JOURNALOFVIROLOGY, June 1992, p.3573-3582 0022-538X/92/063573-10$02.00/0
Copyright ©3 1992, American Society for Microbiology
Localization of cis-Acting Sequence Requirements
in the
Promoter
of
the Latency-Associated Transcript of
Herpes
Simplex Virus
Type
1Required for Cell-Type-Specific Activity
ADRIAN H. BATCHELORAND PETER O'HARE*
Marie Curie ResearchInstitute, The Chart, Oxted, Surrey RH8 OTL, United Kingdom Received 23 December 1991/Accepted 9March 1992
We have previously demonstrated (A. H. Batchelor and P. O'Hare, J. Virol. 64:3269-3279, 1990) the selective activity in human neuroblastoma cells (IMR-32) of a promoter located upstream of the latency-associated transcript of herpes simplex virus type 1. In this work, we provide evidence for the basis of the
selective activity of this latency-associatedpromoter (LAP). Recombinant constructscontaining sequencesup
to -143 (relative to the LAPcap site) linked to the chloramphenicol acetyltransferase gene retain strong
activity in HeLa cells but exhibit extremely weak activity in IMR-32 cells. Sequences mapping within the 108 bp upstream of -143 to position -251 enhance LAP activity by over 15-fold, restoring optimal levels of expression in IMR-32 cells, but have little or no effect (1.5-fold) in HeLa cells. This cell-type-specific enhancement ofpromoter activity took place in two majorsteps, with sequences between -143 and -158 conferringafour-tofivefold effect andsequencesbetween -177 and -251 conferringafurtherthreefold effect. Furthermore, sequences mappingfrom -40 to -258 could transfer the abilitytobe expressed in
neuroblas-tomacellstothenormally inactive immediate-early 110Kpromoter (IEllOK), increasing levels of expression by 35-fold. By comparison, this region had a relatively minor effect (twofold) on the activity of the IEllOK promoter in HeLa cells, even though this promoter is open to activation by other mechanisms. However, neither of the overlapping subregions from -40to -143or-138to -258 could confer efficient IMR-32 cell expressionon the IEllOKpromoter, andwepresentalternative models formultiple element requirementsor the requirement for a critical site around -140 which is not retained in either subfragment. We provide consistent evidence for a site around -140 and demonstrate the presence selectively in IMR-32 cells ofa DNA-binding factor which bindsaprobespanning this region. Weproposethat this element and thecognate
factor (IC-1) maybe involved in the selective activityof the LAP in neuroblastoma cells.
For thealpha neurotropicgroupofherpesviruses (19), the major site of latency, at least for herpes simplex virus (HSV), is within the neuronal-cell population of the sensory ganglia which innervate the site of primary infection (for reviews, seereferences 17, 33, and 47).
Latency results in a dramatic qualitative change in virus
geneexpression from that operating in productively infected cells. Consistent evidence for the abundant expression in ganglionicneuronsofasingle transcription unit, the latency-associated transcript (LAT), and the absence of detectable
amounts ofany other virus-encoded product has been pro-duced from a number of laboratories (5, 7, 14, 22, 35-38). Themajor stable LAT species that have been describedare colinear transcripts of approximately 2.0 and 1.5 kb which are present in the unpolyadenylated RNA fraction and locatedmainly within the neuronal-cell nuclei (22, 36, 42, 43, 45, 46).The5'end of thesetranscripts hasbeenlocalizedto
position 119461 of the HSV genome (by the numbering
systemof McGeochetal. (25, 32)in the internal longrepeat
downstream of the 3' end of the immediate-early IEllOK gene (42, 44, 45). However, a number of lines of evidence
now indicate that the 2.0- and 1.5-kb LAT species are processed products of a large (ca. 8.3- to 9-kb) unstable
primary transcriptwhich initiates further downstream of the
3' end of the IEllOK gene and is transcribed through the
* Correspondingauthor.
longrepeat segment toterminate in the shortrepeatregion of the genome (8, 9, 22, 26-28, 51). Indeed, the major stable LATspecies have been proposedtobe theprocessed introns of this primary transcript (8, 11). Consistent with the iden-tification ofaninitiation site for the large primary transcript atposition 118802, approximately 30 bases downstream ofa consensus TATA box (8, 9), we and others, by using transient expression assays, have demonstrated relatively
strong promoteractivity within the surrounding region (1, 8, 49, 51), and we have localized sequences sufficient for promoteractivity in HeLa cellstowithin143 basesupstream
of the cap site. Although the precise role of LAT remains unclear, one goal of the investigation into latency is to
elucidate the basis for the highly selective expression in neurons. To that end, we previously demonstrated that in human neuroblastomacells, asin latently infectedneurons, themajorityofproductive cyclepromoterswhich we tested wereinactive,while the LATpromoter(LAP)was constitu-tivelyvery strong. Furthermore, wedemonstrated that
se-quencesupstream ofposition -143 hadasignificanteffecton promoteractivity inthe neuroblastoma cells butnoeffect in
HeLa cells.
In this work, we have sought to refine the cis-acting element(s) involved in selective activity of the LAP in
neuroblastoma cells and to examine their interaction with
cellular DNA-binding factors. While multiple elements are
required for optimal transcriptional efficiency, we provide evidence foracriticalcell-type-specificdeterminant located
within 20 bases around -160 toapproximately -140. 3573
Vol. 66, No. 6
on November 9, 2019 by guest
http://jvi.asm.org/
MATERIALSANDMETHODS
Promoter-CAT constructs. Constructs containing the chloramphenicol acetyltransferase (CAT) open reading
frameand the simianvirus40poly(A)site(pBB-2and-3)and their LAP-CATderivatives (pBB-8, -13, -15, and -16) have beendescribed previously (1). Upstreamdeletions ofpBB-8 were made by digestion with SstI (immediately 5' of the LAPsequence) andNaeI (partial), NaeI, SstII, BssHII, or EcoRV. Mungbean nuclease (MBN) treatment andligation generated pBB-22, -21, -20, -19,and -18. Thepositionsofthe resulting promoter fragments are shown in Fig. 2. Internal deletions of pBB-8 were made by digestion with NaeI, BssHII, SstII-EcoRV, and EcoRV-PstI (partial), by MBN
treatment, andbyreligation.
pAB2 is a 5' deletion of the IE1lOK promoter-CAT construct pPO49 (30) and was made by inserting a SmaI fragment from pP049 into pUC19. IE1lOK promoter se-quencesinpAB2extendto-130 and lack theTAATGARAT motifs. Upstream HindIII, PstI, and XbaI sites in the polylinkerwere used forinsertingLAPfragmentsto testfor enhancer function. pBB-29 is a3' deletion ofpBB-8 which lacks the TATA box. pBB-29 was constructed by treating pBB-8 with,inturn,DdeI,MBN, and SstI andinserting the resulting fragment into pBB-2digestedwithSmaI and SstI. pBB-29 treatedwithSstIandMBN(forpBB-51)orSstIIand MBN (forpBB-58), followed by PstI, resulted in two frag-mentswhichwereinserted intopAB2digestedwithHindIll and MBN and then PstItogivepBB-51 and-58.pBB-29cut
with SstI, SstII, or EcoRV followed in turn by MBN and XbaI(3' of theLATsequences) generated fragmentswhich were inserted into pAB2 digested in turn with HindIII, MBN, and XbaI to give pBB-52, -57, and -60. pBB-59was constructedby insertingaPstI-XbaIfragmentfrompBB-29 into pAB2 cut with PstI andXbaI. Double-stranded
oligo-nucleotides B, C, and G (see below) with XbaI overhangs wereinserted intotheXbaI site ofpAB2, giving pBB-61, -62, and -63. The plasmid pBB-64wasconstructed by inserting oligonucleotideS(see below)intopBB-58cutwith PstI and XbaI. Internal deletions of pBB-57 were constructed by digestionwith PstI (pBB-65) orPstI and EcoRV (pBB-66), MBNtreatment, and religation.
