Polymerase Binding and RNA Synthesis
Jesper K. Marklund,aQiaozhen Ye,bJinhui Dong,bYizhi Jane Tao,band Robert M. Kruga
Department of Molecular Genetics and Microbiology, Institute for Cell and Molecular Biology, University of Texas at Austin, Texas, USA,aand Department of Biochemistry and Cell Biology, Rice University, Houston, Texas, USAb
Many proposed mechanisms for influenza A viral RNA synthesis include an interaction of the nucleoprotein (NP) with the viral polymerase. To identify an NP sequence required for this interaction, we used the cryoelectron microscopic structure of an influenza virus miniribonucleoprotein as a guide for choosing promising surface-exposed sequences. We show that three amino acids (R204, W207, and R208) located in a loop at the top of the head domain of NP are required for functional interaction with the viral polymerase. Quantitative reverse transcription-PCR (RT-PCR) measurements of RNAs synthe-sized in minigenome assays established that each of these NP amino acids is required for viral RNA synthesis. The muta-tion of these three amino acids does not affect nuclear localizamuta-tion or RNA-binding and oligomerizamuta-tion activities of NP.
In vitrobinding experiments with purified virus polymerase and NPs established that these three amino acids are required
for NP binding to the viral polymerase.
I
nfluenza A viruses cause a contagious respiratory disease in hu-mans, resulting in annual epidemics and periodic worldwide pandemics. Influenza A viruses transcribe and replicate their eight negative-sense viral RNA (vRNA) segments in the nucleus of in-fected cells. The influenza A virus polymerase, which is comprised of the three polymerase proteins (PB1, PB2, and PA), uses 5= -capped RNA primers derived from cellular pre-mRNAs to tran-scribe the vRNA segments to produce plus-sense polyadenylated viral mRNAs (3,18). In the presence of the viral nucleoprotein (NP), the tripartite polymerase catalyzes the replication of the vRNA segments (20,23). The replication of vRNA consists of two stages. During the first stage, genomic vRNA is replicated into a full-length copy, denoted cRNA, and during the second stage cRNA is copied into vRNA. Both cRNA and vRNA contain a 5=triphosphate end (29), indicating that their synthesis is initiatedde novowithout a primer, in contrast to the primer-dependent initi-ation of viral mRNA synthesis.
Various mechanisms have been proposed for cRNA and vRNA synthesis, but no mechanism has been definitively established (10,
14, 20,22, 25). Many of the proposed mechanisms include an interaction of the NP with the viral polymerase (1,14,19,20). In addition, NP binds to the vRNA segments along their entire lengths at regular intervals to form viral ribonucleoproteins (vRNPs) (5,6). Similar binding of NP to the cRNA segments most likely occurs. The crystal structure of NP shows that it is com-prised of two major domains, denoted the body and head domains (15,28). Between the two domains is a deep groove lined by basic residues that bind single-stranded RNA. On the side of NP oppo-site the RNA binding groove is the tail loop that enables NP to form oligomers by inserting this tail loop into a neighboring NP molecule.
Previous mutational analysis of the NP did not identify a se-quence in NP that is required for its binding to the viral polymer-ase (12,13,16). Here, we identify this NP sequence: it is comprised of several amino acids located in a loop (amino acids 203 to 209) at the top of the head domain. This NP sequence is highly conserved in mammalian and avian influenza A viruses.
MATERIALS AND METHODS
Dual-luciferase reporter assays for viral RNA replication.Transfections were carried out using Mirus transfection reagent. 293T cells in 6-cm plates were transfected with 0.25g each of pcDNA3 plasmids expressing PA, PB1, and PB2, 0.5g of a pcDNA3 plasmid expressing NP, and 0.5g of a pHH21 plasmid that expresses in the negative sense the firefly lucif-erase containing the 5=- and 3=-terminal regions of the NS vRNA of influ-enza A/Udorn/72 (Ud). Where indicated, a different pHH21 plasmid was used, which expresses in the positive sense the firefly luciferase containing the 5=- and 3=-terminal regions of the Ud NS cRNA. In addition, a pcDNA3 plasmid expressing renilla luciferase was transfected to correct for differences in transfection efficiencies. The activities of the two lu-ciferases were determined using a Mithras microplate luminometer.
Measurement of vRNA and viral mRNA using quantitative RT-PCR.
