenge virus on the challenge day. Likewise, no vac-cinees developed neutralizing antibodies (against SIVsm/H4 or SIVsm/E660) by 8 weeks after challenge. In contrast, three of
[image:3.585.47.541.496.658.2]FIG. 2. Antigen-specific T cells in immunized and challenge macaques. Gated CD3⫹CD8⫹T cells from fresh anti-coagulated whole blood were quantified for staining with the p11C, C-M tetramer as previously described (31, 36, 65). Only animals with the Mamu Aⴱ01 immunophenotype were analyzed. For study A, all five vaccinees and two of five control animals were Mamu Aⴱ01⫹. For study B, four of eight vaccinees and two of eight control animals were Mamu Aⴱ01⫹. In study C, two of eight animals in the control DNA group, two of four in the SIV DNA (only) group, and three of four in the SIV DNA plus rAAV2/SIV group were Mamu Aⴱ01⫹. Immunization (black arrowheads [below thexaxis]) and the time of SIV challenge (white arrowhead) are indicated. For study C, the black diamond indicates a final booster dose with either DNA (black squares) or rAAV/SIV (white circles). Data are depicted as means⫾standard errors of the means (error bars).
TABLE 1. Overview of SIV vaccine challenge studies
Study
Vaccine No. of
macaques
Timing (wk) Total study
duration (wk)
Priming Booster Immunizations Challenged
Aa Control rAAV2 Control rAAV2 5 0, 24 38 63
rAAV2/SIV rAAV2/SIV 5
Bb Control rAAV2 Control rAAV2 8 0, 12 15 68
rAAV2/SIV rAAV2/SIV 8
Cc Control DNA Control DNA 4 DNA prime (control or SIV) given at weeks 0, 5, and 14;
28 81
Control rAAV2 4
SIV DNA SIV DNA 4 DNA or rAAV boost per dose (control or SIV given at 24 weeks)
rAAV2/SIV 4
a
rAAV2/SIV vaccine contained a mixture of two rAAV particles, gag-PR-⌬RT and rev-gp160.
b
rAAV2/SIV vaccine contained a mixture of three rAAV particles, gag-PR-⌬RT, RT-IN, and rev-gp160.
c
SIV DNA vaccine was a mixture of three plasmids containing the same genes listed in footnoteb.
d
Challenge virus was SIVsm/E600 given intravenously. The dose in study A was 2 MID50. In studies B and C, the dose was 50 MID50.
on November 8, 2019 by guest
http://jvi.asm.org/
five control animals developed neutralizing antibodies against SIVsm/H4 after challenge, and one of these three animals also neutralized SIVsm/E660.
At 14 weeks after the second vaccination, all macaques were challenged intravenously with SIVsm/E660 (2 MID50). Viral
loads after challenge were monitored by plasma bDNA assays (Fig. 4, study A). In this study, three of five control animals became infected as judged by the bDNA assay; the fact that two animals failed to become infected was predictable because of the low challenge dose (2 MID50). Given that a dose of 1
MID50would be predicted to infect 50% of the animals (by
definition) and that a dilution of 2 MID50is probably within
the experimental error of dilution, one can conclude that ap-proximately half the animals should have become infected. The three control animals that had postchallenge SIV RNA in plasma were the same animals that showed postchallenge in-creases in SIV-specific antibodies. A fourth control animal
(T8) had a transient increase in antigen-specific T cells after challenge. We chose not to call this animal infected because: (i) SIV RNA was not detected in the plasma of this animal; (ii) SIV-specific antibodies were not detected after challenge; and (iii) we were unable to detect SIV DNA sequences in PBMCs collected 10 weeks after challenge or in lymph nodes (axillary and mesenteric) taken at elective necropsy 6 months after challenge. The three macaques with SIV RNA in plasma were all positive for SIV DNA in the same samples.
In contrast, none of the five vaccinated animals became in-fected as judged by the bDNA assay (Fig. 4, study A). Further-more, none of the vaccinees showed an increase in SIV-specific antibodies after challenge (Fig. 3, study A). Finally, we were unable to detect SIV DNA sequences in PBMCs collected 10 weeks after challenge or in lymph nodes (axillary and mesenteric) taken at elective necropsy 6 months after challenge.
When we applied the Fisher exact test to the observations
[image:4.585.50.540.69.224.2]FIG. 3. SIV serum neutralizing antibodies in immunized and challenge macaques. Serum neutralizing antibodies to SIVsm/H4 were determined as previously described (24, 80). The reciprocal serum titer is shown on the ordinate. Immunization (black arrowheads [below thexaxis]) and the time of SIV challenge (white arrowhead) are indicated. For study C, the black diamond indicates a final booster dose with either DNA (black squares) or rAAV/SIV (white circles). Data are depicted as means⫾standard errors of the means (error bars).
FIG. 4. Plasma viral loads after SIV challenge. SIV RNA copies (log10number of copies per milliliter) in plasma were quantified by the Bayer
SIV bDNA assay (97). Week 0 is the challenge day in each study. The values shown at each time point are the mean⫾standard error of the mean (error bar) for the group. A cross indicates that the animal died. The numbers in parentheses are the number of animals alive at the end of the study. For purposes of comparisons between groups, the peak was defined as highest bDNA in the first 4 weeks, while the set point was defined as the highest bDNA between 9 and 12 weeks.
on November 8, 2019 by guest
http://jvi.asm.org/
(zero of five infected animals versus three of five infected animals), thePvalue (P⫽0.617 in a two-sided test) failed to reach statistical significance. However, if we had included in the infected group the animal (T8) in the control group that had a transient T-cell response after challenge, then the P
value (zero of five animals versus four of five animals) would have reached statistical significance (P⫽0.048 in a two-sided test).
Study B (high-dose challenge).Sixteen macaques were im-munized as described in Table 1. In study B, 6 of the 16 macaques (2 controls and 4 vaccinees) were Mamu Aⴱ01⫹and could thus be analyzed by tetramer staining (Fig. 2). There were no p11C, C-M tetramer-positive cells in the control group prior to challenge. After challenge, both Mamu Aⴱ01⫹control macaques had significant tetramer-positive populations that persisted for at least 4 months after challenge. In the SIV vaccine group, all four Mamu Aⴱ01⫹macaques responded to the initial vaccine dose. Responses peaked at 4 weeks and then were intermittently positive up to the time of challenge. An anamnestic response was not observed after the second vacci-nation at week 12. This observation was consistent with study A where we also failed to see a booster effect on T-cell re-sponses after a second dose of vaccine. After challenge, all vaccinees showed significant increases in tetramer-positive cells.
Tetramer staining for two other Mamu Aⴱ01 restricted epitopes, p54m in Env and p68A in Pol, was also evaluated. All rAAV/SIV-vaccinated animals responded in a fashion similar to that observed for p11C, C-M (data not shown). Also, as for study A, pools of fresh PBMCs were expanded and tested for CTL. As expected, each animal had epitope-specific, Mamu Aⴱ01 restricted cytotoxic activity.
Serum neutralizing antibodies against the vaccine virus (SIVsm/H4) were also measured (Fig. 3, study B). As expected, control animals responded only after the challenge infection. All of the SIV vaccine recipients responded to the primary vaccination. After the second vaccination, no animal had a fourfold increase in titer indicative of an anamnestic response. These data are consistent with the lack of an anamnestic re-sponse in antigen-specific T cells. After challenge, an anam-nestic response was seen in a single animal, while titers either declined (three of eight animals) or remained stable (four of eight animals) in the remaining monkeys. As in study A, none of the sera from vaccinated animals (or control animals) sig-nificantly neutralized the SIVsm/E660 challenge virus on the challenge day. However, by 9 weeks after challenge, almost all animals (all eight vaccinees and six of eight controls) had demonstrable serum neutralizing activity against SIVsm/E660. At 3 weeks after the second vaccination (Table 1), all ma-caques were challenged intravenously with SIVsm/E660 (50 MID50), and all 16 animals became infected. However, there
were significant differences in the restriction of viral replication between the control and SIV vaccine groups (Fig. 4, study B). At peak virus replication, there was an average 1.3 log10
re-duction in the vaccine group compared to the control group (P ⫽0.03); at the set point, there was an average 1.9 log10
reduc-tion (P⫽0.04). By 16 weeks after challenge, three macaques in the control group had died of SIV-related illnesses, while none of the macaques in the SIV vaccine group died in the first
4 months. One macaque (H42) in the vaccine group died after routine anesthesia at 6 months.