Thestructures of allconstructswere confirmedbydirect sequencing of denatured plasmids as described previously (1), and precise 5' and 3' endpoints of the constructs and positions of the fragmentinserts are indicated in summary figures 2,4, and 6.
Cells,transfectionprocedures,and CATassays.HeLacells weregrown inDulbecco modified Eagle medium with 10% newborn calf serum. Cells were plated the day before transfectionintocluster dishes(6 by 35 mm) at 106 cellsper well. IMR-32 cells (41), a human neuroblastoma cell line
obtainedfrom the AmericanTissueCultureCollection,were grown in Dulbecco modified Eagle medium with 10% fetal calf serum. IMR-32 cells were split 1:2 into cluster dishes and allowed to grow to confluence (3 to 5 days) prior to
transfection. DNA transfections were carried out by the calciumphosphateprecipitationmethod(16,31)modifiedby the use of BES [N,N-bis(2-hydroxethyl)-2-amino-ethane-sulfonic
acid]-buffered
saline (4) in place of HEPES (N-2-hydroxyethylpiperazine-N'-2 ethanesulfonic acid)-buffered saline. Thetotalamountof DNAtransfectedwas equalized withpUC19to2.5 pginallcases. Extractsweremade 42to48 haftertransfection,andCATassays wereperformedand quantitatedasdescribed previously (15, 31). In eachfigure,
the amounts of radioactivity
appearing
in theacetylated
products aregiven below each lane
(10-3 cpm).
Oligonucleotides.
Oligonucleotides
were made byusing
an Applied Biosystems DNA synthesizer, and complementary strandswereannealed. Probes used in this workare asfollows. B: CTAGAGTGCCCGCGAGATATCAATCCGTTAAGTGCTTCACGGGCGCTCTATAGTTAGGCAATTCACGAGATC C: CTAGATATCAATCCGTTAAGTGCTCTGCAGACAGGT
TATAGTTAGGCAATTCACGAGACGTCTGTCCAGATC G: CTAGAGGCCTGTTTTTGCTGCGTCATCTGAGCCTTT
TCCGGACAAAAACGACGCAGTAGACTCGGAAAGATC
CCAAT: CTGATGGGGTAGGAACCAATGAAATGAAAGGTGACA ACCCCATCCTTGGTTACTTTACTTTCCACTGTAGTC
Spl: CTAGAGACTACCACGCCCACGAGAT
TCTGATGGTGCGGGTGCTCTAGATC
T24: CCATGGAGATCTCGTGCATGCTAATGATATTCTT
TCTAGAGCACGTACGATTACTATAAGAAGGTACC
S: GACAGGGGCACCGT ACGTCTGTCCCCGTGGCAGATC
The positions of oligonucleotides B, C, and G within the LAP are shown in Fig. 4b. CCAATcontains therat albumin promoterCCAAT site. Spl contains anadenovirus ElI late Splbinding site. T24 containsan
IEllOK
promoter octamer-GARAT element(30).
The annealed probes were radiola-beled with[a-32P]dCTP
by using the Klenow fragment ofEscherichia
coli DNApolymerase
andpurified
by electro-phoresis in 12%polyacrylamide
gels.Cell extracts and gel retardation analysis. Whole-cell ex-tracts weremadeasdescribedby Wu
(48),
exceptthat 3pellet
volumes ofextraction buffer were added to the frozen cell pellet.Supplemental protease inhibitors (2 ,g of leupeptin per ml, 1 ,g of
pepstatin
Aperml,2,g ofaprotinin
perml, and 0.2mMN-a-p-tosyl-L-lysine
chloromethylketone)
werealso used in addition to 0.5 mMphenylmethylsulfonyl
fluoride. Protein concentrations were determined by the method of Bradford(2)
andequalized
forgel retardation studies.Gel retardation
analyses
wereperformed essentially
as describedpreviously (12,
13,30) except that 20RI
ofbinding
reaction mixture contained the protease inhibitors detailed above, 1 ,ug of
poly(dI-dC),
4,ul
of whole-cell extract (approximately 5 ,ug of total protein), andnoadditional salt. Extracts werepreincubated withpoly(dI-dC)
for 5 minand then for 20 min withprobe
at room temperature. Forcompetition
studies, coldcompetitor oligonucleotides
were added to the mixture for 20 min prior to the addition of radiolabeled probe.DNA-binding specieswereseparated from the free probes by
electrophoresis
in 5%polyacrylamide gels
(19:1 cross-linkerratio) in 0.5x Tris-borate buffer.RESULTS
Localization ofupstream sequences required forpromoter activity in neuroblastoma cells. For the sake ofclarity, we have
changed
our labeling system from that used in our previouspublication (1)toindicate the location of sites with referencetothe5' end of the primary LAT, position 118802 ofthe HSVtype1(HSV-1) genome (9), instead ofthe5' end of the 2.0-kb species. We have previously shown that the constructpBB-8, which contains thePvuI
fragment spanning positions -608 to +2, exhibits maximal activity in both HeLacells andthe neuroblastomacell lineIMR-32 and that sequencesbetween -608 and -138 areselectively required for promoteractivity
in the neuroblastoma cells. Torefine
on November 9, 2019 by guest
http://jvi.asm.org/
LATENCY-ASSOCIATED PROMOTER OF HSV-1 3575
CN V- ID Ocm co LO
co cN c -
-m m m m a: m m
E:L CL m: CL QL QL Q
* * * * * *
-* e
0
177 174 140 1 1 2 9 8
..0
0
73 72
*a
95 88 94 99 29 15 3
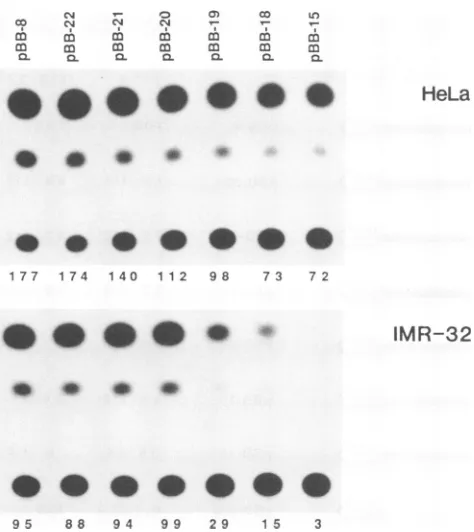
FIG. 1. Typical results from transfection ofaseries of 5'
dele-tions into HeLa and IMR-32cells.Equal amountsof the indicated plasmidDNAs (50 ngfor HeLacells and 100ngfor IMR-32cells)
weretransfected into the indicated cell typestogetherwithcarrier DNAasindicated in Materials andMethod. Cellswereharvested 44
h after transfection, and equal amounts were assayed for CAT
activity.The amountsofradioactivity appearing in the acetylated productsaregivenbelow the lanes(10-3cpm).
thisanalysis,aseries of restrictionenzymesiteswasusedto makeprogressivedeletionsof theupstreamregion of pBB-8. Typical results for the relative levels of CAT expression
after transfection of these constructs into HeLa cells or neuroblastoma cellsare shown inFig. 1 and summarized in Fig. 2. Figure 2 also includes a schematic diagram of the endpointsof each variant.
