Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Induction of a Mucosal Cytotoxic T-Lymphocyte Response by
Intrarectal Immunization with a Replication-Deficient
Recombinant Vaccinia Virus Expressing Human
Immunodeficiency Virus 89.6 Envelope Protein
IGOR M. BELYAKOV,1LINDA S. WYATT,2JEFFREY D. AHLERS,1PATRICIA EARL,2
C. DAVID PENDLETON,1BRIAN L. KELSALL,3WARREN STROBER,3
BERNARD MOSS,2ANDJAY A. BERZOFSKY1*
Molecular Immunogenetics and Vaccine Research Section, Metabolism Branch, National Cancer Institute,1and
Laboratory of Viral Diseases2and Mucosal Immunity Section, Laboratory of Clinical Investigation,3National
Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, Maryland 20892
Received 29 April 1998/Accepted 18 June 1998
To improve the safety of recombinant vaccinia virus vaccines, modified vaccinia virus Ankara (MVA) has been employed, because it has a replication defect in most mammalian cells. Here we apply MVA to human immunodeficiency virus type 1 (HIV-1) vaccine development by incorporating the envelope protein gp160 of HIV-1 primary isolate strain 89.6 (MVA 89.6) and use it to induce mucosal cytotoxic-T-lymphocyte (CTL) immunity. In initial studies to define a dominant CTL epitope for HIV-1 89.6 gp160, we mapped the epitope to a sequence, IGPGRAFYAR (from the V3 loop), homologous to that recognized by HIV MN loop-specific CTL and showed that HIV-1 MN-specific CTLs cross-reactively recognize the corresponding epitope from strain 89.6 presented by H-2Dd. Having defined the CTL specificity, we immunized BALB/c mice intrarectally with recombinant MVA 89.6. A single mucosal immunization with MVA 89.6 was able to elicit long-lasting antigen-specific mucosal (Peyer’s patch and lamina propria) and systemic (spleen) CTL responses as effective as or more effective than those of a replication-competent vaccinia virus expressing 89.6 gp160. Immunization with MVA 89.6 led to (i) the loading of antigen-presenting cells in vivo, as measured by the ex vivo active presentation of the P18-89.6 peptide to an antigen-specific CTL line, and (ii) the significant production of the proinflammatory cytokines (interleukin-6 and tumor necrosis factor alpha) in the mucosal sites. These results indicate that nonreplicating recombinant MVA may be at least as effective for mucosal immunization as replicating recombinant vaccinia virus.
Current estimates indicate that the present 15 million hu-man immunodeficiency virus (HIV) infections will increase to over 40 million by the end of the millennium (37). For most countries a safe and effective vaccine offers the only hope of controlling the spread of this disease (25). In North America and Europe homosexual transmission is still the most common route by which infection with HIV type 1 (HIV-1) is acquired (37), and thus an effective vaccine against AIDS will need to confer protection against mucosal challenge (3, 9, 21). Our recent data show the possibility of induction of long-lasting mucosal P18-specific cytotoxic-T-lymphocyte (CTL) responses after mucosal immunization with synthetic peptide HIV vac-cine constructs (3).
Vaccinia virus represents an alternative vector for mucosal immunization. However, there are safety concerns regarding the use of standard vaccinia virus strains in immunodeficient individuals (13, 25, 27). Modified vaccinia virus Ankara (MVA) was originally developed as an attenuated smallpox vaccine by more than 500 passages in chick embryo fibroblasts, during which it suffered multiple deletions and lost the ability to replicate in human and most other mammalian cells (4, 15–17, 22–24, 30, 38). Recent studies indicated that the block
in replication of MVA in human cells occurs at a step in virion assembly rather than at an early stage of infection as occurs with other poxvirus host range mutants (29). An important consequence of the late-stage block is that viral or recombinant gene expression is unimpaired, making MVA an efficient as well as a safe live vector (29). Protective immune responses have been induced by recombinant MVA expressing influenza virus, parainfluenza virus, and simian immunodeficiency virus (SIV) proteins (4, 18, 38).
Little is known about approaches for inducing a CTL re-sponse in the mucosa with a recombinant MVA virus express-ing HIV Env protein. Furthermore, the mechanism of regula-tion of mucosal CTL responses (roles of the antigen-presenting cells [APC] and cytokines) is still unknown (6). In the present work we address all these issues with studies of mucosal CTL responses to recombinant MVA expressing HIV-1 envelope protein of strain 89.6 (MVA 89.6). Strain 89.6 was chosen because it is a primary isolate that is both T cell tropic and macrophage tropic (10), although the ability of the HIV-1 89.6 envelope protein to induce CTL response is unknown.