Study C (priming with naked DNA and high-dose chal-lenge).Sixteen macaques were immunized as described in Ta-ble 1. This experiment included a series of three priming vac-cinations with naked DNA. Eight macaques were primed with control DNA, while another eight received DNA representing the same SIV sequences in the rAAV/SIV vaccine used in study B (SIV DNA). At 24 weeks, a single dose of rAAV/SIV was given as a booster vaccine to four macaques that had received SIV DNA; all other animals were given a fourth dose of DNA (SIV or control [Table 1]). In this study, 7 of the 16 macaques were Mamu Aⴱ01⫹ and were analyzed for p11C, C-M, and p54m tetramer responses: 2 were in the control DNA group, 2 were in the SIV DNA (only) group, and 3 were in the SIV DNA plus rAAV2/SIV group (Fig. 3, study C). None of the DNA vaccines (control or SIV) elicited significant p11C, C-M tetramer responses after any of the four doses. After the single dose of rAAV2/SIV at week 24, responses were observed in the three Mamu Aⴱ01⫹animals by the chal-lenge day. Interestingly, these responses were not anamnestic in nature as might have been expected after the SIV DNA priming. After challenge, three of four animals in the DNA group showed spikes in tetramer-positive cells that persisted through week 40 (12 weeks after challenge). Both animals that had received only SIV DNA spiked early after challenge in-fection, while a third animal that received control DNA peaked slightly later. In contrast, the responses after challenge in the group given a rAAV2/SIV booster dose were more gradual and plateaued out to week 40. Staining with the p54m-specific (Env) tetramer yielded results similar to those for p11C, C-M (data not shown).
For serum neutralizing antibodies (Fig. 3, study C), the con-trol and SIV DNA monkeys responded only after the challenge infection. In contrast, the animals in the group given a booster dose of rAAV2/SIV all had measurable titers by the challenge day. None of the sera from vaccinated animals (or controls) significantly neutralized the SIVsm/E660 challenge virus on the challenge day. After challenge, responses in the controls and SIV DNA groups were indistinguishable through 10 weeks after challenge. Responses in the group given a booster dose of rAAV2/SIV after challenge were less dramatic. By 8 weeks after challenge, 15 of 16 animals had demonstrable serum neutralizing activity against SIVsm/E660.
In this study, all 16 animals became infected after challenge (Fig. 4, study C). However, as in study B, there were significant differences in the restriction of viral replication among the groups. Because there was no statistical difference between the animals vaccinated with DNA (control and SIV DNA), these groups were combined for purposes of analysis. At peak virus replication, there was on average a 2.3 log10reduction in the
group given a booster dose of rAAV2/SIV compared to the combined group (P⫽0.001), and at the set point, there was an average 2.2 log10 reduction (P ⫽ 0.02). By 20 weeks after
challenge, two macaques in the control group had died of SIV-related illnesses. The rest of the animals remained healthy until the study was terminated at week 81 (53 weeks after challenge).
Neutralizing antibody responses to the rAAV vector. To ascertain the pre- and postimmunization immune responses to
on November 8, 2019 by guest
http://jvi.asm.org/
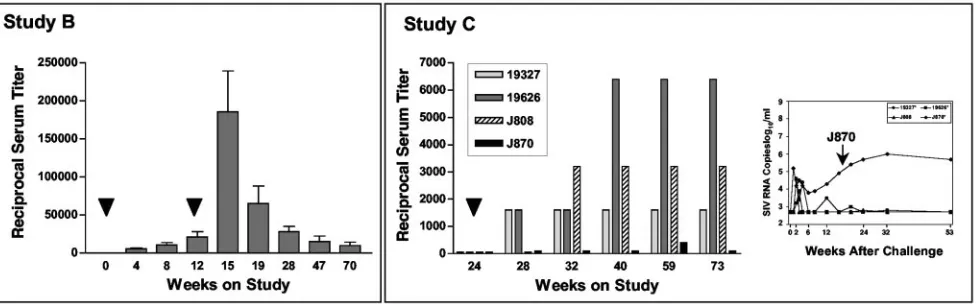
the rAAV vector, we measured serum neutralization titers against AAV2 at selected time points from studies B and C (Fig. 5). In study B, after the first dose of vaccine, titers rose gradually until week 12 (left panel). A second dose of vaccine at week 12 led to dramatic increases in the titer in all animals by week 15 that gradually diminished over the ensuing year.
In study C, a single dose of rAAV vaccine was given to four macaques at study week 24. Interestingly, three of four animals responded with the expected increase in AAV serum neutral-ization titer (Fig. 5, study C). The animal that failed to mount a significant anti-AAV response (J870) was also the only ani-mal in this group that failed to control virus replication after challenge (Fig. 5, study C, insert). These data suggested that either the injection into the muscle was incomplete or that this animal resisted full transduction for unknown reasons.
Effect of the Mamu Aⴱ01 allele.Recently published studies have suggested that Mamu Aⴱ01⫹macaques fare better after SIV and recombinant SIV-HIV (SHIV) infections than Mamu Aⴱ01⫺animals do (79, 115). Because each of our studies con-tained Mamu Aⴱ01⫹animals, we examined the potential in-fluence of Mamu Aⴱ01 on our results. In study A, all five vaccinees and two of five controls were Mamu Aⴱ01⫹(Fig. 2). Of the control animals, one of the two Mamu Aⴱ01 animals and two of three Mamu Aⴱ01⫺animals became infected. The small numbers of animals in each group make meaningful analysis difficult.
In study B, two of eight control animals and four of eight SIV vaccinees were Mamu Aⴱ01⫹. When segregated according to Mamu Aⴱ01 status, the overall bDNA levels of the Mamu Aⴱ01⫹and Mamu A01⫺animals were not distinguishable. In study C, three groups were analyzed. In the control animals (that received either non-SIV DNA or rAAV), there were two Mamu Aⴱ01⫹macaques and six Mamu Aⴱ01⫺macaques. Be-tween weeks 10 and 20, the Mamu Aⴱ01⫹animals appeared to fare better than the Mamu Aⴱ01⫺ animals (10- to 30-fold difference in SIV RNA). The same general observation was made for the animals that received the SIV DNA vaccine (two Mamu Aⴱ01⫹ animals and two Mamu Aⴱ01⫺ animals). In animals that received SIV DNA booster dose of rAAV/SIV
(three Mamu Aⴱ01⫹animals and one Mamu Aⴱ01⫺animal), the single animal that failed to control SIV replication was Mamu Aⴱ01⫹ (animal J870). Thus, for study C, if one re-stricted attention to only Mamu Aⴱ01⫺macaques, the data are difficult to interpret. This is because there was only one Mamu Aⴱ01⫺ animal in the group immunized with SIV DNA and given a booster dose of rAAV/SIV, and this animal effectively controlled SIV replication (while one of three Mamu Aⴱ01⫹ animals failed to control SIV replication).