RNA was TRIzol extracted from a dual-luciferase reporter assay in which the positive-sense firefly luciferase RNA (cRNA) served as the template. The RNA was then treated with 1 U of RNase-free DNase for 30 min at 37°C to degrade any residual DNA, followed by phenol-chloroform ex-traction. RNA was ethanol precipitated and dissolved in 40l of water. Oneg of RNA, which corresponds to equal cell equivalents, was reverse transcribed using primers specific for firefly luciferase vRNA-sense RNA (gacgtcatgaataggatgaatcgAGCAAAAGCAGGGTGACAAAG), firefly lu-ciferase mRNA (cgcagatcgttcgagtcgTTTTTTTTTTTTTTTTTTTTTTAT CATTAC), or renilla luciferase mRNA (cgcagatcgttcgagtcgTTTCATCAG GTGCATCTTCTTGC). The sequences shown in lowercase are not complementary to the RNA and serve as tags that are used in the PCR step (11). The Universal Probe Library 142 (Roche) (GCCAAGA) was used for both renilla luciferase and firefly luciferase RNAs. The PCR was carried out with the FastStart TaqMan Probe Master with Rox (Roche) and prim-ers specific for the particular RNA: firefly luciferase vRNA-sense RNA (gacgtcatgaataggatgaatcg and CATTACACGGCGATCTTTCC), firefly luciferase mRNA (cgcagatcgttcgagtcg and CGACGCAAGAAAAATCAG
Received4 January 2012 Accepted10 April 2012 Published ahead of print24 April 2012
Address correspondence to Robert M. Krug, [email protected].
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.00014-12
on November 7, 2019 by guest
http://jvi.asm.org/
AGAGA), or renilla luciferase mRNA (cgcagatcgttcgagtcg and CGCAGA TCGTTCGAGTCGT). Real-time PCR analysis was carried out using the Perkin-Elmer/Applied Biosystems 7900HT sequence detector. The threshold cycle (CT) values for the luciferase RNAs were normalized to
theCTvalues of renilla luciferase mRNA. The quantitative reverse
tran-scription-PCRs (RT-PCRs) were performed in triplicate. Error bars are shown inFig. 3.
Immunofluorescence and confocal microscopy. HeLa cells were grown in 4-chamber covered glass-bottom microscopic slides and were transfected with pcDNA3 plasmids expressing either wild-type (wt) or triple-mutant NP. After 12 h, the cells were washed with phosphate-buff-ered saline (PBS) and fixed with 4% paraformaldehyde for 20 min at room temperature. Following three washes with PBS, the cells were permeabil-ized in 0.5% Triton X-100 in PBS for 10 min on ice. Cells were then washed three times with PBS and incubated in blocking solution (PBS-NGS-gelatin; 0.5 ml of 100% normal goat serum and 0.2 ml of 10% gelatin in 1⫻PBS) for 30 min at room temperature, followed by incubation with NP mouse monoclonal antibody (1:200 dilution in PBS-NGS-gelatin blocking solution) for 1 h at room temperature. The cells then were washed 3 times with PBST (PBS with 0.1% Tween 20) and incubated with fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibody (1: 200 dilution in blocking buffer) in the dark for 30 min at room tempera-ture. The cells were washed as described above, stained with 4= ,6-diamidino-2-phenylindole (DAPI; 1l of 1 mg/ml stock concentration in 15 ml PBS), washed with PBS, and then observed using the 40⫻oil im-mersion objective in a Leica confocal microscope.
RNA-binding assay using fluorescence polarization.The wt and tri-ple-mutant (R204A, W207A, and R208A) NP of influenza A/WSN/33 (WSN) virus were produced and purified as described previously (28). A 5=-fluoresceinated 20-base RNA oligonucleotide (5=-AGUAGAACAGGGU GACAAAG-3=with the conserved 5=vRNA sequence) was chemically synthesized (Dharmacon). The wt or triple-mutant NP was serially titrated into a 1-ml reaction mixture containing 20 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5 mM 2-mercaptoethanol, 1 mM EDTA, and 0.2 nM fluoresceinated oligonucleotide. Samples were excited at 490 nm and emission was measured at 520 nm using a PanVera Beacon 2000 FP system at room temperature. Each protein titration was repeated in triplicate. The data were fitted to a sigmoidal curve using Igor Pro (Wavemetrics, CA).