Consistent with ourpreviouswork (1),overall deletion of the LAP to the PstI site at -138 (pBB-15) had a two- to threefold effect on activity in HeLa cells (Fig. 1 and 2). Although this effect was reproducible, it could not be as-signed toanysingle regionbut resulted from thecumulative
minor effects of several deletionstowhich itwasdifficultto attribute significance. In contrast, deletion to -138 had a dramatic 30-fold effect in IMR-32 cells(Fig. 1),andoverthe courseof this work, the effect averaged 16-fold (Fig. 2).
Moreover, the reduction in expression in the neuroblas-toma cells could be attributed specifically to two regions.
While each of the progressive deletions to -251 had little
effect, deletion ofsequencesbetween -251 and -177 (pBB-19) resulted in a decrease in promoter activity of approxi-mately threefold, and deletion of 15 bases from -158 to -143 (pBB-15) resulted in a further fivefold reduction in promoter activity in IMR-32 cells only. Removal of the
sequencesfrom -177to -158 hadaslight effectinboth cell types. Together, these results indicate that sequence ele-mentswithinoraround the 74bpfrom -251to -177 and the 15bp from -158 to -143 contributetopromoter activityof LAP selectivelyin neuroblastoma cells.
Consistent result were obtained with internal deletion mutantswhich lackedsequence from -262 to -160 or -162 to -132 and exhibited three-tofivefoldless activity than the parent construct in neuroblastoma cells but near normal levels in HeLa cells (data not shown). Generally, however, theeffects of internal deletionswereless dramatic than the effects of 5' deletions, possibly because of redundancy within thepromoter. Torefine the identification of cis-acting regulatory elements selectively active in neuroblastoma cells, we performed a series of experiments to determine whether upstream sequences could confer neuroblastoma-specific expression on a normally weak or inactive promoter. Selective andefficient expressionof an inactive promoter in neuroblastoma cellsby transfer of upstream LAP regulatory regions. We havepreviously shown that theimmediate-early promoter-regulatory region of the HSV-1 IEllOK gene is extremely weak in neuroblastoma cells (1). (Detectable levelsofexpression can, however,be achieved by transfec-tion ofthe IEllOK-CATconstruct into neuroblastomacells in 10- to 20-fold-greater amounts than those required for expression in HeLa cells.)
Variousupstreamfragments of theLAP were linkedtothe IEllOK promoter (truncated at the 5' end at -130) and comparedwith theparental IEllOKpromoterinHeLa cells orneuroblastoma cells(summarized in Fig. 4 and 6). In the first constructs, we linked the entire region of the LAP upstream of the TATA box from positions -40 to -608 (pBB-52) or from positions -138 to -608 (pBB-51) to the test promoter. Typical results of comparison of these chi-mericpromoterswith the parental IEllOKpromoter(pAB2) areshown inFig. 3. Insertion of the entire upstream region ofLAP resulted in astriking 22-fold increase inexpression compared with the IEllOKpromoteralone in the neuroblas-toma cells while having a 2- to 3-fold effect in HeLacells. The results illustrated in Fig. 3 were obtained by using greater amountsof DNA in IMR-32 cellsthanin HeLacells to facilitate detection ofexpression from the IEllOK pro-moter (pAB2) at levels significantly above background. Identical resultswereobtained, i.e., 30-fold enhancement in IMR-32cellsversus2-foldenhancement in HeLa cells, when equal amounts ofDNA were used (e.g., 50 ngin both cell types) except that basal expression for pAB2 was not significantly above background in the neuroblastoma cells. The lack ofenhancement in HeLa cells is not because the IEllOK promoter is already working at optimal efficiency, since it can be transactivated by either Vmw65 or the IEllOKprotein itself byup to50-fold (24, 30, 34). Moreover, certaincis-actingelements(see Fig. 4)canenhance IEllOK promoter
activity
in HeLacellsby 10- to 12-fold. Over the courseof thiswork, the construct pBB-52 exhibited 20- to 40-fold-higherlevelsofactivitythan the parent inneuroblas-toma cells while
giving
onlymarginally higher
levels ofactivity
(2-fold) in HeLa cells(Fig.
4). Since a similarconstructwhich contains the-40to-608
region
of LATbut lacks theIEBlOK
promoter iscompletely inactive in either celltype,these results indicatethat upstream elements of the LAP can selectively increase activity of a heterologous promoter in neuroblastomacclls.This conclusion isentirely consistent withthe resultsfrom5' deletion of the LAPitself,
and together, the data support the proposal for a
cell-type-specific
enhancer element upstreamofthe LAP.Transfer ofafragment
containing
sequences from -138to-608 (pBB-51) resulted in a3- to4-fold increase in expres-sion
(compared
with a 30-fold increase with the sequences from -40 to -608),indicating
that sequences from -40to -138 were
required
forneuroblastoma-specific
enhance-VOL.66, 1992QF
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.61.298.79.345.2](A(A C,,~~~~~~~~~(
I _ I
II
_I I I I I 1
I ~~~~~~~~II I
0D 0 01 )
0 CNJ U) N w C
I? CO CQ
> (
TATAC.
0-I I
U) N% CD
CN + CD
L- -
M=g-L~~~~~~~~~~~~~~~~~C
ATE CAl
Relive promoteractivity± s.d.
HeLa IMR-32
pBB-8
pBB-22
100 100
79 i16 89 i13
pBB-21
E -251
E -177
-1 58
72 i12 92 i12
pBB-20 57 ±10 98 ±15
pBB-19 60 ±20 37 ±10
pBB-18 46 ±10 24 ±6
E
-143
WE!>
pBB-15 36 i6 6 i4 [image:4.612.66.554.79.335.2]-E>
pBB-2/3 0.4 i0.1 0.5 ±0.2FIG. 2. Summary of5' deletionconstructsand their relative activities in HeLa and IMR-32 cells. Thetop line isarepresentationof the
LAPregionmapping from the PvuI site at118194 tothePstI site at118867, whichmapatpositions -608 and+66, respectively,inrelation
tothe startsite of the primary LAT (9). The virussequencescontained in the recombinant CATplasmidareindicatedbysolidthicklines, and their respective end pointsarenumbered.Forrelativepromoteractivityin HeLa and IMR-32cells,theparentconstructpBB-8has been
assigned avalue of 100 and the 5' deletionsaregiven avalue(+ thestandard deviation [s.d.]) relative topBB-8inthat celltype. Average valuesweredetermined fromatleast fourindependent experiments. Inabsoluteactivity, pBB-8wasabouttwo-tofourfoldhigherin HeLa
cells than in IMR-32 cells(see,e.g., Fig. 1),andtransfectionswereperformedwith theappropriateamount of DNA(usually50ngin HeLa
cells and 100nginIMR-32cells)tokeep the CATassays linear withrespectto DNAand enzymeactivity.