DISCUSSION
rAAV/SIV vaccines are efficacious. In these studies, we showed that SIV vaccines based on rAAV2 vectors elicited robust immune responses (antibodies and antigen-specific CD8⫹T cells) after a single dose of vaccine. Moreover, bio-logic relevance was evident in three separate studies in which immunized animals demonstrated significant restriction of SIV replication after intravenous challenge compared to control animals. Study A was unusual in that none of the rAAV/SIV vaccine recipients became infected, whereas 60% of the con-trol animals did. This result was likely attributable to the low-dose challenge. Even though the results (zero of five vaccinees versus three of five control animals infected) did not reach statistical significance, this sort of experiment might reflect a more realistic view of the situation to be encountered in large-scale vaccine efficacy trials. Not surprisingly, when the chal-lenge dose was increased (study B), all the animals became infected. Nonetheless, the rAAV/SIV vaccine recipients fared significantly better than the control animals in terms of survival and total viral burden (Fig. 4). The best restriction of postchal-lenge SIV replication was observed in study C where naked DNA was tested as a priming vaccine. Even though the DNA vaccine was weakly immunogenic, postchallenge virus loads (compared to study B) suggested that the DNA priming doses had an effect. A group that received only the rAAV/SIV vac-cine was not included in study C, thereby hindering further analysis.
[image:6.585.51.539.72.224.2]It was not possible to precisely define immune correlates of
FIG. 5. AAV serum neutralizing antibodies in immunized macaques. Serum antibodies that neutralize AAV2 were measured before and after vaccination for animals in studies B and C. (Left) In study B, bars shown at each time point are the mean and standard deviation for the group of eight animals. Immunizations (black arrowheads) were given at 0 and 10 weeks; challenge was at week 15. (Right) Bars for the animals in study C are shown. The single rAAV2/SIV immunization (black arrowhead) was given at week 24; challenge was at week 28. The insert to the right shows the corresponding plasma SIV RNA levels for the same animals after SIV challenge. These bDNA values are also depicted in Fig. 4.
on November 8, 2019 by guest
http://jvi.asm.org/
protection in these studies. Since serum antibodies that neu-tralized the challenge virus stock were not detected in any of the vaccinated animals on the challenge day, it was tempting to assign the complete effect of the vaccines to antigen-specific CTL. While this is plausible for studies B and C, it was more difficult to attribute the sterilizing immunity in study A to CTL. However, given the low-dose challenge employed, it remains formally possible that CTL eliminated target cells that were infected after challenge.
Comparisons with other SIV vaccines. Our results can be most readily compared to other SIV vaccine studies where the challenge viruses were matched. In two related studies of SIV vaccines based on modified vaccinia virus Ankara (MVA) vec-tors, monkeys were challenged with the same stock of SIVsm/ E660 used in our studies (80, 91). In the first study, MVA/ SIVgag-pol (four doses) was used as the sole immunogen, while the second study compared four doses of MVA/SIVgag-pol alone, MVA/SIVenv alone, or MVA/SIVgag-MVA/SIVgag-pol-env. Analysis of viral loads after challenge in the second study (the first study had too few animals for meaningful analysis) showed that all of the vaccines effected a reduction in viral loads similar to the reduction we observed in studies B and C. In both of the MVA studies, increased immune responses were observed after the second, third, and fourth doses. In our studies A and B, no effect of a second dose was seen. Although not formally proven, it is likely that antivector immune re-sponses blocked the effect of a second dose of the same rAAV serotype.
A third published study using this same challenge virus eval-uated the efficacy of a plasmid DNA vaccine expressing the SIV gag gene; four doses (5 mg each) were given over 6 months (30). After intravenous challenge, all animals were infected, but those that received the SIV DNA appeared to restrict viral replication better than control animals, although the numbers of animals in each group prevented meaningful statistical anal-ysis.
It is more difficult to present meaningful comparisons with other published nonhuman primate vaccine studies. The major conundrum is how to account for differences in the challenge viruses. SHIV89.6P has been used by a number of investigators who have reported remarkably similar success in the reduction of viral loads and protection from acute death, CD4 T-cell loss, and AIDS (2, 3, 7, 9, 11–13, 25, 29, 57, 76, 84, 85, 95, 106, 108). The vaccine approaches used in these studies have included subunit proteins, DNA, MVA, adenovirus vectors, vesicular stomatitis virus vectors, and live attenuated SIVmac239⌬nef. Moreover, the vaccine constellations have varied widely, rang-ing from Tat protein to the entire SIV or SHIV genome. The remarkable consistency of efficacy in the face of such widely various regimens has caused some to question the SHIV89.6P model itself in terms of vaccine development (67, 81).
When contemplating a model system, one must consider the naturally occurring conditions that serve as the basis for mod-eling. Because most (⬎90%) HIV strains isolated during acute infection have the R5 phenotype, it makes sense that a vaccine will need to work against an R5 virus. SIVs use CCR5 almost exclusively, whereas SHIV89.P is a dual-tropic (R5X4) virus with a strong bias to X4 usage (39, 43–47, 81). The R5X4 phenotype is also typical of other highly pathogenic SHIV (43–47, 54, 93). Moreover, these dual-tropic SHIV display
sig-nificant biologic, pathological, and clinical differences com-pared to natural infection with SIV or HIV. It is also instruc-tive to consider the physical properties of the challenge virus, especially with respect to neutralization. The neutralization properties of pathogenic SIVs (including SIVsm/E660) are more typical of HIV-1 field isolates compared to SHIV89.6P, which is considered to be intermediate between T-cell-line-adapted TCLA strains and primary isolates (64, 80). Thus, any comparison between vaccine studies where the challenge vi-ruses differ so dramatically (SIV versus SHIV89.6P) is fraught with numerous and significant caveats.
rAAV vectors share features with plasmid DNA.rAAV vec-tors are unusual among engineered viruses used for gene trans-fer. In fact, for two important reasons, they are more like plasmid DNA vectors than other viral vectors. First, rAAV vectors do not contain any AAV genes. The only viral se-quences retained in the vector genome are the noncoding ITRs (145 nucleotides). Thus, no native viral proteins are synthe-sized after transduction, and the ensuing immune response is mostly driven by the characteristics of the transgene. Although there is clearly an immune response to the AAV capsid (Fig. 5), it is relatively noninflammatory (114), especially compared to other vectors like adenovirus where innate immune re-sponses to the capsid appear quickly (114). Interestingly, this latter observation suggests that rAAV vector vaccines might benefit from a chemical or molecular adjuvant like those used to improve plasmid DNA vaccines (5–7, 23, 41, 73, 89).
Second, like plasmid DNA, rAAV vectors do not integrate in vivo as part of their life cycle (26, 90). Vector genomes in vivo mostly take the form of concatameric circular episomes that comprise active transcriptional units (26, 90). These ob-servations are in contrast to the dogma that rAAV vectors integrate in vivo either randomly or in the AAVS1 locus. This concept is derived from work done over a decade ago which showed that wild-type AAV can integrate at a specific locus on human chromosome 19 in cultured transformed cells (58–61, 88). Importantly, this unusual and unique biologic property has never been demonstrated in human tissues. Further com-pounding the confusion is the finding that rAAV vector DNA has been shown to randomly integrate in cultured cells. The vast majority of these studies were performed with vectors encoding drug resistance markers that demonstrated integra-tion of rAAV vector in drug-resistant clones (48, 62, 74, 75, 87, 101, 103). Where rAAV vector persistence has been analyzed in the absence of selection in vitro, it appears that rAAV vectors integrate at a very low frequency. In fact, in the absence of selection, transformed cells in culture transduced with rAAV vectors have shown a loss of transgene expression over time (10, 69). The loss of transgene expression corresponded to a loss of rAAV vector DNA, suggesting that rAAV vectors persisted predominately as episomes that declined in a dividing cell population. A somewhat analogous situation appears to exist for plasmid DNA transfected into transformed cells in culture. There is a loss of plasmid DNA copies in cells over time due to the episomal nature of this DNA (33). However, under selective conditions, plasmid DNA has been shown to readily integrate in vitro, predominantly in head-to-tail arrays (107). Conversely, several in vivo studies have demonstrated that plasmid DNA persists only at very low levels after intra-muscular injection in mouse tissue (63, 70, 104). Thus, in the
on November 8, 2019 by guest
http://jvi.asm.org/
antigen) or some other mechanism, such as direct transduction of professional antigen-presenting cells.