Assay for NP oligomerization.The wt or triple-mutant NP was equil-ibrated in 50 mM Tris-HCl, pH 7.5, 200 mM NaCl, 5 mM 2-mercapto-ethanol, and 10% glycerol and then loaded onto a Superdex 200 HR 10/60 column equilibrated with the same buffer. The molecular weight of the major NP species was estimated using a molecular weight marker kit (Sigma).
Assays for the binding of NP to the polymerase. (i) Transfection assay.293T cells in a 6-cm dish were transfected with pcDNA3 plasmids expressing Ud PB1, PB2, and PA with a C-terminal 3⫻Flag tag and either the Ud wt NP or the Ud double-mutant (W207A and R208A) NP. Extracts were prepared from cells at 24 h posttransfection by disrupting the cells with 600l of 100 mM Tris-HCl (pH 7.5), 250 mM NaCl, 0.5% NP-40, and 1 mM phenylmethylsulfonyl fluoride. After centrifugation at 18,000⫻gfor 2 min to remove cell debris, the extract was mixed with Flag M2 monoclonal antibody for 18 h at 4°C. Sepharose A and Sepharose G beads (GE Healthcare) then were added, and the mixture was incubated for 1 h at 4°C. The beads were washed 3 times for 10 min with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.5% NP-40, and protein was eluted from the beads using 0.625 mM 3⫻Flag peptide (Sigma) in a buffer of 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1.2% Triton X-100, 10% glycerol, and 0.2 mM EDTA. The immunoprecipitate was analyzed by immunoblotting using the antibody directed against the Ud NP.
(ii) Binding assay using purified proteins.The tripartite viral poly-merase containing PB1, PB2, and PA with an N-terminal His tag was expressed using the baculovirus expression system and purified as de-scribed previously (14). The polymerase (0.5g) was incubated with 2g of either wt or triple-mutant NP for 30 min at 30°C in a buffer containing
50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.5% NP-40. The reactions then were immunoprecipitated with NP antibody, and the immunopre-cipitates were analyzed by immunoblots probed with either PB1, PB2, His, or NP antibody.
RESULTS
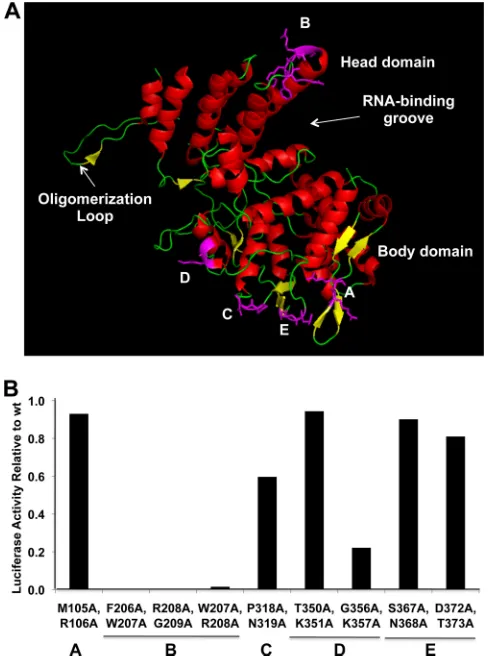
Identification of an NP sequence that is required for viral RNA synthesis as determined in minigenome assays.We used the cryoelectron microscopic (cyro-EM) structure of an influenza vi-rus mini-RNP (5) as a guide for our mutational analysis of NP to identify a sequence that is required for viral RNA synthesis. The X-ray crystal structure of NP has been fitted into the cryo-EM NP structure using the best statistics (5), thereby indicating which NP sequences might be directed toward the tripartite polymerase. Of these NP sequences, we chose those that are surface exposed in the NP crystal structure and are not close to the RNA-binding groove (15,28). These sequences are denoted A through E inFig. 1A. We changed two residues of the NP of influenza A/Udorn/72 (Ud) virus in each of these regions to alanines and tested the ability of the resulting mutant NPs to support viral RNA synthesis, which
FIG 1Identification of an NP sequence required for viral RNA synthesis. (A) The crystal structure of NP (28). The head and body domains, the RNA-binding groove, and the oligomerization loop are denoted. The locations of the double-alanine mutations are shown in magenta. The B region corresponds to the loop between helix 4 and 5, which is located at the top of the head domain. (B) The activities of the mutant NPs in the minigenome dual-luciferase assay described in the text. The firefly/renilla luciferase activities of the mutants are shown relative to the activity of wt NP. Because this was an initial screen, this assay was carried out only once.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:2.585.299.543.67.395.2]was measured using the dual-luciferase minigenome assay. We cotransfected 293T cells with pcDNA3 plasmids which, under the control of a polymerase II promoter, express the PA, PB1, and PB2 proteins, and either the wt or a mutated NP. The vRNA template was provided by transfecting a pHH21 plasmid, which, under the control of a polymerase I promoter, expresses in the negative sense the firefly luciferase containing the 5=- and 3=-terminal regions of the Ud NS vRNA. In addition, a polymerase II plasmid express-ing the renilla luciferase was transfected to correct for differences in transfection efficiencies. Of the five mutated NPs that were tested, only the mutant NP containing double-alanine mutations in the B sequence lost all activity in this minigenome assay
(Fig. 1B).