Thus, in preparation for use of this virus for mucosal immu-nization, we first demonstrate that the HIV-1 89.6 Env protein is able to induce a CTL response restricted by H-2Dd, and we
map the minimal CTL epitope. We also show cross-reactive recognition between an HIV-1 MN V3 loop-specific CTL line and the corresponding epitope from HIV-1 89.6. Using re-sponse to this epitope as a measure of CTL activity, we then show the mucosal immunogenicity of the MVA virus
express-* Corresponding author. Mailing address: Molecular Immunogenet-ics and Vaccine Research Section, Metabolism Branch, National Can-cer Institute, Building 10, Room 6B-12 (MSC #1578), NIH, Bethesda, MD 20892-1578. Phone: (301) 496-6874. Fax: (301) 496-9956. E-mail: [email protected].
8264
on November 9, 2019 by guest
http://jvi.asm.org/
ing HIV-1 89.6 Env protein. We find that a single intrarectal (i.r.) immunization with MVA 89.6 induced long-lasting, anti-gen-specific CTL memory in both the inductive and effector mucosal sites, as well as in systemic immune tissue, at least as efficiently as a replication-competent recombinant vaccinia vi-rus did. We show that mucosal immunization with MVA 89.6 can induce local production of proinflammatory cytokines and effective antigen presentation, both as measured ex vivo after mucosal administration of the virus, which can be important factors for induction and maintenance of the mucosal CTL responses. These mucosal P18 HIV 89.6-specific CTLs may be protective against mucosal challenge with virus expressing HIV antigen (as we showed recently with HIV peptide immuniza-tions [3]).
MATERIALS AND METHODS
Animals.Female BALB/c mice (H-2d) were purchased from Frederick Cancer
Research Center (Frederick, Md.). Mice used in this study were 6 to 12 weeks old. Mice were maintained in a specific-pathogen-free environment.
Viruses.The recombinant MVA 89.6 Env virus was constructed in the follow-ing manner. Plasmid pSVK3, containfollow-ing an HIV 89.6 subclone obtained from R. Collman (University of Pennsylvania School of Medicine, Philadelphia, Pa.), was cut with SalI and BclI. The DNA fragment containing the env, tat, rev, and vpu genes was digested with KpnI, and a subfragment containing the 89.6 env coding sequence minus the first 120 bp of the env gene was isolated. The env gene was reconstructed by ligation with a PCR fragment made from the first 120 bp of the 89.6 env (containing SalI- and KpnI-digested ends), and the SalI- and BclI-digested original vector. The coding sequence of the 89.6 env was confirmed by sequencing. The only early vaccinia transcription termination signal (TTTTTNT) in the 89.6 env was removed by site-directed mutagenesis (12) without changing the encoded amino acids. The 89.6 env fragment was excised from the plasmid by SalI-BclI digestion, blunt ended with Klenow enzyme, and cloned into the SmaI site of pLW-17 (39), a plasmid transfer vector containing the modified H5 promoter (38) and MVA deletion 11 flanking sequences. This plasmid, pLW30, was used to make recombinant MVA expressing the 89.6 envelope by homolo-gous recombination with MVA as previously described (38). Stocks of MVA (22, 23) and recombinant MVA were prepared in secondary chick embryo fibroblasts as previously described (30).
vBD3, a recombinant vaccinia virus (strain WR) expressing HIV 89.6 envelope regulated by the strong synthetic promoter (7), was obtained from R. Doms (10) and grown in BS-C-1 and HeLa cells as previously described (11). For clarity, we will refer to vBD3 as WR 89.6 hereafter in this work.
Peptides. P18-89.6R10 peptide (IGPGRAFYAR), P18-89.6A9 peptide (IGPGRAFYA), 10-mer peptide P18-MNT10 from the V3 loop of the HIV-1 MN Env protein (IGPGRAFYTT), and 10-mer peptide I10 from the V3 loop of the HIV-1 IIIB Env protein (RGPGRAFVTI) were synthesized on an auto-mated peptide synthesizer (Symphony Multiplex; Rainin, Boston, Mass.) by utilizing 9-fluourenylmethoxycarbonyl chemistry. These sequences correspond to residues 311 to 320 of HIV-1 gp160 in the numbering of the Los Alamos database (26). The peptides were cleaved from the resin with trifluoroacetic acid and initially purified by preparative high-performance liquid chromatography (P4 BioGel; Bio-Rad Laboratories, Mountain View, Calif.). Purification to single peaks was achieved by reverse-phase high-performance liquid chromatography onmBondapack reverse-phase C18analytical and preparative columns (Waters Associates, Milford, Mass.).