Second-generation rAAV vaccine vectors. While our first-generation vaccines based on rAAV2 vectors are obviously useful, a number of enhancements are likely to improve im-munogenicity in second-generation vaccines. As noted above, selected adjuvants could be introduced either as exogenous agents or within the genetic constitution of the vector genome. Another prospective enhancement is augmentation of trans-duction by changing the AAV capsid used in the vaccine. The first-generation vaccine is based on the AAV serotype 2 capsid, but emerging data suggest that at least two other serotypes are superior to AAV2 at directing gene delivery and expression in muscle (14, 38, 50, 83, 86, 109). In fact, two groups have recently broken tolerance to a self protein after gene transfer using vectors based on the AAV1 capsid (15, 37). Our own preliminary experiments confirm that immunogenicity of rAAV vaccines is enhanced when an alternate capsid is used to package the vector genome.
A third type of enhancement for a second-generation vac-cine might be achieved by using a priming-booster strategy like that tested in study C. Our plasmid DNA vaccine was admit-tedly not optimized but still apparently provided some measure of priming. Other potential modalities for priming include other AAV serotypes (e.g., rAAV2 followed by rAAV1) or other unrelated viral vectors.
Preexisting immunity to AAV. In this study, we have not addressed the important issue of naturally occurring immunity to AAV that might block transduction by rAAV vectors. For some viral vectors, this issue has proven to be problematic (4, 40, 94). Although infection with AAV2 is ubiquitous, the actual levels of serum neutralizing antibodies appears to be relatively low in most individuals (38, 78; our unpublished data) The effect of such serum antibodies in clinical trials is currently unknown. For other serotypes (e.g., AAV1 or AAV5), sero-prevalence is dramatically lower, suggesting that these viruses do not circulate widely in humans. Thus, rAAV vectors based on these less prevalent serotypes provide a viable alternative to AAV2 if it appears that natural immunity in the general pop-ulation blocks vector transduction (42, 82).
rAAV vectors carrying a gene of interest can now be con-sidered viable vaccine modalities, not only for HIV, but also for other conditions where robust stimulation of the immune system is necessary for treatment or prevention of maladies, such as infectious diseases and cancer. Continued modifica-tions to the overall vector system will enhance the ability to engender biologically relevant immune responses.
Hulsey, J. Miller, H. M. McClure, J. M. McNicholl, B. Moss, and H. L. Robinson.2001. Control of a mucosal challenge and prevention of AIDS by
a multiprotein DNA/MVA vaccine. Science292:69–74.
3.Amara, R. R., F. Villinger, S. I. Staprans, J. D. Altman, D. C. Montefiori, N. L. Kozyr, Y. Xu, L. S. Wyatt, P. L. Earl, J. G. Herndon, H. M. McClure, B. Moss, and H. L. Robinson.2002. Different patterns of immune responses but similar control of a simian-human immunodeficiency virus 89.6P mu-cosal challenge by modified vaccinia virus Ankara (MVA) and DNA/MVA
vaccines. J. Virol.76:7625–7631.
4.Aste-Amezaga, M., A. J. Bett, F. Wang, D. R. Casimiro, J. M. Antonello, D. K. Patel, E. C. Dell, L. L. Franlin, N. M. Dougherty, P. S. Bennett, H. C. Perry, M. E. Davies, J. W. Shiver, P. M. Keller, and M. D. Yeager.2004. Quantitative adenovirus neutralization assays based on the secreted alka-line phosphatase reporter gene: application in epidemiologic studies and in
the design of adenovector vaccines. Hum. Gene Ther.15:293–304.
5.Barouch, D. H., T. M. Fu, D. C. Montefiori, M. G. Lewis, J. W. Shiver, and N. L. Letvin. 2001. Vaccine-elicited immune responses prevent clinical
AIDS in SHIV(89.6P)-infected rhesus monkeys. Immunol. Lett.79:57–61.
6.Barouch, D. H., and N. L. Letvin.2000. Cytokine-induced augmentation of DNA vaccine-elicited SIV-specific immunity in rhesus monkeys. Dev. Biol.
104:85–92.
7.Barouch, D. H., S. Santra, J. E. Schmitz, M. J. Kuroda, T. M. Fu, W. Wagner, M. Bilska, A. Craiu, X. X. Zheng, G. R. Krivulka, K. Beaudry, M. A. Lifton, C. E. Nickerson, W. L. Trigona, K. Punt, D. C. Freed, L. Guan, S. Dubey, D. Casimiro, A. Simon, M. E. Davies, M. Chastain, T. B. Strom, R. S. Gelman, D. C. Montefiori, M. G. Lewis, E. A. Emini, J. W. Shiver, and N. L. Letvin.2000. Control of viremia and prevention of clinical AIDS in
rhesus monkeys by cytokine-augmented DNA vaccination. Science290:
486–492.
8.Bartlett, R. J., and J. M. McCue.2000. Adeno-associated virus based gene
therapy in skeletal muscle. Methods Mol. Biol.133:127–156.
9.Bertley, F. M., P. A. Kozlowski, S. W. Wang, J. Chappelle, J. Patel, O. Sonuyi, G. Mazzara, D. Montefiori, A. Carville, K. G. Mansfield, and A. Aldovini.2004. Control of simian/human immunodeficiency virus viremia and disease progression after IL-2-augmented DNA-modified vaccinia virus
Ankara nasal vaccination in nonhuman primates. J. Immunol.172:3745–
3757.
10.Bertran, J., J. L. Miller, Y. Yang, A. Fenimore-Justman, F. Rueda, E. F. Vanin, and A. W. Nienhuis.1996. Recombinant adeno-associated virus-mediated high-efficiency, transient expression of the murine cationic amino acid transporter (ecotropic retroviral receptor) permits stable transduction
of human HeLa cells by ecotropic retroviral vectors. J. Virol.70:6759–6766.
11.Cafaro, A., A. Caputo, C. Fracasso, M. T. Maggiorella, D. Goletti, S. Baroncelli, M. Pace, L. Sernicola, M. L. Koanga-Mogtomo, M. Betti, A. Borsetti, R. Belli, L. Akerblom, F. Corrias, S. Butto, J. Heeney, P. Verani, F. Titti, and B. Ensoli.1999. Control of SHIV-89.6P-infection of
cynomol-gus monkeys by HIV-1 Tat protein vaccine. Nat. Med.5:643–650.
12.Cafaro, A., A. Caputo, M. T. Maggiorella, S. Baroncelli, C. Fracasso, M. Pace, A. Borsetti, L. Sernicola, D. R. Negri, P. Ten Haaft, M. Betti, Z. Michelini, I. Macchia, E. Fanales-Belasio, R. Belli, F. Corrias, S. Butto, P. Verani, F. Titti, and B. Ensoli.2000. SHIV89.6P pathogenicity in cynomol-gus monkeys and control of viral replication and disease onset by human
immunodeficiency virus type 1 Tat vaccine. J. Med. Primatol.29:193–208.
13.Cafaro, A., F. Titti, C. Fracasso, M. T. Maggiorella, S. Baroncelli, A. Caputo, D. Goletti, A. Borsetti, M. Pace, E. Fanales-Belasio, B. Ridolfi, D. R. Negri, L. Sernicola, R. Belli, F. Corrias, I. Macchia, P. Leone, Z. Michelini, P. ten Haaft, S. Butto, P. Verani, and B. Ensoli.2001. Vaccina-tion with DNA containing tat coding sequences and unmethylated CpG motifs protects cynomolgus monkeys upon infection with simian/human
immunodeficiency virus (SHIV89.6P). Vaccine19:2862–2877.