The B sequence is located in a loop at the top of the head domain (Fig. 1A). It was resolved in some, but not all, of the NP crystal structures (15,28), suggesting that it has a relatively flexible structure. The sequence alignment of available NP sequences of avian and mammalian influenza A viruses showed that the se-quence in the loop, DRNFWRG (amino acids 203 to 209), is highly conserved (⬎98%).
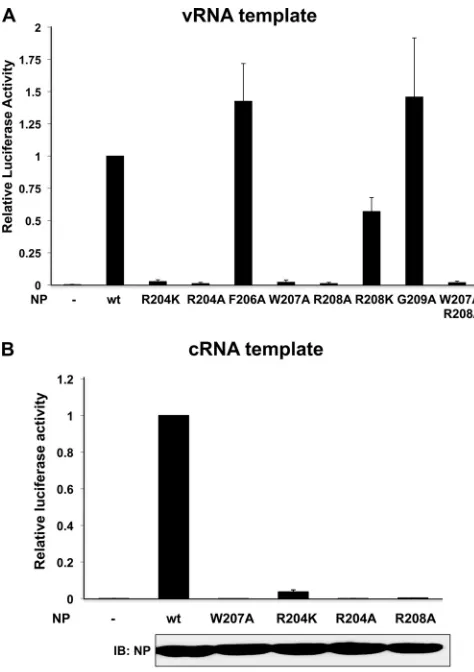
To determine the role of individual amino acids in the 203 to 209 loop, we tested Ud NP mutants containing alanine substitu-tions at position R204, F206, W207, R208, or G209 in the minige-nome assay (Fig. 2A). The R204A, W207A, and R208A NP mutant proteins lost essentially all activity in the minigenome assay, whereas the F206A and G209A NP mutant proteins were fully active in the minigenome assay. These results show that R204, W207, and R208 are required for this activity.
We also changed either R204 or R208 to K to determine the effect of a smaller basic amino acid at these positions. The R204K NP mutant protein lost essentially all activity, providing further evidence that R204 is required for activity. In contrast, the R208K NP mutant protein possessed approximately 50% of the activity of the wt NP.
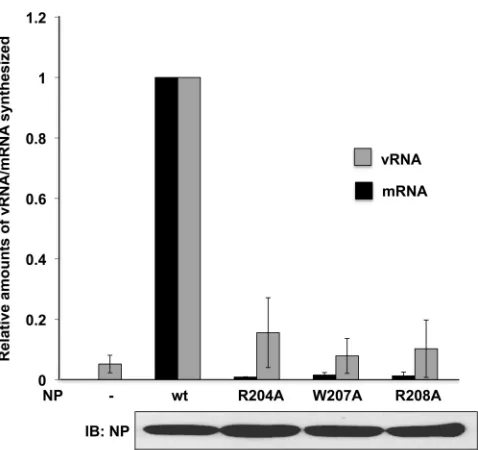
We carried out a second minigenome assay in which the vRNA template was replaced with a cRNA template, specifically a pHH21 plasmid that expresses in the positive sense the firefly luciferase containing the 5=- and 3=-terminal regions of the Ud NS cRNA. Because the viral polymerase must replicate cRNA to produce vRNA molecules before mRNAs can be synthesized, this luciferase reporter is dependent on vRNA replication. As shown inFig. 2B, R204, W207, and R208 NP mutant proteins were also inactive in the cRNA-templated minigenome assay. The amount of both vRNA and mRNA synthesized in the cRNA-templated minige-nome assay was measured directly using quantitative RT-PCR
(Fig. 3). The assays for vRNA showed a low level of vRNA
synthe-sis in the absence of NP (approximately 4% of that produced with wt NP) and only 2- to 4-fold more vRNA synthesis with the R204A, W207A, and R208A NP mutant proteins (corresponding to approximately 8 to 16% of that produced with wt NP). In ad-dition, little or no mRNA synthesis was detected in the absence of NP or in the presence of these three NP mutant proteins. We conclude that these three NP mutant proteins have little or no activity in viral RNA synthesis. Similar results were obtained using the NP from another influenza A virus strain, H1N1 influenza A/WSN/33 (WSN) (data not shown).