IMR-32
m co m
CL n
HeLa
CM
co
:L
0
_ N
an an
aI
co m
co CQ
.*
*
*
9 47 202 4 5 11
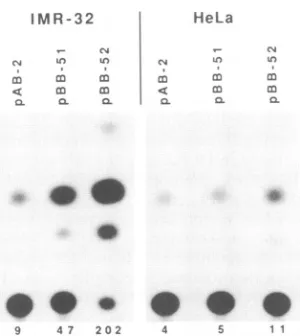
FIG. 3. The upstream region of the LAP acts as a
cell-type-specific enhancer, augmenting activity ofan immediate-early
pro-moterselectivity inneuroblastoma cells. Equal amounts (25 ngfor
HeLa cellsand250ngforIMR-32cells) of theparent IEllOK-CAT
construct (pAB2) or constructs containing additional upstream
fragments from the LAP region (pBB-51 and pBB-52;seeFig. 4 for
maps)weretransfected into each celltype,and cellswereharvested for CATactivity 44 h later. Theamountsofradioactivity appearing
in theacetylated products are givenbelowthe lanes(10-3cpm).
ment of promoter activity. The plasmid pBB-57, which contained sequences from -40 to -258, was as active as pBB-51, indicatingthat sequencesfrom -258 to -608 have little effect, and we therefore refined our analysis within pBB-57.Twoderivatives,pBB-59 (-40to-143)andpBB-58 (-138 to -259), which together span the entire insert in pBB-57, were constructed. Surprisingly, neither of these
constructsexhibited asignificantincrease in promoter activ-ity in neuroblastoma cells compared with the parental IEl1OK promoter construct (Fig. 4 and 5). This result indicated that there are elements within both the -138 to
-259 and -40to -143 regionsthat arerequired togetherto
reconstitute theneuroblastoma-specific activityof theentire fragment from -40 to -258. Alternatively, if a single
ele-ment is critical, it may havebeen that cleavage at the PstI site at -138 disrupted the element so that neither
subfrag-ment contained it intact. To examine this possibility, we madeaplasmidwhichextended the 5' end of the fragmentin
pBB-59toposition -161togivetheconstructpBB-60 (Fig. 6a), inwhich thesequence around the PstI sitewas intact. However, pBB-60 exhibited little increase in activity over that of the parent construct (Fig. 6b). Conversely,
down-streamextension of thefragmentinpBB-58toposition-126
to yield the construct pBB-64 (Fig. 6a) did not restore
efficient expression in the neuroblastoma cells (Fig. 6b). Together, these results indicate that for transfer of neuro-blastoma-cell-specific enhancementoftheIEllOKpromoter activity, ifa single element is critical, the element spans at
least theboundaries from -161 to -126sothat itwouldnot
be contained inanyofthe derivatives ofpBB-57 despitethe a.
0
co 0
-608
-389
I
E
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.114.264.482.650.2]LATENCY-ASSOCIATED PROMOTER OF HSV-1 3577
(A) Wa.
I I I
C. . m
in _ Xl
C? 7 7
2
TATAo00
Xl CY
Relativepromoter activity±s.d.
HeLa IMR-32
IE11OK
pAB-2
-138
-40
-258 -40
-259 -138
-143 -40
mzzZ21n>
pBB-51EZZZD> pBB-52
mrzmm pBB-57
pBB-58
=MrE>
pBB-592.1 ±0.7 3.5 ±1
2.5 ±0.2 3 2 ±8
1.3 ±0.6 35 ±16
0.8 ±0.4 1 .1 ±0.5
1.5 ±0.8 1 .4 ±0.2
!zzm> pBB-61
ezzm4 pBB-62
1 .1 ±0.2 0.6 ±0.2
0.6 ±0.3 0.5 ±0.1
-59 -28
mn
[image:5.612.73.541.77.357.2]pBB-63 11 ±3 1.4 ±0.4
FIG. 4. Summary of the results of transfer of fragmentsupstreamof the LAPtoatestpromoter.Thetestpromoter,IEll1K, isillustrated
bya striped line, and fragments from the LAP are represented by solid lines. The upstream and downstream coordinates ofall inserted fragmentsoroligonucleotides (pBB-61, pBB-62, and pBB-63)are indicated. Theparentplasmid pAB2 has been assigned anactivity of1 in
both HeLa and IMR-32 cellssothat foreach construct,the valuegiven representsthe fold enhancement conferredbythe corresponding fragment in each celltype. In mostcases, 100ngof each constructwastransfected intoIMR-32 cellsand50 ngwastransfected into HeLa
cells.Average values (+ standard deviation [s.d.])weredeterminedfrom at least three independent experiments.
overlap in the corresponding fragments. Alternatively, a -138 was involved in the increased expression of the more likely explanation in our view is that while elements IEllOK promoter in IMR-32 cells, two internal deletions of around -138 may be required, additional elements from the pBB-57 fragment were constructed, resulting in the
upstreamand/or downstream of the site are also necessary plasmids pBB-65 and pBB-66, which contain 4- and 26-bp
for efficientexpression in neuroblastoma cells. deletions, respectively, around this region, as illustrated in
To demonstrate that the region around the PstI site at Fig. 6a.
Removal of the 4-bp TGCA had a modest (but
reproduc-ible) effect on IMR-32 cell expression but no effect on
r- co CD° ° N
en
Xexpression
in HeLa cells (Fig. 6b). Consistent resultswereaim m m m m m m m m obtained with pBB-66, where removal of the 26 bp from
<: m m m mm mm C -162to -137selectively reduced expressionto20to25% of
0 0
control values only in the neuroblastoma cells (Fig. 6b). Thus, the fragment from -258 to -40 that contained this
deletionwasstill able to augmentexpression ofthe IEllOK
e promoter in the neuroblastoma cells but at a significantly
reduced efficiencycomparedwith that of the intact fragment
(7-fold increase compared with 33-fold increase). Overall, these results are consistent with those from the 5' deletion
* * * * * * * analysis and the internal deletionseries, andour
interpreta-tion of this series ofexperiments is asfollows.
5 1 1 3 6 7 8 3 3 5 13 1 (i) Judging from the series ofexperimentssummarized in
FIG. 5. Comparison of the abilities of varioussubfragmentsfrom Fig. 4 and 6 (compare pBB-57 and pBB-60), sequences theupstreamregionof the LAP toenhanceactivityof the IEllOK upstream of -161 are unequivocally necessary forefficient
promoter in neuroblastoma cells. Equal amounts (100 ng) of the activity of thenormallyinactive IEllOKpromoterin neuro-parent construct pAB2 orof recombinants containingvarious up- blastoma cells. This is consistentwith the 5' deletion
analy-stream fragments from the LAP(see Fig. 6)were transfected into
IMR-32 cells. The cells were harvested 46 h later, and equal slsofthe IntactLATpromoter-regulatory region,where the
amounts wereassayed forCAT activity. The amountsofradioac- deletion to-158 (pBB-18) resulted in a significant decrease
tivityappearingin theacetylatedproductsaregivenbelowthelanes in expression (Fig. 1 and 2). From comparison of pBB-20,
(10-3cpm). pBB-19, and pBB-18, this effect could largely be attributed
(a)
-6068
-608
(b)
P1
-174 -144
I.