14.Chao, H., Y. Liu, J. Rabinowitz, C. Li, R. J. Samulski, and C. E. Walsh.
2000. Several log increase in therapeutic transgene delivery by distinct
adeno-associated viral serotype vectors. Mol. Ther.2:619–623.
15.Chenuaud, P., T. Larchet, J. E. Rabinowitz, N. Provost, Y. Cherel, N.
on November 8, 2019 by guest
http://jvi.asm.org/
Casadevall, R. J. Samulski, and P. Moullier.2004. Autoimmune anemia in
macaques following erythropoietin gene therapy. Blood103:3303–3304.
16.Clark, K. R.2002. Recent advances in recombinant adeno-associated virus
vector production. Kidney Int.61(Suppl. 1):9–15.
17.Clark, K. R., and P. R. Johnson. 2001. Gene delivery of vaccines for
infectious disease. Curr. Opin. Mol. Ther.3:375–384.
18.Clark, K. R., X. Liu, J. P. McGrath, and P. R. Johnson.1999. Highly purified recombinant adeno-associated virus vectors are biologically active and free of detectable helper and wild-type viruses. Hum. Gene Ther.
10:1031–1039.
19.Clark, K. R., T. J. Sferra, and P. R. Johnson.1997. Recombinant adeno-associated viral vectors mediate long-term transgene expression in muscle.
Hum. Gene Ther.8:659–669.
20.Clark, K. R., F. Voulgaropoulou, D. M. Fraley, and P. R. Johnson.1995. Cell lines for the production of recombinant adeno-associated virus. Hum.
Gene Ther.6:1329–1341.
21.Clark, K. R., F. Voulgaropoulou, and P. R. Johnson.1996. A stable cell line carrying adenovirus-inducible rep and cap genes allows for infectivity
titra-tion of adeno-associated virus vectors. Gene Ther.3:1124–1132.
22.Conrad, C. K., S. S. Allen, S. A. Afione, T. C. Reynolds, S. E. Beck, M. Fee-Maki, X. Barrazza-Ortiz, R. Adams, F. B. Askin, B. J. Carter, W. B. Guggino, and T. R. Flotte.1996. Safety of single-dose administration of an adeno-associated virus (AAV)-CFTR vector in the primate lung. Gene
Ther.3:658–668.
23.Craiu, A., D. H. Barouch, X. X. Zheng, M. J. Kuroda, J. E. Schmitz, M. A. Lifton, T. D. Steenbeke, C. E. Nickerson, K. Beaudry, J. D. Frost, K. A. Reimann, T. B. Strom, and N. L. Letvin.2001. An IL-2/Ig fusion protein
influences CD4⫹T lymphocytes in naive and simian immunodeficiency
virus-infected rhesus monkeys. AIDS Res. Hum. Retrovir.17:873–886.
24.Davis, N. L., I. J. Caley, K. W. Brown, M. R. Betts, D. M. Irlbeck, K. M. McGrath, M. J. Connell, D. C. Montefiori, J. A. Frelinger, R. Swanstrom, P. R. Johnson, and R. E. Johnston.2000. Vaccination of macaques against pathogenic simian immunodeficiency virus with Venezuelan equine
en-cephalitis virus replicon particles. J. Virol.74:371–378.
25.Doria-Rose, N. A., C. Ohlen, P. Polacino, C. C. Pierce, M. T. Hensel, L. Kuller, T. Mulvania, D. Anderson, P. D. Greenberg, S. L. Hu, and N. L. Haigwood.2003. Multigene DNA priming-boosting vaccines protect
ma-caques from acute CD4⫹-T-cell depletion after simian-human
immunode-ficiency virus SHIV89.6P mucosal challenge. J. Virol.77:11563–11577.
26.Duan, D., P. Sharma, J. Yang, Y. Yue, L. Dudus, Y. Zhang, K. J. Fisher, and J. F. Engelhardt.1998. Circular intermediates of recombinant adeno-asso-ciated virus have defined structural characteristics responsible for long-term
episomal persistence in muscle tissue. J. Virol.72:8568–8577. (Erratum
73:861, 1999.)
27.Duan, D., Z. Yan, Y. Yue, W. Ding, and J. F. Engelhardt.2001. Enhance-ment of muscle gene delivery with pseudotyped adeno-associated virus type
5 correlates with myoblast differentiation. J. Virol.75:7662–7671.
28.During, M. J., C. W. Symes, P. A. Lawlor, J. Lin, J. Dunning, H. L. Fitzsimons, D. Poulsen, P. Leone, R. Xu, B. L. Dicker, J. Lipski, and D. Young.2000. An oral vaccine against NMDAR1 with efficacy in
experimen-tal stroke and epilepsy. Science287:1453–1460.
29.Earl, P. L., L. S. Wyatt, D. C. Montefiori, M. Bilska, R. Woodward, P. D. Markham, J. D. Malley, T. U. Vogel, T. M. Allen, D. I. Watkins, N. Miller, and B. Moss.2002. Comparison of vaccine strategies using recombinant env-gag-pol MVA with or without an oligomeric Env protein boost in the
SHIV rhesus macaque model. Virology294:270–281.
30.Egan, M. A., W. A. Charini, M. J. Kuroda, J. E. Schmitz, P. Racz, K. Tenner-Racz, K. Manson, M. Wyand, M. A. Lifton, C. E. Nickerson, T. Fu, J. W. Shiver, and N. L. Letvin.2000. Simian immunodeficiency virus (SIV) gag DNA-vaccinated rhesus monkeys develop secondary cytotoxic T-lym-phocyte responses and control viral replication after pathogenic SIV
infec-tion. J. Virol.74:7485–7495.
31.Egan, M. A., M. J. Kuroda, G. Voss, J. E. Schmitz, W. A. Charini, C. I. Lord, M. A. Forman, and N. L. Letvin.1999. Use of major
histocompati-bility complex class I/peptide/2M tetramers to quantitate CD8⫹cytotoxic
T lymphocytes specific for dominant and nondominant viral epitopes in simian-human immunodeficiency virus-infected rhesus monkeys. J. Virol.
73:5466–5472.
32.Favre, D., N. Provost, V. Blouin, G. Blancho, Y. Cherel, A. Salvetti, and P. Moullier.2001. Immediate and long-term safety of recombinant adeno-associated virus injection into the nonhuman primate muscle. Mol. Ther.
4:559–566.
33.Felgner, P. L., and G. M. Ringold.1989. Cationic liposome-mediated
trans-fection. Nature337:387–388.
34.Fisher, K. J., K. Jooss, J. Alston, Y. Yang, S. E. Haecker, K. High, R. Pathak, S. E. Raper, and J. M. Wilson.1997. Recombinant
adeno-associ-ated virus for muscle directed gene therapy. Nat. Med.3:306–312.
35.Flotte, T. R., P. L. Zeitlin, T. C. Reynolds, A. E. Heald, P. Pedersen, S. Beck, C. K. Conrad, L. Brass-Ernst, M. Humphries, K. Sullivan, R. Wetzel, G. Taylor, B. J. Carter, and W. B. Guggino.2003. Phase I trial of intranasal and endobronchial administration of a recombinant adeno-associated virus
serotype 2 (rAAV2)-CFTR vector in adult cystic fibrosis patients: a
two-part clinical study. Hum. Gene Ther.14:1079–1088.
36.Furchner, M., A. L. Erickson, T. Allen, D. I. Watkins, A. Sette, P. R. Johnson, and C. M. Walker. 1999. The simian immunodeficiency virus
envelope glycoprotein contains two epitopes presented by the Mamu-Aⴱ01
class I molecule. J. Virol.73:8035–8039.