Nuclear localization, RNA-binding, and oligomerization ac-tivities of NP are not affected by the R204A, W207A, and R208A mutations.NP has a functional nonconventional nuclear local-ization signal (NLS) near its amino terminus (7,26). It was
pos-tulated that NP also has a bipartite NLS extending from amino acids 198 to 216 (27), but this sequence does not bind importin-␣ and plays at best a limited role in the nuclear import of NP (7,16). To compare the localization of the wt and triple-mutant (R204A, W207A, and R208A) NPs, plasmids expressing either the WSN wt NP or the WSN triple-mutant NP were transfected into HeLa cells, and immunofluorescence was performed at 12 h using an NP monoclonal antibody (Fig. 4A). Stained cells were observed using a confocal microscope. Both the wt and triple mutant NP were consistently localized in the nucleoplasm, and no difference in localization between the wt and triple-mutant NPs was observed. Several experiments involving the examination of numerous transfected cells at various times after transfection by confocal microscopy did not detect the transient nucleolar localization of wt NP reported by others (16).
To compare the RNA-binding activities of the wt and triple-mutant NP, we purified the wt and triple-triple-mutant NP (WSN virus) proteins. The binding of these two NPs to a 20-base single-stranded RNA was measured using fluorescence polarization (Fig. 4B). The binding of wt NP was found to have aKd(dissociation
constant) of 3.6⫾0.5 nM, and the binding of the triple mutant NP
FIG 2Activity of the NP loop mutants in the vRNA- and cRNA-templated minigenome assays. Shown are firefly/renilla luciferase activities of NP loop mutants relative to wt NP activity in minigenome assays using a vRNA-sense firefly luciferase template (A) or a cRNA-sense firefly luciferase template (B). Error bars show the standard deviations from three independent experiments. The levels of NP synthesis were assayed using an immunoblot probed with the NP antibody.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.300.537.64.400.2]was found to have aKdof 5.0⫾1.4 nM. Although theKdof the
triple-mutant NP appeared to be slightly higher than that of the wt NP, this difference is within the margin of error and is unlikely to be the cause of the dramatic decrease in the ability of the triple-mutant NP to support viral RNA replication. Our results are con-sistent with a previous study that used a qualitative measurement to show that a single alanine substitution at position 204, 207, or 208 did not reduce NP RNA-binding activity (9).
NP forms oligomers by inserting a tail loop into another NP molecule. In the absence of single-stranded RNA, wt NP primarily forms trimers (28). Because the oligomerization loop and its in-sertion site are located far from positions 204, 207, and 208, it was unlikely that mutations at these positions would affect oligomer-ization. As verification, the oligomerization of the wt and triple-mutant NPs was analyzed using size-exclusion chromatography
(Fig. 4C). A predominant species of approximately 150 kDa, the
expected size of a trimer, was found for both the wt and triple-mutant NPs, demonstrating that mutating positions 204, 207, and 208 does not alter the ability of NP to oligomerize. In addition, a previous study using a pulldown assay showed that a single alanine substitution at position 204, 207, or 208 did not significantly re-duce NP-NP interactions (8).
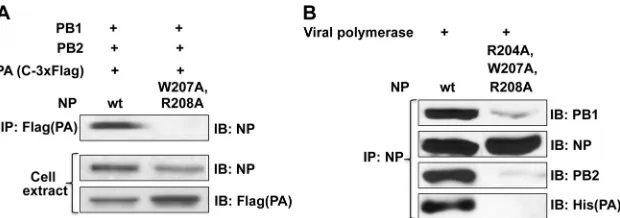
The double-mutant (W207A and R208A) and triple-mutant (R204A, W207A, and R208A) NPs do not bind to the tripartite viral polymerase.Two approaches were taken to assay the bind-ing of the wt and mutant NPs to the tripartite viral polymerase. First, we assayed polymerase binding using a transfection assay. 293T cells were transfected with plasmids expressing Ud PB1, PB2, and PA with a C-terminal 3⫻Flag tag and either the Ud wt NP or
the Ud double-mutant (W207A and R208A) NP (Fig. 5A). Ex-tracts were immunoprecipitated with Flag antibody, followed by immunoblotting using NP antibody. The wt NP, but not the dou-ble-mutant NP, was coimmunoprecipitated with the Flag-tagged PA protein, indicating that the double mutation (W207A and R208A) eliminated polymerase binding.