-165 -133
L x
VOL. 66,1992
F-D0
I
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.63.296.518.646.2](a)
A:-AATATCrAAT"'CCG-TAA3T(;CTCt^Z!1TC-,AC-ACACGt:C,ACj probeC
C; C_ATVCA T Ct
-4 ---C:."(-AIAAeTCTCACTTr1 S1AxCvS'T vICATC-AG>A-t7{;CA14tCC'-C - -4
(b)
tN 13 fLn 4n 4
1 ~"?
co co co co co co
m m o, a. co
pBB-57
HeLa
pBB-6C
pBB-64
-CcA A - -- - ^..-AIAGEG8: A,TGCoA:1 C
2:s-F--TC: ...
--4.
*
*
*
*
1 2 1 2 7.3 996 1 19
pBB-65
pE3B-66
1, 2.a 2.1 64 26 1 4
c:zz::1 H
FIG. 6. Overlapping subfragments of the enhancer regiondonotconfercell-type-specific activity. (a)Thesequencebetween thecritical EcoRV-to-PstI regionisillustratedtogetherwithasummaryoftheoverlapping subfragmentsordeleted variants ofthe intactfragmentwhich weretransferred upstreamof the IEllOKpromoter.Asterisks below theprimarysequenceindicatehomologywiththecorresponding region ofthe HSV-2genome (see text). Thesequenceofaprobe, probe C, spanning this region andused ingel retardationassaysisgivenatthe corresponding positionabove thefragmentsequence.(b)Theparentplasmid, pAB2,and variousderivativesweretransfected into HeLacells (50 ng)orIMR-32 cells (100 ng)andharvested 44 h later. Theamountsofradioactivity appearingintheacetylated productsaregivenbelow thelanes(10-3cpm). (c) Schematic representationofmodels for therequirementtoreconstitute enhancement of promoteractivity selectivity in neuroblastoma cells.Combined results indicate thatmultipleelementsare required.Inmodel1,wehaveproposedtherequirementfora
downstream promoterproximalfactor(shaded circle), indicatingUSFonlyasanexample (i.e.,this factor wouldnotbindtothe basalIEllOK promoter). In this case, the factorwhich is required around -143 can lie entirelyupstream of -138 (see text). In model 2, there is no
requirementforanadditional downstreampromoterproximal factor,and thedata therefore indicate that in additiontoarequirementupstream of-177, thereisarequirement forafactor whichoverlaps sequencesfrom -143to -126.
to sequences upstream of -177. However, pBB-18, unlike pBB-60, was still active in neuroblastoma cells, and in
absolute terms,pBB-18wasfive- tosevenfold strongerthan pBB-60. Therefore, thesequencesupstreamof-161seemto augment the downstream LAP sequences, which indepen-dently retainsomeactivity in neuroblastoma cells, while the regionupstreamof-161 isabsolutely required for activity of the IEllOKpromoter, which independently is virtually in-active in thesecells.
(ii) The cell-type-specific activities of sequences
down-streamof-158aredemonstrated from the 5' deletion series,
in whichremoval ofsequencesfrom-158to -143 (compare
pBB-18 and pBB-15, Fig. 1 and 2) results inasignificant and specific four-tofivefold reduction in expression only in the neuroblastoma cells. The signal removed by the -158 to
-143 deletion may overlap the -143 site rather than be containedentirelyupstreamof-143, because in the transfer experiments, plasmid pBB-65, which contains a4-bp
dele-tion from -142to -139, isreproducibly less active than the
parentpBB-57.
Confirmation that one of thesignals required for optimal levels of expression in neuroblastoma cells lies within (or overlaps) the -162 to -137 region was obtained from the
internal deletion mutant andpBB-66, which exhibited
four-to fivefold less activity than the parentconstruct in neuro-blastoma cells but similar levels of activity in HeLa cells (Fig. 6).
(iii) The absence of activity of pBB-64 compared with pBB-57 (Fig. 6) indicates thatsequences from -126to -40 are required forthe enhancement of the IEllOKpromoter.
A schematic representation of these conclusions with
referencetotheproposal ofatleasttwosites within the LAT regulatory region is given in Fig. 6c.
ComparisonofDNA-bindingactivity inHeLa and IMR-32 cells to a region required for selective LAP activity. One
explanation for the selective activity ofaDNAsequencein augmenting promoteractivityinoneparticular celltypeand
not in another is the corresponding selective presence (or activity) in those cells of a positively acting transcription
factor which recognizes the sequence. To examine this possibility, wemadewhole-cell extractsofHeLa or neuro-blastomacells andexamined thebinding profile of each cell
type to aprobe which spanned the -161 to -138 region of
the LAP (Fig. 6a, probe C). As controls, we used probes containing consensus motifs for a number of known
tran-scription factors (e.g., SPI and CCAAT box binding sites)
andanumber ofprobesfromareasinthe LATregion which exhibited no selective effect on promoter activity. Typical
(c)
1.
2.
IMR-32
0
0
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.70.546.76.361.2]LATENCY-ASSOCIATED PROMOTER OF HSV-1 3579
(a)
probebpe C Spi CCAAT
H H IH
.
(b)
CCAAT
H I
competitor (pmoles)
C CCAAT Spi T 2 4
08 4 0.8 4 0.8 4 0.8 4
I
AL I
C -1 -o
HC--1
H C -2
--Ic-1
I I
nr
[image:7.612.71.285.82.387.2]I
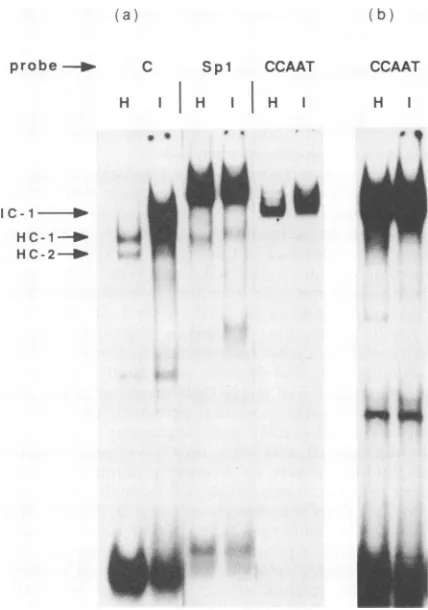
FIG. 7. Identification ofaDNA-binding factor which ispresent
in IMR-32 cells but absent fromHeLacells. (a)Whole-cellextracts
wereprepared from HeLa (H) or IMR-32 (I) cellsas described in
Materials and Methods. Binding assays were performed with the
probes indicated in the presence of nonspecific competitor DNA,
and theresultingcomplexes were separated on 5% nondenaturing
polyacrylamide gels. Qualitatively similar profileswereobtainedfor eachprobeinextractsof bothcelltypeswith the exception of probe C, with which complex IC-1 was observed only in IMR-32 cell
extracts.(b) Independent comparison of theextractsfromHeLa and IMR-32 cellswith the CCAATprobe inexcess.