37.Gao, G., C. Lebherz, D. J. Weiner, R. Grant, R. Calcedo, B. McCullough, A. Bagg, Y. Zhang, and J. M. Wilson.2004. Erythropoietin gene therapy leads
to autoimmune anemia in macaques. Blood103:3300–3302.
38.Gao, G. P., M. R. Alvira, L. Wang, R. Calcedo, J. Johnston, and J. M. Wilson.2002. Novel adeno-associated viruses from rhesus monkeys as
vec-tors for human gene therapy. Proc. Natl. Acad. Sci. USA99:11854–11859.
39.Glushakova, S., Y. Yi, J. C. Grivel, A. Singh, D. Schols, E. De Clercq, R. G. Collman, and L. Margolis.1999. Preferential coreceptor utilization and cytopathicity by dual-tropic HIV-1 in human lymphoid tissue ex vivo.
J. Clin. Investig.104:R7–R11.
40.Graham, B. S.2002. Clinical trials of HIV vaccines. Annu. Rev. Med.
53:207–221.
41.Gurunathan, S., D. M. Klinman, and R. A. Seder.2000. DNA vaccines:
immunology, application, and optimization. Annu. Rev. Immunol.18:927–
974.
42.Halbert, C. L., E. A. Rutledge, J. M. Allen, D. W. Russell, and A. D. Miller.
2000. Repeat transduction in the mouse lung by using adeno-associated
virus vectors with different serotypes. J. Virol.74:1524–1532.
43.Harouse, J. M., A. Gettie, T. Eshetu, R. C. Tan, R. Bohm, J. Blanchard, G. Baskin, and C. Cheng-Mayer.2001. Mucosal transmission and induction of simian AIDS by CCR5-specific simian/human immunodeficiency virus SHIVSF162P3. J. Virol.75:1990–1995.
44.Harouse, J. M., A. Gettie, R. C. Tan, J. Blanchard, and C. Cheng-Mayer.
1999. Distinct pathogenic sequela in rhesus macaques infected with CCR5
or CXCR4 utilizing SHIVs. Science284:816–819.
45.Harouse, J. M., A. Gettie, R. C. Tan, T. Eshetu, M. Ratterree, J. Blanchard, and C. Cheng-Mayer. 2001. Pathogenic determinants of the mucosally transmissible CXCR4-specific SHIV(SF33A2) map to env region. J.
Ac-quir. Immune Defic. Syndr.27:222–228.
46.Harouse, J. M., R. C. Tan, A. Gettie, P. Dailey, P. A. Marx, P. A. Luciw, and C. Cheng-Mayer.1998. In vitro infection of primate PBMC with simian/ human immunodeficiency virus, SHIV(SF33A): correlation to in vivo
out-come. J. Med. Primatol.27:81–86.
47.Harouse, J. M., R. C. Tan, A. Gettie, P. Dailey, P. A. Marx, P. A. Luciw, and C. Cheng-Mayer.1998. Mucosal transmission of pathogenic
CXCR4-utiliz-ing SHIVSF33A variants in rhesus macaques. Virology248:95–107.
48.Hermonat, P. L., and N. Muzyczka.1984. Use of adeno-associated virus as a mammalian DNA cloning vector: transduction of neomycin resistance
into mammalian tissue culture cells. Proc. Natl. Acad. Sci. USA81:6466–
6470.
49.Herzog, R. W.2004. AAV-mediated gene transfer to skeletal muscle.
Meth-ods Mol. Biol.246:179–194.
50.Hildinger, M., A. Auricchio, G. Gao, L. Wang, N. Chirmule, and J. M. Wilson.2001. Hybrid vectors based on adeno-associated virus serotypes 2
and 5 for muscle-directed gene transfer. J. Virol.75:6199–6203.
51.Hirsch, V. M., R. A. Olmsted, M. Murphey-Corb, R. H. Purcell, and P. R. Johnson.1989. An African primate lentivirus (SIVsm) closely related to
HIV-2. Nature339:389–392.
52.Hirsch, V. M., P. M. Zack, A. P. Vogel, and P. R. Johnson.1991. Simian immunodeficiency virus infection of macaques: end-stage disease is char-acterized by widespread distribution of proviral DNA in tissues. J. Infect.
Dis.163:976–988.
53.Hunt, S.2001. Technology evaluation: anticancer gene therapy, MediGene.
Curr. Opin. Mol. Ther.3:509–514.
54.Igarashi, T., C. R. Brown, Y. Endo, A. Buckler-White, R. Plishka, N. Bischofberger, V. Hirsch, and M. A. Martin.2001. Macrophage are the principal reservoir and sustain high virus loads in rhesus macaques after the
depletion of CD4⫹T cells by a highly pathogenic simian immunodeficiency
virus/HIV type 1 chimera (SHIV): implications for HIV-1 infections of
humans. Proc. Natl. Acad. Sci. USA98:658–663.
55.Johnson, P. R., D. C. Montefiori, S. Goldstein, T. E. Hamm, J. Zhou, S. Kitov, N. L. Haigwood, L. Misher, W. T. London, J. L. Gerin, A. Allison, R. H. Purcell, R. M. Chanock, and V. M. Hirsch.1992. Inactivated whole-virus vaccine derived from a proviral DNA clone of simian immunodefi-ciency virus induces high levels of neutralizing antibodies and confers
pro-tection against heterologous challenge. Proc. Natl. Acad. Sci. USA 89:
2175–2179.
56.Kessler, P. D., G. M. Podsakoff, X. Chen, S. A. McQuiston, P. C. Colosi, L. A. Matelis, G. J. Kurtzman, and B. J. Byrne.1996. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a
therapeutic protein. Proc. Natl. Acad. Sci. USA93:14082–14087.
57.Kim, J. J., J. S. Yang, L. K. Nottingham, D. J. Lee, M. Lee, K. H. Manson, M. S. Wyand, J. D. Boyer, K. E. Ugen, and D. B. Weiner.2001. Protection from immunodeficiency virus challenges in rhesus macaques by
multicom-ponent DNA immunization. Virology285:204–217.
on November 8, 2019 by guest
http://jvi.asm.org/
63.Ledwith, B. J., S. Manam, P. J. Troilo, A. B. Barnum, C. J. Pauley, T. G. Griffiths II, L. B. Harper, C. M. Beare, W. J. Bagdon, and W. W. Nichols.
2000. Plasmid DNA vaccines: investigation of integration into host cellular
DNA following intramuscular injection in mice. Intervirology43:258–272.
64.Letvin, N. L., S. Robinson, D. Rohne, M. K. Axthelm, J. W. Fanton, M. Bilska, T. J. Palker, H. X. Liao, B. F. Haynes, and D. C. Montefiori.2001. Vaccine-elicited V3 loop-specific antibodies in rhesus monkeys and control of a simian-human immunodeficiency virus expressing a primary patient
human immunodeficiency virus type 1 isolate envelope. J. Virol.75:4165–
4175.
65.Letvin, N. L., J. E. Schmitz, H. L. Jordan, A. Seth, V. M. Hirsch, K. A. Reimann, and M. J. Kuroda.1999. Cytotoxic T lymphocytes specific for the
simian immunodeficiency virus. Immunol. Rev.170:127–134.
66.Lewis, A. D., R. Chen, D. C. Montefiori, P. R. Johnson, and K. R. Clark.
2002. Generation of neutralizing activity against human immunodeficiency
virus type 1 in serum by antibody gene transfer. J. Virol.76:8769–8775.
67.Lifson, J. D., and M. A. Martin.2002. One step forwards, one step back.
Nature415:272–273.
