Leishmania genome dynamics during environmental adaptation reveals strain-specific differences in gene copy number variation, karyotype instability, and telomeric amplification
41
0
0
Full text
(2)
(3)
(4)
(5)
(6)
(7)
(8)
(9)
(10)
(11)
(12)
(13)
(14)
(15)
(16)
(17)
(18)
(19)
(20)
(21)
(22)
(23)
(24)
(25)
Figure
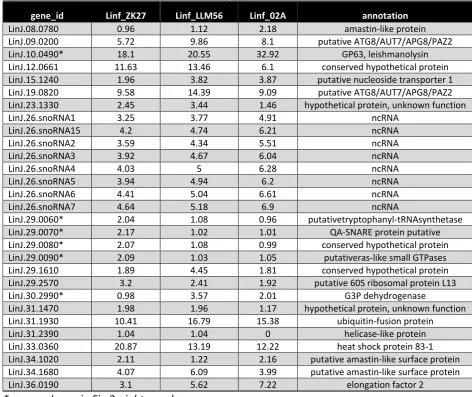
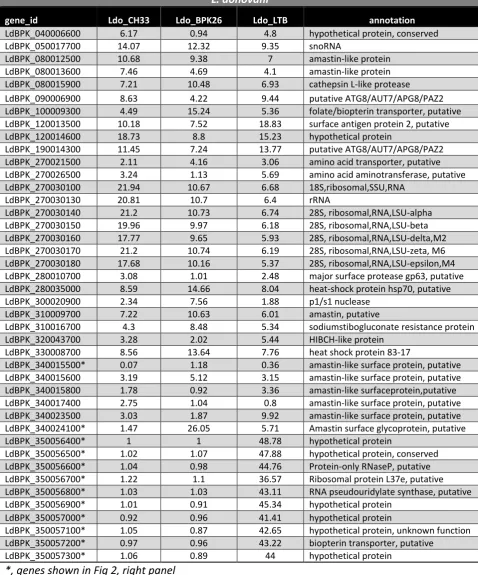
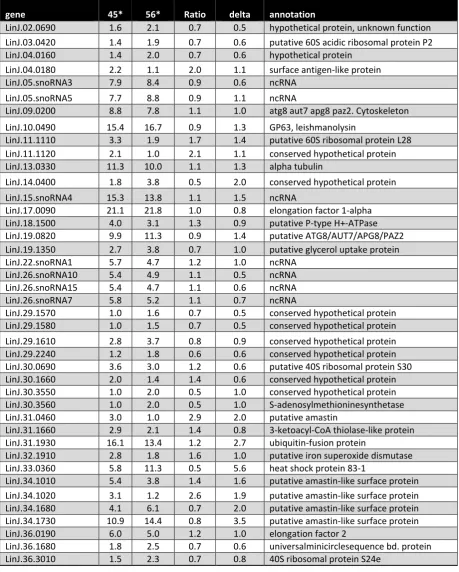
Related documents
One route is through conventional legal consulting organs (xinfang) that were set up in county level women’s federations. These organs provide women with
Abbreviations: ABRAEAD, Brazilian Statistical Yearbook of Distance Education; DE, Distance education; FHP, Family Health Program; ICTs, Information and communication technologies;
Enzyme entrapped polyacrylamide and agar/agarose gels were fragile and could not withstand repeated use whereas enzyme entrapped in large calcium alginate beads was used
Experimental data indicate that the adsorption efficiency is dependent on operating variables such as adsorbent mass, pH, shaking speed, particle size, contact time
Various classical as well as soft computing based search and optimization methods are available in literature for the training of neural networks and are shown
In addition to developing a robust regime for its export of uranium, Greenland must also seek to ensure that indigenous groups are incorpo- rated into the mining decision-making
Aurora’s husband is willing to tolerate the fact that his wife keeps the house like a pigsty, that she doesn’t know how to make a sorry pot of beans, and that she can’t take care
This study was undertaken to (1) determine in- stitutional criteria to enable prediction of mortality based on Pa02 and A-aDo2 and then maximize the criteria with regard to