results fromsuch comparisonsareshown in Fig. 7. Virtually
identicalprofileswereobserved for HeLa (H) and IMR-32 (I) cells with both SP1 and CCAAT box probes (Fig. 7). Similarly,nodifferencewasobserved withanumber of LAP probes and a number ofadditional probes (including those forUSF andOct-1; datanotshown). In contrast, adistinct
qualitative difference between the two cell types was
ob-served withprobe C. WhiletwoHeLa cell complexeswere observed (HC-1 and HC-2)in the IMR-32cells, these com-plexeswereonlyaminorfraction of the total probe C-bind-ing activity, and a major complex of lower mobility (IC-1) that partly obscured the cell-type-common doublet was observed. Alterations in binding conditions, competitor
type, or overall protein-competitor ratio and analysis of complex formation in protein dose-response experiments never resulted in the appearance of this complex in HeLa cells.
Specificity of the IC-1 complex was demonstrated in
[image:7.612.328.545.82.413.2]competition experiments (Fig. 8), in whicha40-to 200-fold excessof theSPI(SP1)orOct-I (T24)-binding sitesresulted in littlereduction inIC-1 formationonprobe C. Although the CCAATbox, andtosome extentthe Oct-i-binding site, did
FIG. 8. Specificity of binding of IC-1. Aliquots of the IMR-32 cell extract were incubated withincreasing amounts of cold competitor oligonucleotides in addition to the nonspecific competitor
poly(dI-dC)atapproximately40- and 200-fold molar excess over the amount ofradiolabeled probe. After 20 min of preincubation with competitor DNAs, radiolabeled probe C was added for a further 20 min, and
complexeswerethen separated on a5% nondenaturing polyacryl-amidegel.Themajorprobe C-binding complex, IC-1, is indicated by an arrow.
compete for the IC-1 complex at higher doses, competition was less efficient than that observed with the homologous site (Fig. 7, competitor C). Furthermore, there was little difference between HeLa cells and IMR-32 cells when the CCAATboxortheOct-1sitewasusedasprobe (Fig.7and datanot shown). Overall, the results indicate that the IC-1 factor and theCCAATboxfactorortheOct-I factordonot
specifically
bind identical sites and that the competition observed with the CCAAT box site for IC-1 was due toless-specific
similarities.Our current data demonstrate that the IC-1 factor is selectively present in the human neuroblastoma cells IMR-32 (compared with HeLa cells) and binds a DNA sequencewhichis
selectively
required foroptimal
activity
in that cell type. Weanticipate
thatbinding
of this factor is involved inaugmenting
expression
in the neuroblastoma cells, and we arecurrently
characterizing
thebinding site,
cell type, and tissue location in orderto
provide
evidence for the involvement ofIC-1in the selectiveactivity
of the LAT promoterin neuronal cells.VOL. 66,1992
on November 9, 2019 by guest
http://jvi.asm.org/
DISCUSSION
During a latent infection with HSV, there is a dramatic
alteration in the pattern of virus gene expression from that found in a productively infected cell. There is a complete
absence of detectable amounts of any of the virus gene products normally expressed during a productive infection
and abundant expression of a single transcription unit (LAT). Although stabilization ofan RNAspecies may
con-tribute to the selective detection of LAT (11), it has been demonstrated that insertion of theLAPregionupstreamofa
target reporter gene will allow selective expression of the target gene in latently infected neurons (9, 18). The most
reasonableinterpretation oftheseresultsisthatthe selective expression of LAT is mediated by the selective activityof the LAP in neurons and that this promoter in turn is regulated by specificcis-acting sequenceswhich are absent
in other HSV promoters and which bind positively acting transcription factorspresent inneuronal cells. Other
possi-bilities, including, for example, the precise location of the
promoter in the HSV genome and/or the presence of
se-quences which mediate the absence of genomic repression (butnotby bindingpositively acting factors), whilepossible, seem intuitively less likely.
In thiswork, wehave extended ourprevious
demonstra-tionofselectiveactivity of theLAPin human neuroblastoma cells (1) with results which demonstrate the presence of upstream regulatory sequences which are required for pro-moteractivityintheneuroblastoma cells but dispensablefor activityin human epithelial cells (HeLa). Those regulatory sequences were contained within or overlapped 100 bases mapping fromapproximately -258 to -138 and selectively exertedadramatic 16-fold effectonexpression in neuroblas-tomacells.Ourpresent evidenceindicates (i) that theremay
bemore thanoneelement within this region contributing to
expressionintheneuroblastoma cells and (ii) that the region between the EcoRV and PstI sites from -161 to -138 encompasses at least one of these elements. Furthermore, we have identified a factor, present in the neuroblastoma cellsandnotin HeLacells, which bindstothe -161to-138 region. Taken together, these results indicate that the
ele-mentwe haveidentifiedand the factor which binds toitare
involvedinthecell-type-specific activityof the LAP. Within the probe used to detect the IC-1 factor, there are two
palindromes 6 and 8 bp in length and an element which is
wellconserved in the corresponding regionupstream ofthe HSV-2LAT(3, 6,24a,26,40,46). Wearecurrently refining
precise sequence requirements for IC-1 binding by contact
analysis andgel retardation studies with specifically altered probes.
Consistent results have recently been publishedby Devi-Raoetal. (8), who showed thatsequenceelements between -258 and -161wereselectively required forLAPactivityin
the murine neuroblastoma cell line Neura-2A. Deletion of
this sequence resulted in a sixfold decrease in Neura-2A
cellsbutatwofoldincrease in rabbit skin cells.Zwaagstraet
al. (50)alsorecentlyreporteda4-fold increasein expression
specifically in neuronal cells that wasconferred by aregion
encompassing -283 to -162, but these workers have also
reported that sequences from between -608 and -283
increased expression by up to 3-fold in cells of neuronal origin but repressed expression by 3- to 12-fold in cells of nonneuronal origin(51). We have noevidencethat either of
theselattereffectsoperatesin IMR-32orHeLa cells. Zwaag-stra etal. (50) furtherdemonstrated thepresenceofafactor
which bound to a site between -72 and -65 in the LAP.
However, this factor, which may be related to USF/MLTF, a helix-loop-helix protein which binds to the consensus sequence CACGTG, was present in both HeLa cells and neuroblastoma cells, and the site was equally important for promoter activity in both cell types. Nonetheless, it is possible that the upstream cell-type-specific element which we have identified could act upon or in concert with cell-type-common downstream elements.