68.Liu, D. W., Y. P. Tsao, J. T. Kung, Y. A. Ding, H. K. Sytwu, X. Xiao, and S. L. Chen.2000. Recombinant adeno-associated virus expressing human papillomavirus type 16 E7 peptide DNA fused with heat shock protein
DNA as a potential vaccine for cervical cancer. J. Virol.74:2888–2894.
69.Malik, P., S. A. McQuiston, X. J. Yu, K. A. Pepper, W. J. Krall, G. M. Podsakoff, G. J. Kurtzman, and D. B. Kohn.1997. Recombinant adeno-associated virus mediates a high level of gene transfer but less efficient
integration in the K562 human hematopoietic cell line. J. Virol.71:1776–
1783.
70.Manam, S., B. J. Ledwith, A. B. Barnum, P. J. Troilo, C. J. Pauley, L. B. Harper, T. G. Griffiths II, Z. Niu, L. Denisova, T. T. Follmer, S. J. Pac-chione, Z. Wang, C. M. Beare, W. J. Bagdon, and W. W. Nichols.2000. Plasmid DNA vaccines: tissue distribution and effects of DNA sequence, adjuvants and delivery method on integration into host DNA. Intervirology
43:273–281.
71.Manning, W. C., X. Paliard, S. Zhou, M. P. Bland, A. Y. Lee, K. Hong, C. M. Walker, J. A. Escobedo, and V. Dwarki.1997. Genetic immunization with adeno-associated virus vectors expressing herpes simplex virus type 2
gly-coproteins B and D. J. Virol.71:7960–7962.
72.Manno, C. S., A. J. Chew, S. Hutchison, P. J. Larson, R. W. Herzog, V. R. Arruda, S. J. Tai, M. V. Ragni, A. Thompson, M. Ozelo, L. B. Couto, D. G. Leonard, F. A. Johnson, A. McClelland, C. Scallan, E. Skarsgard, A. W. Flake, M. A. Kay, K. A. High, and B. Glader.2003. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B.
Blood101:2963–2972.
73.McKay, P. F., J. E. Schmitz, D. H. Barouch, M. J. Kuroda, M. A. Lifton, C. E. Nickerson, D. A. Gorgone, and N. L. Letvin.2002. Vaccine protection against functional CTL abnormalities in simian human immunodeficiency
virus-infected rhesus monkeys. J. Immunol.168:332–337.
74.McLaughlin, S. K., P. Collis, P. L. Hermonat, and N. Muzyczka.1988. Adeno-associated virus general transduction vectors: analysis of proviral
structures. J. Virol.62:1963–1973.
75.Miller, D. G., E. A. Rutledge, and D. W. Russell.2002. Chromosomal effects
of adeno-associated virus vector integration. Nat. Genet.30:147–148.
76.Miyake, A., T. Akagi, Y. Enose, M. Ueno, M. Kawamura, R. Horiuchi, K. Hiraishi, M. Adachi, T. Serizawa, O. Narayan, M. Akashi, M. Baba, and M. Hayami.2004. Induction of HIV-specific antibody response and protection against vaginal SHIV transmission by intranasal immunization with
inacti-vated SHIV-capturing nanospheres in macaques. J. Med. Virol.73:368–
377.
77.Monahan, P. E., K. Jooss, and M. S. Sands.2002. Safety of adeno-associ-ated virus gene therapy vectors: a current evaluation. Expert Opin. Drug
Safety1:79–91.
78.Moss, R. B., D. Rodman, L. T. Spencer, M. L. Aitken, P. L. Zeitlin, D. Waltz, C. Milla, A. S. Brody, J. P. Clancy, B. Ramsey, N. Hamblett, and A. E. Heald.2004. Repeated adeno-associated virus serotype 2 aerosol-mediated cystic fibrosis transmembrane regulator gene transfer to the lungs
against vaginal challenge with a pathogenic R5 simian/human immunode-ficiency virus at serum levels giving complete neutralization in vitro. J.
Vi-rol.75:8340–8347.
82.Peden, C. S., C. Burger, N. Muzyczka, and R. J. Mandel.2004. Circulating anti-wild-type adeno-associated virus type 2 (AAV2) antibodies inhibit recombinant AAV2 (rAAV2)-mediated, but not rAAV5-mediated, gene
transfer in the brain. J. Virol.78:6344–6359.
83.Rabinowitz, J. E., F. Rolling, C. Li, H. Conrath, W. Xiao, X. Xiao, and R. J. Samulski.2002. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction
with broad specificity. J. Virol.76:791–801.
84.Ramsburg, E., N. F. Rose, P. A. Marx, M. Mefford, D. F. Nixon, W. J. Moretto, D. Montefiori, P. Earl, B. Moss, and J. K. Rose.2004. Highly effective control of an AIDS virus challenge in macaques by using vesicular stomatitis virus and modified vaccinia virus Ankara vaccine vectors in a
single-boost protocol. J. Virol.78:3930–3940.
85.Rose, N. F., P. A. Marx, A. Luckay, D. F. Nixon, W. J. Moretto, S. M. Donahoe, D. Montefiori, A. Roberts, L. Buonocore, and J. K. Rose.2001. An effective AIDS vaccine based on live attenuated vesicular stomatitis
virus recombinants. Cell106:539–549.
86.Rutledge, E. A., C. L. Halbert, and D. W. Russell.1998. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other
than AAV type 2. J. Virol.72:309–319.
87.Rutledge, E. A., and D. W. Russell.1997. Adeno-associated virus vector
integration junctions. J. Virol.71:8429–8436.
88.Samulski, R. J., X. Zhu, X. Xiao, J. D. Brook, D. E. Housman, N. Epstein, and L. A. Hunter.1991. Targeted integration of adeno-associated virus
(AAV) into human chromosome 19. EMBO J.10:3941–3950.
89.Sasaki, S., R. R. Amara, A. E. Oran, J. M. Smith, and H. L. Robinson.2001. Apoptosis-mediated enhancement of DNA-raised immune responses by
mutant caspases. Nat. Biotechnol.19:543–547.
90.Schnepp, B. C., K. R. Clark, D. L. Klemanski, C. A. Pacak, and P. R. Johnson.2003. Genetic fate of recombinant adeno-associated virus vector
genomes in muscle. J. Virol.77:3495–3504.
91.Seth, A., I. Ourmanov, J. E. Schmitz, M. J. Kuroda, M. A. Lifton, C. E. Nickerson, L. Wyatt, M. Carroll, B. Moss, D. Venzon, N. L. Letvin, and V. M. Hirsch.2000. Immunization with a modified vaccinia virus expressing simian immunodeficiency virus (SIV) Gag-Pol primes for an anamnestic Gag-specific cytotoxic T-lymphocyte response and is associated with
reduc-tion of viremia after SIV challenge. J. Virol.74:2502–2509.
92.Sferra, T. J., G. Qu, D. McNeely, R. Rennard, K. R. Clark, W. D. Lo, and P. R. Johnson.2000. Recombinant adeno-associated virus-mediated cor-rection of lysosomal storage within the central nervous system of the adult
mucopolysaccharidosis type VII mouse. Hum. Gene Ther.11:507–519.
93.Shibata, R., F. Maldarelli, C. Siemon, T. Matano, M. Parta, G. Miller, T. Fredrickson, and M. A. Martin.1997. Infection and pathogenicity of chi-meric simian-human immunodeficiency viruses in macaques: determinants
of high virus loads and CD4 cell killing. J. Infect. Dis.176:362–373.
94.Shiver, J. W., and E. A. Emini.2004. Recent advances in the development of HIV-1 vaccines using replication-incompetent adenovirus vectors. Annu.
Rev. Med.55:355–372.