Immunization.Mice were i.r. immunized with recombinant MVA 89.6 or WR 89.6. Viruses were diluted to the appropriate titer (PFU) in sterile phosphate-buffered saline (PBS), and 150ml of the virus inoculum was i.r. injected through an umbilical catheter inserted;4 cm deep while mice were under inhalation anesthesia (methoxyflurane; Pitman-Moore, Inc., Mundelein, Ill.). We used ei-ther a single dose of virus for immunization (intraperitoneal [i.p.] or i.r.) or one single dose plus one boosting dose (of 13107, 53107, or 13108PFU) for i.r. immunization. A control group of mice were immunized i.p. with a dose of 13
108PFU.
Cell purification. After the immunization dose (2 weeks, 4 weeks, or 6 months), antigen-specific T cells were isolated from the Peyer’s patches (PPs), lamina propria (LP), and spleen (SP) of each mouse. The PPs were carefully excised from the intestinal wall and dissociated into single cells by use of colla-genase type VIII (300 U/ml; Sigma, St. Louis, Mo.) as described previously (3). Our data showed that most PP CD31T cells isolated from normal mice were CD41, while CD31CD81T cells were less frequent. Further, collagenase did not alter the expression of CD3, CD4, or CD8 on splenic T cells treated with this enzyme. Lamina propria lymphocyte (LPL) isolation was performed as described previously (3). The small intestines were dissected from individual mice, and the mesenteric and connective tissues were carefully removed. Fecal material was flushed from the lumen with unsupplemented medium (RPMI). After the PPs were identified and removed from the intestinal wall, the intestines were opened longitudinally, cut into short segments, and washed extensively in RPMI-1640
containing 2% fetal bovine serum (FBS). To remove the epithelial cell layer, tissues were placed into 100 ml of 1 mM EDTA and incubated twice (first for 40 min and then for 20 min) at 37°C with stirring. After the EDTA treatment, tissues were washed in RPMI-1640 with 2% fetal calf serum for 10 min at room temperature and then placed in 50 ml of RPMI-1640 containing 10% fetal calf serum and incubated for 15 min at 37°C with stirring. The tissues and medium were transferred to a 50-ml tube and shaken vigorously for 15 s, and then the medium containing epithelial cells was removed. This mechanical removal of cells was repeated twice more, with fresh medium each time, in order to com-pletely remove the epithelial cell layer. Histologic examination revealed that the structures of the villi and LP were preserved. To isolate LPL, tissues were cut into small pieces and incubated in RPMI-1640 containing collagenase type VIII (300 U/ml; Sigma) for 50 min at 37°C with stirring. Supernatants containing cells were collected, washed, and then resuspended in complete RPMI-1640. This collagenase dissociation procedure was repeated two times, and the isolated cells were pooled and washed again. Cells were passed through a glass wool column to remove dead cells and tissue debris and then layered onto a discontinuous gradient containing 75% and 40% Percoll (Pharmacia Fine Chemicals, Pharma-cia Inc., Uppsala, Sweden). After centrifugation (4°C, 6003g, 20 min), the interface layer between the 75% and 40% Percoll was carefully removed and washed with incomplete medium. This procedure provided.90% viable lym-phocytes with a cell yield of 1.53106to 23106lymphocytes/mouse (3). The SPs were aseptically removed, and single-cell suspensions were prepared by gently teasing them through sterile screens. The erythrocytes were lysed in Tris-buffered ammonium chloride, and the remaining cells were washed extensively in RPMI-1640 containing 2% FBS.
For the purification of the dendritic cell APC population from the PPs and SPs, mouse anti-CD11c (N418) Magnetic Cell Sorting (MACS) MicroBeads (Miltenyi Biotec, Bergisch-Gladbach, Germany) were used, according to the manufacturer’s instructions. To obtain high purities of dendritic cells (DC), Fc receptor-mediated magnetic labeling of macrophages was blocked by adding mouse immunoglobulin (1 mg per 500-ml volume) to the cell suspension before adding CD11c MicroBeads. The magnetically retained CD11c1 APC were eluted, by washing with buffer, as the positively selected cell fraction.