As a second approach,in vitrobinding experiments were car-ried out with purified NP and tripartite viral polymerase (Fig. 5B).
FIG 3Relative amounts of vRNA and mRNA produced by the NP mutants in the cRNA-templated minigenome assay. RT-PCR was performed on RNA extracted from the indicated minigenome assays, using the primers described in Materials and Methods, to determine the levels of firefly luciferase vRNA, firefly luciferase mRNA, and renilla luciferase mRNA. TheCTvalues for the
luciferase RNAs were normalized to theCTvalues of renilla luciferase mRNA.
The amounts of the vRNAs and mRNAs produced by the mutant NPs are shown relative to the amounts produced by the wt NP. The error bars show the standard deviations from three independent experiments. The levels of NP synthesis were assayed using an immunoblot probed with the NP antibody.
FIG 4Triple-mutant NP behaves likes wild-type NP in nuclear localization, RNA binding, and oligomerization. (A) Nuclear localization of wt and mutant NPs. HeLa cells were transfected with a plasmid expressing either wt or mutant NP. At 12 h posttransfection, cells were fixed, incubated with NP antibody (Ab), processed for immunofluorescence as described in Materials and Meth-ods, and visualized with a confocal microscope. White arrowheads in upper panels point to nucleoli. Lower panels show the DAPI staining of nuclei. (B) RNA-binding activities of the wt and triple-mutant NPs are similar. Fluores-cence polarization was used to measure the binding of single-stranded RNA to purified wt and triple-mutant NP, as described in Materials and Methods. (C) The oligomerization of wt and triple-mutant NP was analyzed using size-exclusion chromatography.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.44.283.62.287.2] [image:4.585.317.518.63.499.2]The purified tripartite viral polymerase containing PB1, PB2, and PA with an N-terminal His tag was incubated for 30 min at 30°C with either the wt NP or the triple-mutant (R204A, W207A, and R208A) NP, followed by immunoprecipitation with NP antibody. Immunoblotting using PB1, PB2, and His(PA) antibodies showed that the wt NP, but not the triple-mutant NP, coimmunoprecipi-tated with the three proteins comprising the viral polymerase, verifying that amino acids R204, W207, and R208 of NP are re-quired for its binding to the viral polymerase.
DISCUSSION
We have shown that three amino acids (R204, W207, and R208) in the loop at the top of the head domain of NP are required for both its binding to the viral polymerase and its ability to support viral RNA synthesis catalyzed by the viral polymerase. The mutation of these amino acids does not have any significant effects on other functions of the NP, namely, nuclear localization, RNA binding, and oligomerization. The lack of effect on nuclear localization is consistent with previous results that the postulated bipartite NLS extending from amino acids 198 to 216 does not bind importin-␣ and plays at best a limited role in the nuclear import of NP (7,16), whereas the nonconventional NLS near its amino terminus is largely, if not totally, responsible for the nuclear import of NP (7,
26). Because the sequence containing these three amino acids is structurally disordered in two of the three subunits of the H1N1 NP trimer crystal structure, it is apparent that this region is struc-turally flexible, and it is unlikely that the alanine substitution of any of the three residues mentioned above would result in major structural distortion or structural misfolding. Indeed, the triple-mutant (R204A, W207A, and R208A) NP exhibited behaviors identical to those of the wt protein in all chromatography purifi-cation steps (i.e., heparin affinity, gel filtration, and ion exchange). Our results also suggest that the residue 204 to 208 loop sequence of NP interacts with the viral polymerase through direct protein-protein interaction, as one of our binding assays used purified viral polymerase and NP samples, thus ruling out the participa-tion of other viral or host factors. Because these three amino acids are highly conserved in the NPs of mammalian and avian influ-enza A viruses, it is likely that these three NP amino acids are required for a functional interaction with the viral polymerase in all influenza A virus strains. Hence, this NP-polymerase interac-tion would be a possible target for the development of antiviral drugs.