95.Shiver, J. W., T. M. Fu, L. Chen, D. R. Casimiro, M. E. Davies, R. K. Evans, Z. Q. Zhang, A. J. Simon, W. L. Trigona, S. A. Dubey, L. Huang, V. A. Harris, R. S. Long, X. Liang, L. Handt, W. A. Schleif, L. Zhu, D. C. Freed, N. V. Persaud, L. Guan, K. S. Punt, A. Tang, M. Chen, K. A. Wilson, K. B. Collins, G. J. Heidecker, V. R. Fernandez, H. C. Perry, J. G. Joyce, K. M. Grimm, J. C. Cook, P. M. Keller, D. S. Kresock, H. Mach, R. D. Troutman, L. A. Isopi, D. M. Williams, Z. Xu, K. E. Bohannon, D. B. Volkin, D. C. Montefiori, A. Miura, G. R. Krivulka, M. A. Lifton, M. J. Kuroda, J. E. Schmitz, N. L. Letvin, M. J. Caulfield, A. J. Bett, R. Youil, D. C. Kaslow, and E. A. Emini.2002. Replication-incompetent adenoviral vaccine vector
elicits effective anti-immunodeficiency-virus immunity. Nature 415:331–
335.
96.Snyder, R. O., S. K. Spratt, C. Lagarde, D. Bohl, B. Kaspar, B. Sloan, L. K. Cohen, and O. Danos.1997. Efficient and stable adeno-associated
on November 8, 2019 by guest
http://jvi.asm.org/
mediated transduction in the skeletal muscle of adult immunocompetent
mice. Hum. Gene Ther.8:1891–1900.
97.Sodora, D. L., F. Lee, P. J. Dailey, and P. A. Marx.1998. A genetic and viral load analysis of the simian immunodeficiency virus during the acute phase in macaques inoculated by the vaginal route. AIDS Res. Hum. Retrovir.
14:171–181.
98.Song, S., M. Morgan, T. Ellis, A. Poirier, K. Chesnut, J. Wang, M. Brantly, N. Muzyczka, B. J. Byrne, M. Atkinson, and T. R. Flotte.1998. Sustained secretion of human alpha-1-antitrypsin from murine muscle transduced
with adeno-associated virus vectors. Proc. Natl. Acad. Sci. USA95:14384–
14388.
99.Song, S., M. Scott-Jorgensen, J. Wang, A. Poirier, J. Crawford, M. Camp-bell-Thompson, and T. R. Flotte.2002. Intramuscular administration of recombinant adeno-associated virus 2 alpha-1 antitrypsin (rAAV-SERPINA1) vectors in a nonhuman primate model: safety and
immuno-logic aspects. Mol. Ther.6:329–335.
100.Sun, J. Y., S. Chatterjee, and K. K. Wong, Jr.2002. Immunogenic issues concerning recombinant adeno-associated virus vectors for gene therapy.
Curr. Gene Ther.2:485–500.
101.Tratschin, J. D., I. L. Miller, M. G. Smith, and B. J. Carter.1985. Adeno-associated virus vector for high-frequency integration, expression, and
res-cue of genes in mammalian cells. Mol. Cell. Biol.5:3251–3260.
102.Wagner, J. A., A. H. Messner, M. L. Moran, R. Daifuku, K. Kouyama, J. K. Desch, S. Manley, A. M. Norbash, C. K. Conrad, S. Friborg, T. Reynolds, W. B. Guggino, R. B. Moss, B. J. Carter, J. J. Wine, T. R. Flotte, and P. Gardner.1999. Safety and biological efficacy of an adeno-associated virus vector-cystic fibrosis transmembrane regulator (AAV-CFTR) in the cystic
fibrosis maxillary sinus. Laryngoscope109:266–274.
103.Walsh, C. E., J. M. Liu, X. Xiao, N. S. Young, A. W. Nienhuis, and R. J. Samulski.1992. Regulated high level expression of a human gamma-globin gene introduced into erythroid cells by an adeno-associated virus vector.
Proc. Natl. Acad. Sci. USA89:7257–7261.
104.Wang, Z., P. J. Troilo, X. Wang, T. G. Griffiths, S. J. Pacchione, A. B. Barnum, L. B. Harper, C. J. Pauley, Z. Niu, L. Denisova, T. T. Follmer, G. Rizzuto, G. Ciliberto, E. Fattori, N. L. Monica, S. Manam, and B. J. Ledwith.2004. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther.
11:711–721.
105.Watchko, J., T. O’Day, B. Wang, L. Zhou, Y. Tang, J. Li, and X. Xiao.2002. Adeno-associated virus vector-mediated minidystrophin gene therapy im-proves dystrophic muscle contractile function in mdx mice. Hum. Gene
Ther.13:1451–1460.
106.Willey, R. L., R. Byrum, M. Piatak, Y. B. Kim, M. W. Cho, J. L. Rossio, Jr., J. Bess, Jr., T. Igarashi, Y. Endo, L. O. Arthur, J. D. Lifson, and M. A. Martin.2003. Control of viremia and prevention of simian-human immu-nodeficiency virus-induced disease in rhesus macaques immunized with recombinant vaccinia viruses plus inactivated simian immunodeficiency
vi-rus and human immunodeficiency vivi-rus type 1 particles. J. Virol.77:1163–
1174.
107.Wurm, F. M., and C. J. Petropoulos.1994. Plasmid integration, amplifica-tion and cytogenetics in CHO cells: quesamplifica-tions and comments. Biologicals
22:95–102.
108.Wyand, M. S., K. Manson, D. C. Montefiori, J. D. Lifson, R. P. Johnson, and R. C. Desrosiers.1999. Protection by live, attenuated simian
immuno-deficiency virus against heterologous challenge. J. Virol.73:8356–8363.
109.Xiao, W., N. Chirmule, S. C. Berta, B. McCullough, G. Gao, and J. M. Wilson.1999. Gene therapy vectors based on adeno-associated virus type 1.
J. Virol.73:3994–4003.
110.Xiao, X., J. Li, and R. J. Samulski.1996. Efficient long-term gene transfer into muscle tissue of immunocompetent mice by adeno-associated virus
vector. J. Virol.70:8098–8108.
111.Xin, K. Q., T. Ooki, H. Mizukami, K. Hamajima, K. Okudela, K. Hashi-moto, Y. Kojima, N. Jounai, Y. KumaHashi-moto, S. Sasaki, D. Klinman, K. Ozawa, and K. Okuda.2002. Oral administration of recombinant adeno-associated virus elicits human immunodeficiency virus-specific immune
re-sponses. Hum. Gene Ther.13:1571–1581.
112.Xin, K. Q., M. Urabe, J. Yang, K. Nomiyama, H. Mizukami, K. Hamajima, H. Nomiyama, T. Saito, M. Imai, J. Monahan, K. Okuda, and K. Ozawa.
2001. A novel recombinant adeno-associated virus vaccine induces a long-term humoral immune response to human immunodeficiency virus. Hum.
Gene Ther.12:1047–1061.
113.Yuasa, K., M. Sakamoto, Y. Miyagoe-Suzuki, A. Tanouchi, H. Yamamoto, J. Li, J. S. Chamberlain, X. Xiao, and S. Takeda.2002. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal mus-cles evokes enhanced immune response against the transgene product.
Gene Ther.9:1576–1588.
114.Zaiss, A. K., Q. Liu, G. P. Bowen, N. C. Wong, J. S. Bartlett, and D. A. Muruve.2002. Differential activation of innate immune responses by
ade-novirus and adeno-associated virus vectors. J. Virol.76:4580–4590.
115.Zhang, Z. Q., T. M. Fu, D. R. Casimiro, M. E. Davies, X. Liang, W. A. Schleif, L. Handt, L. Tussey, M. Chen, A. Tang, K. A. Wilson, W. L. Trigona, D. C. Freed, C. Y. Tan, M. Horton, E. A. Emini, and J. W. Shiver.
2002. Mamu-Aⴱ01 allele-mediated attenuation of disease progression in
simian-human immunodeficiency virus infection. J. Virol.76:12845–12854.