In another study examining the regulation of the LAP, Leib et al. (23) demonstrated that a cyclic AMP response element, located around -42, binds purified CREB factor and possibly an additional factor present in PC12 cells. Although the relevance of this site to neuronal-cell-specific expression was not addressed, the cyclic AMP response element was shown to be functional in mediating a three- to fourfold response to cyclic AMP by the LAP. Surprisingly, our results show (Fig. 4) that an oligonucleotide which encompasses the cyclic AMP response element, while hav-ing little effect in IMR-32 cells, significantly increases con-stitutive expression of the IEllOK promoter in HeLa cells. Generally,cis-acting elements and regulatory factors gov-erning nerve-cell-specific gene expression have not been well characterized, and when they have been examined in detail, multiple motifs are frequently required to generate efficient selective transcription, for example, in the human calcitonin ordopa-decarboxylase genes (21, 39). In this latter gene, multiple elements within a 600-bp distal enhancer in conjunction with a promoter proximal element at -70 are required to confer cell-type-specific neuronal expression. Our computer analysis of the sequence of the LAP has revealed potential sites in addition to those described above for anumber of factors, including sites for NTF-1, a factor thatregulates homeotic geneexpression in neurons (10), and CFla, a POU-domain protein involved in selective expres-sion in dopaminergic neurons (20). Since present results frommany systems indicate that selective genetranscription is mediated by aspecific complex combination of factors, it may be that several additional sites are required for LAT expression in neurons. We propose that the sites involved in selective activity of LAP in neuroblastoma cells may be required for expression in neurons and that the factor binding one of these sites may regulate promoter activity in combination with other factors.
Even when the identification of cis-acting elements pro-posed to be required for specific neuronal expression is confirmed by analysis of in vivo LAT expression of specif-ically altered viruses, the analysis of the corresponding regulatory factors cannot easily proceed without the use of neuronal cell lines or extracts of neuronal tissue. Our current studies are directed towards identification of other factors which are present only in neuroblastoma cells or neuronal cells and are involved in LAP activity. In addition, the ability to extract enough protein from small numbers of trigeminal ganglia for characterization of DNA-binding ac-tivity and specificity (29) should further facilitate the identi-fication of factors likely to be relevant to in vivo promoter activity.
REFERENCES
1. Batchelor, A. H., and P. O'Hare. 1990. Regulation and cell-type-specific activity of a promoter located upstream of the latency-associatedtranscript of herpes simplex virus type 1. J. Virol. 64:3269-3279.
2. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. 3. Burke, R.L., K. Hartog, K. D. Croen, and J. M. Ostrove. 1991.
on November 9, 2019 by guest
http://jvi.asm.org/
LATENCY-ASSOCIATED PROMOTER OF HSV-1 3581
Detection and characterization of latent HSV RNA by in situ and Northern blot hybridization in guinea pigs. Virology 181: 793-797.
4. Chen, C., and H. Okayama. 1987. High efficiency transforma-tion of mammalian cells by plasmid DNA. Mol. Cell. Biol. 7:2745-2752.
5. Croen, K. D., J. M. Ostrove, L. J. Dragovic, J. E. Smialek, and S. E. Straus. 1987. Latent herpes simplex virus in human trigeminal ganglia. N. Engl. J. Med. 317:1427-1432.
6. Croen, K. D., J. M. Ostrove, L. J. Dragovic, and S.E. Straus. 1991. Characterization of herpes simplex virus type 2 latency-associated transcription in human sacral ganglia and in cell culture. J. Infect. Dis. 163:23-28.
7. Deatly, A. M., J. G. Spivack, E. Lavi, and N. W. Fraser. 1987. RNA from an immediate-early region of the type 1 herpes simplex virus genome is present in the trigeminal ganglia of latently infected mice. Proc. NatI. Acad. Sci. USA 84:3204-3208.
8. Devi-Rao, G. B., S. A. Goodart, L. M. Hecht, R. Rochford, M. K. Rice, and E. K. Wagner. 1991. Relationship between polyadenylated and nonpolyadenylated herpes simplex virus type 1latency-associated transcripts. J. Virol. 65:2179-2190. 9. Dobson, A. T., F. Sedarati, G. Devi-Rao, W. M. Flanagan, M. J.
Farrell, J.G. Stevens, E. K. Wagner, and L. T. Feldman. 1989.
Identification of the latency-associated transcript promoter by expression of rabbit beta-globin mRNA in sensory nerve ganglia latently infected with a recombinant herpes simplex virus. J. Virol. 63:3844-3851.
10. Dynlacht,B. D., L. D.Attardi, A. Admon, M. Freeman, and R. Tjian. 1989. Functional analysis of NTF-1, a developmentally regulated Drosophila transcription factor that binds neuronal cis elements. Genes Dev. 3:1677-1688.
11. Farrell, M. J., A. T. Dobson, and L. T.Feldman. 1991. Herpes simplexvirus latency-associated transcript is a stable intron. Proc.Natl. Acad. Sci. USA 88:790-794.
12. Fried, M., and D. M. Crothers. 1981. Equilibria and kinetics of lacrepressor-operator interactions by polyacrylamide gel
elec-trophoresis.NucleicAcids Res. 9:6505-6525.
13. Garner, M. M., and A. Revzin. 1981. A gel electrophoresis methodfor quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids. Res. 9:3047-3060.
14. Gordon, Y. J., B. Johnson, E. Romanowski, and T. Araullo-Cruz. 1988. RNAcomplementary to herpes simplex virus type 1
ICPOdemonstrated in neurons of human trigeminal ganglia. J. Virol. 62:1832-1835.
15. Gorman, C. M.,L.F. Moffat, and B. Howard. 1982. Recombi-nantgenomeswhich express chloramphenicol acetyltransferase inmammalian cells. Mol. Cell. Biol. 2:1044-1051.
16. Graham, F. L., andA.J.Vander Eb.1973. A newtechnique for the assayofinfectivityof humanadenovirus 5 DNA. Virology 52:456-457.
17. Hill, T. J. 1985.Herpessimplex virus latency, p. 175-252.In B. Roizman (ed.), The herpesviruses, vol. 3. Plenum Publishing
Corp., New York.
18. Ho,D.Y.,and E.S.Mocarski. 1989. Herpessimplexvirus latent RNA (LAT) isnot required for latent infection in the mouse. Proc.Natl. Acad. Sci. USA86:7596-7600.
19. Honess, R.W.,and D. H.Watson. 1977.Unity and diversityin the herpesviruses. J. Gen. Virol. 37:15-37.
20. Johnson, W. A., andJ. Hirsh. 1990. Binding ofa Drosophila POU-domain protein to a sequence element regulating gene expression inspecificdopaminergic neurons. Nature(London) 343:467-470.
21. Johnson,W. A., C. A. McCormick, S. J. Bray, and J.Hirsh. 1989.Aneuron-specificenhancer of theDrosophila dopa
decar-boxylasegene. GenesDev. 3:676-686.
22. Krause, P. R., K. D.Croen, S. E. Straus, andJ. M. Ostrove. 1988. Detection and preliminary characterization of herpes
simplex virus type 1 transcripts in latently infected human
trigeminal ganglia. J. Virol. 62:4819-4823.
23. Leib, D.A., K. C. Nadeau, S. A.Rundle, and P. A. Schaffer.
1991. The promoter of the latency-associated transcripts of herpes simplex virus type 1 contains a functional cAMP-re-sponse element: role of the latency-associated transcripts and cAMP in reactivation of viral latency. Proc. Natl. Acad. Sci. USA 88:48-52.