These three amino acids may be part of a larger binding site that includes other amino acids located nearby in the three-di-mensional structure. In a previous mutational analysis of con-served amino acids of NP (12), the mutations that strongly re-duced activity in the minigenome assay were located in four regions: (i) in internal regions of the structure (e.g., A260, A337, and A387), where it would be expected to denature the protein; (ii) in regions that would be expected to inhibit oligomerization (e.g., E339, Q405, and F412), which is required for vRNA replica-tion (4); (iii) in areas implicated in RNA binding (e.g., R150, K273, and R355); or (iv) near, or at the top of, the head domain (e.g., R208 and E254). The latter result suggests that a region sur-rounding the loop at positions 204 to 208 in the three-dimen-sional structure is also part of the binding site.
Our experiments do not rule out the participation of amino acids in additional regions of NP in functional binding with the viral polymerase. Evidence for the participation of nonconserved NP amino acids in polymerase binding comes from the observa-tions that NP-viral polymerase interacobserva-tions are important deter-minants of influenza A virus host range (2,21). In a recent study it was shown that some H5N1 NPs preferentially support efficient vRNA replication by viral polymerases whose PB2 proteins con-tain K at position 627, whereas other H5N1 NP preferentially support vRNA replication by viral polymerases whose PB2 pro-teins contain E at position 627 (2). Both H5N1 NP contain the consensus R204, W207, and R208 amino acids and differ from each other at only 14 of the 498 total positions in NP.
Several roles for the NP-polymerase interaction have been pro-posed, but no role has been definitively established (10,14,20,25). The recent identification of short (approximately 20 bases in length) vRNA-sense fragments (denoted svRNAs) in infected cells (17,24) provides support for one of the proposed roles for the NP-polymerase interaction. In this model, the viral polymerase has the inherent capacity to initiate unprimed vRNA synthesis, but it synthesizes only short transcripts (approximately 20 bases long) that do not extend past the surface of the polymerase (10). When the viral NP binds to the polymerase, it also binds to the emerging nascent transcript, enabling the elongation of these transcripts, a process analogous to the promoter clearance mech-anisms of other polymerases. In this model, the svRNAs identified in infected cells, comprising less than 1% of full-length vRNA chains, would correspond to nascent transcripts that have failed to
FIG 5Mutant NP does not bind to the viral polymerase. (A) Transfection assay. 293T cells were transfected with the indicated PB1, PB2, and PA plasmids and either the wt or double-mutant (W207A and R208A) plasmid. Extracts were immunoprecipitated with Flag(PA) antibody, and the immunoprecipitate was analyzed in an immunoblot probed with NP antibody (top panel). The bottom two panels show the cell extract analyzed by immunoblots probed with either NP or Flag(PA) antibody. (B)In vitrobinding experiments with purified, baculovirus-expressed virus polymerase (in which PA contains an N-terminal His tag) and wt or triple-mutant (R204A, W207A, and R208A) NP. After incubation, the mixture was immunoprecipitated with NP antibody and the immunoprecipitates were analyzed by immunoblots probed by PB1, NP, PB2, and His(PA) antibodies.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.138.448.65.174.2]elongate. Future experiments will determine whether this model is viable.
ACKNOWLEDGMENTS
This research was supported by Public Health Service grant AI-077785 to R.M.K. and Y.J.T. and the Welch Foundation grant C1565 to Y.J.T.
REFERENCES
1.Biswas SK, Boutz PL, Nayak DP.1998. Influenza virus nucleoprotein interacts with influenza virus polymerase proteins. J. Virol.72:5493–5501. 2.Bogs J, et al.2011. Reversion of PB2-627E to -627K during replication of an H5N1 clade 2.2 virus in mammalian hosts depends on the origin of the nucleoprotein. J. Virol.85:10691–10698.
3.Bouloy M, Plotch SJ, Krug RM.1978. Globin mRNA are primers for the transcription of influenza viral RNA in vitro. Proc. Natl. Acad. Sci. U. S. A.
75:4886 – 4890.
4.Chan WH, et al.2010. Functional analysis of the influenza virus H5N1 nucleoprotein tail loop reveals amino acids that are crucial for oligomer-ization and ribonucleoprotein activities. J. Virol.84:7337–7345. 5.Coloma R, et al.2009. The structure of a biologically active influenza
virus ribonucleoprotein complex. PLoS Pathog.5:e1000491. doi:10.1371/ journal.ppat.1000491.
6.Compans RW, Content J, Duesburg PH.1972. Structure of the ribonu-cleoprotein of influenza virus. J. Virol.10:795– 800.
7.Cros JF, Palese P.2003. Trafficking of viral genomic RNA into and out of the nucleus: influenza, Thogoto and Borna disease viruses. Virus Res.
95:3–12.
8.Elton D, Medcalf E, Bishop K, Digard P.1999. Oligomerization of the influenza virus nucleoprotein: identification of positive and negative se-quence elements. Virology260:190 –200.
9.Elton D, Medcalf L, Bishop K, Harrison D, Digard P.1999. Identifica-tion of amino acid residues of influenza virus nucleoprotein essential for RNA binding. J. Virol.73:7357–7367.
10. Kawaguchi A, Momose F, Nagata K.2011. Replication-coupled and host factor-mediated encapsidation of the influenza virus genome by viral nu-cleoprotein. J. Virol.85:6197– 6204.
11. Kawakami E, et al.2011. Strand-specific real-time RT-PCR for distin-guishing influenza vRNA, cRNA, and mRNA. J. Virol. Methods173:1– 6. 12. Li Z, et al.2009. Mutational analysis of conserved amino acids in the
influenza A virus nucleoprotein. J. Virol.83:4153– 4162.
13. Mena I, et al.1999. Mutational analysis of influenza A virus nucleopro-tein: identification of mutations that affect RNA replication. J. Virol.73: 1186 –1194.
14. Newcomb LL, et al.2009. Interaction of the influenza A virus nucleocap-sid protein with the viral RNA polymerase potentiates unprimed viral RNA replication. J. Virol.83:29 –36.
15. Ng AK, et al.2008. Structure of the influenza virus A H5N1 nucleopro-tein: implications for RNA binding, oligomerization, and vaccine design. FASEB J.22:3638 –3647.
16. Ozawa M, et al.2007. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol.81:30 – 41. 17. Perez JT, et al.2010. Influenza A virus-generated small RNAs regulate the
switch from transcription to replication. Proc. Natl. Acad. Sci. U. S. A.
107:11525–11530.
18. Plotch SJ, Bouloy M, Ulmanen I, Krug RM. 1981. A unique cap (m7GpppXm)-dependent influenza virion endonucleaase cleaves
capped RNAs to generate the primers that initiate viral RNA transcrip-tion. Cell23:847– 858.
19. Poole E, Elton D, Medcalf L, Digard P.2004. Functional domains of the influenza A virus PB2 protein: identification of NP- and PB1-binding sites. Virology321:120 –133.
20. Portela A, Digard P.2002. The influenza virus nucleoprotein: a multi-functional RNA-binding protein pivotal to virus replication. J. Gen. Virol.
83:723–734.
21. Rameix-Welti MA, Tomoiu A, Dos Santos Afonso E, van der Werf S, Naffakh N.2009. Avian influenza A virus polymerase association with nucleoprotein, but not polymerase assembly, is impaired in human cells during the course of infection. J. Virol.83:1320 –1331.
22. Resa-Infante P, Jorba N, Coloma R, Ortin J.2011. The influenza virus RNA synthesis machine: advances in its structure and function. RNA Biol.
8:207–215.
23. Shapiro GI, Krug RM.1988. Influenza virus RNA replication in vitro: synthesis of viral template RNAs and virion RNAs in the absence of an added primer. Virology62:2285–2290.
24. Umbach JL, Yen HL, Poon LL, Cullen BR.2010. Influenza A virus expresses high levels of an unusual class of small viral leader RNAs in infected cells. mBio1:e00204 –10. doi:10.1128/mBio.00204-10. 25. Vreede FT, Jung TE, Brownlee GG.2004. Model suggesting that
repli-cation of influenza virus is regulated by stabilization of replicative inter-mediates. J. Virol.78:9568 –9572.
26. Wang P, Palese P, O’Neill RE.1997. The NPI-1/NPI-3 (karyopherin alpha) binding site on the influenza A virus nucleoprotein NP is a non-conventional nuclear localization signal. J. Virol.71:1850 –1856. 27. Weber F, Kochs G, Gruber S, Haller O.1998. A classical bipartite nuclear
localization signal on Thogoto and influenza A virus nucleoproteins. Vi-rology250:9 –18.
28. Ye Q, Krug RM, Tao YJ.2006. The mechanism by which influenza A virus nucleoprotein forms oligomers and binds RNA. Nature444:1078 – 1082.
29. Young RJ, Content J.1971. 5=Terminus of the influenza virus RNA. Nature230:140 –142.