24. Mackem, S., and B. Roizman. 1982. Structural features of the herpes simplex virus alpha gene 4, 0, and 27 promoter-regula-tory sequences which confer alpha regulation on chimeric thymidine kinasegenes. J. Virol. 44:939-949.
24a.McGeoch, D. Personalcommunication.
25. McGeoch, D. J., M. A. Dalrymple, A. J. Davison, A. Dolan, M. C.Frame, D. McNab, L. J. Perry, J. E. Scott, and P. Taylor. 1988. The completeDNAsequence of the long unique region in the genome of herpes simplex virus type 1. J. Gen. Virol. 69:1531-1574.
26. Mitchell, W. J., S. L.Deshmane,A. Dolan, D. J.McGeoch, and N. W. Fraser. 1990. Characterization of herpes simplex virus type2transcription during latent infection of mousetrigeminal ganglia. J.Virol. 64:5342-5348.
27. Mitchell, W. J., R. P. Lirette, and N. W. Fraser. 1990. Mapping of low abundance latency-associated RNA in the trigeminal ganglia of mice latentlyinfected with herpes simplex virus type 1. J. Gen. Virol. 71:125-132.
28. Mitchell, W. J., I.Steiner, S. M.Brown, A. R. MacLean, J. H. Subak-Sharpe,and N. W.Fraser. 1990. A herpes simplex virus type 1 variant, deleted in the promoter region of the latency-associated transcripts, does notproduce any detectable minor RNAspeciesduring latency in the mouse trigeminal ganglia. J. Gen. Virol. 71:953-957.
29. O'Hare, P., and S. Efstathiou. Unpublished data.
30. O'Hare, P., and C. R. Goding. 1988. Herpes simplex virus regulatory elements and the immunoglobulin octamer domain bind acommonfactor and areboth targetsforvirion transacti-vation. Cell 52:435-445.
31. O'Hare, P., and G. S.Hayward. 1984. Expression of recombi-nant genes containing herpes simplex virus delayed-early and immediate-early regulatory regions andtrans-activationby her-pesvirus infection. J.Virol.52:522-531.
32. Perry, L. J., and D. J. McGeoch.1988. The DNAsequencesof the long repeat region and adjoining parts ofthe long unique region in the genome ofherpes simplex virus type 1. J. Gen. Virol.69:2831-2846.
33. Price, R. W. 1985. Herpes simplex viruslatency:adaptation to the peripheral nervous system. II. Cancer Invest.3:389-403.
34. Roberts, M. S., A.Boundy, P. O'Hare, M. C. Pizzorno, D. M. Ciufo, and G. S. Hayward. 1988. Direct correlation between a negative autoregulatory response element at the cap site of the herpes simplex virus type 1 IE175 (alpha 4) promoter and a specific binding site for the IE175 (ICP4) protein. J. Virol. 62:4307-4320.
35. Rock,D.L.,A. B.Nesburn,H.Ghiasi,J.Ong,T. L.Lewis, J. R. Lokensgard, and S. L. Wechsler. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J. Virol.
61:3820-3826.
36. Spivack, J. G., and N. W. Fraser. 1987. Detection of herpes simplex virustype1 transcripts duringlatent infection in mice. J. Virol. 61:3841-3847.
37. Steiner, I., J.G. Spivack, D. R. O'Boyle, E. Lavi, and N. W. Fraser.1988.Latentherpes simplexvirus type 1transcriptionin human trigeminalganglia. J. Virol. 62:3493-3496.
38. Stevens, J.G.,E. K.Wagner, G. B.Devi-Rao,M. L.Cook,and L. T. Feldman. 1987. RNA complementary to a herpes virus alpha gene mRNA is prominent in latently infected neurons. Science 235:1056-1059.
39. Stolarsky-Fredman, L., S. E.Leff, E. S.Klein,E. B. Crenshaw, J. Yeakley, and M. G. Rosenfeld. 1990. A tissue-specific en-hancer in therat-calcitonin/CGRP gene isactiveinboth neural andendocrine celltypes. Mol. Endocrinol. 4:497-5t)4.
40. Tenser, R.B., W. A.Edris,K. A.Hay,and B. E. de Galan. 1991. Expression of herpes simplex virus type 2 latency-associated
transcript in neuronsand nonneurons.J. Virol.65:2745-275(0. 41. Tumilowicz,J. J.,W. W.Nichols, J. J.Cholon,and A. E.Green. VOL. 66, 1992
on November 9, 2019 by guest
http://jvi.asm.org/
1970. Definition ofacontinuous cell line derived from
human-neuroblastoma. Cancer Res. 30:2100-2118.
42. Wagner, E. K., G. Devi-Rao, L. T.Feldman, A. T. Dobson, Y. F. Zhang, W. M. Flanagan, and J. G. Stevens. 1988. Physical characterization of the herpes simplex virus latency-associated transcript inneurons.J. Virol. 62:1194-1202.
43. Wagner, E. K., W. M. Flanagan, G. Devi-Rao, Y. F. Zhang, J. M.Hill, K. P.Anderson, and J. G. Stevens. 1988. The herpes simplex viruslatency-associated transcript is spliced during the latent phase of infection. J. Virol. 62:4577-4585.
44. Wechsler, S.L., A. B. Nesburn, R. Watson, S. M. Slanina, and H.Ghiasi. 1988. Finemapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J. Gen. Virol. 69:3101-3106.
45. Wechsler, S.L.,A. B. Nesburn,R.Watson, S. M.Slanina, and H. Ghiasi. 1988. Fine mapping of the latency-related gene of herpes simplex virustype 1: alternative splicing produces dis-tinct latency-related RNAs containingopen reading frames. J.
Virol.62:4051-4058.
46. Wechsler, S. L., A. B. Nesburn,J. Zwaagstra, and H. Ghiasi. 1989. Sequence of the latency-related geneofherpes simplex
virustype 1.Virology168:168-172.
47. Wildy, P., H. Field, and A. A. Nash. 1982. Classical herpes latencyrevisited.Soc. Gen. Microbiol. Symp.33:133-167. 48. Wu,C. 1984. Twoprotein-bindingsites inchromatinimplicated
in theactivation ofheat-shockgenes.Nature(London) 309:229-334.
49. Zwaagstra,J., H. Ghiasi, A. B.Nesburn, andS. L. Wechsler. 1989. In vitro promoter activity associated with the latency-associated transcript gene ofherpes simplex virus type 1. J. Gen. Virol. 70:2163-2169.
50. Zwaagstra,J. C., H. Ghiasi, A. B. Nesburn, and S. L. Wechsler. 1991. Identification of a major regulatory sequence in the
latencyassociatedtranscript (LAT)promoterofherpessimplex
virustype 1(HSV-1). Virology 182:287-297.
51. Zwaagstra,J. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, S. C.
Wheatley,K.Lillycrop,J.Wood, D. S. Latchman, K. Patel, and S. L.Wechsler. 1990. Activity of herpes simplex virus type1 latency-associated transcript (LAT)promoterinneuron-derived cells: evidence for neuron specificity and for a large LAT
transcript. J. Virol. 64:5019-5028.