CTL lines.CTL lines specific for P18MN and P18-89.6 were prepared as described previously (34). Briefly, mice were immunized intravenously with 107 PFU of recombinant vaccinia virus expressing gp160 of strain MN or 89.6. Three weeks later, immune SP cells were restimulated in vitro with irradiated syngeneic SP cells which were pulsed before the irradiation with 5mM peptide P18MN or P18-89.6R10. The cell lines were maintained by weekly restimulation at 53105 per well in a 24-well plate with 53106syngeneic SP cells pulsed with 5mM peptide, in the presence of 10% T-Stim (Collaborative Biomedical Products, Bedford, Mass.) as a source of interleukin 2 (IL-2).
CTL assay.Immune cells from SPs, PPs, and LPs were cultured at 53106/ml in 24-well culture plates in complete T-cell medium (CTM): RPMI-1640 con-taining 10% FBS, 2 mMD-glutamine, penicillin (100 U/ml), streptomycin (100 mg/ml), and 531025M 2-mercaptoethanol (1, 32–34). Three days later we added 10% concanavalin A supernatant as a source of IL-2 (T-Stim). LPLs were studied after 7 days of stimulation with 1mM P18-89.6R10 Env peptide together with 43106of 3,300-rad-irradiated syngeneic SP cells. SP and PP cells were stimulated in vitro similarly for two 7-day culture periods before assay. Cytolytic activity of CTL lines was measured by a 4-h assay with51Cr-labeled targets. The P815 cell line was used as a target cell. For testing the peptide specificity of CTLs, 51Cr-labeled P815 targets were pulsed for 2 h with peptide at the beginning of the assay (1, 32–34). The percent specific51Cr release was calculated as 1003 (experimental release2spontaneous release)/(maximum release2spontaneous release). Maximum release was determined from supernatants of cells that were lysed by addition of 5% Triton X-100. Spontaneous release was determined from target cells incubated without added effector cells.
Cytokine ELISA.Levels of gamma interferon (IFN-g) were determined in culture supernatants by using a murine cytokine immunoassay, MiniKit (Endo-gen, Cambridge, Mass.), according to the manufacturer’s instructions. Concen-trations of IL-6 and tumor necrosis factor alpha (TNF-a) were studied by using a cytokine enzyme-linked immunosorbent assay (ELISA) kit (PharMingen, San Diego, Calif.) according to the manufacturer’s instructions.
Antibody ELISA.ELISA was used to determine the presence of anti-P18-89.6R10 antibody in serum and rectal wash samples. P18-anti-P18-89.6R10 peptide was suspended in coating buffer (PBS) at a concentration of 30mg/ml and plated in 96-well microtiter plates (Nunc, Roskilde, Denmark) at 50ml/well. After over-night incubation at 4°C, the contents of the wells were discarded and blocking buffer (PBS with 2% bovine serum albumin [BSA]–0.01% thimerosal [pH 7.2 to 7.4]) was added at 200ml/well. After incubating at room temperature for 2 h, plates were washed three times with wash buffer (50 mM Tris, 0.2% Tween 20) before addition of the samples. All samples were diluted in serum diluent and added to ELISA plates at 100ml/well. After overnight incubation at 4°C, plates were washed three times with wash buffer. Peroxidase-conjugated goat anti-mouse immunoglobulin G (IgG), IgM, and IgA (Sigma) were diluted 1:2,000 (in PBS with 2% BSA–0.01% thimerosal [pH 7.2 to 7.4]) and used as the detection antibody (100ml/well). After incubation at room temperature for 2 h, plates were washed three times with wash buffer. Horseradish peroxidase-streptavidin (PharMingen) was diluted 1:1,000 (PBS with 2% BSA–0.01% thimerosal [pH 7.2 to 7.4]) and added to ELISA plates at 100ml/well. After 30 min, plates were
VOL. 72, 1998 REPLICATION-DEFICIENT VACCINIA VIRUS INDUCES MUCOSAL HIV CTL 8265
on November 9, 2019 by guest
http://jvi.asm.org/
washed three times with wash buffer and reacted with ABTS [2,29 -azinobis(3-ethylbenzthiazolinesulfonic acid)] peroxidase substrate (Kirkegaard & Perry Laboratories, Gaithersburg, Md.). After a 10-min incubation, plates were read at 405 nm on a plate reader (Molecular Devices Corp., Menlo Park, Calif.).
RESULTS
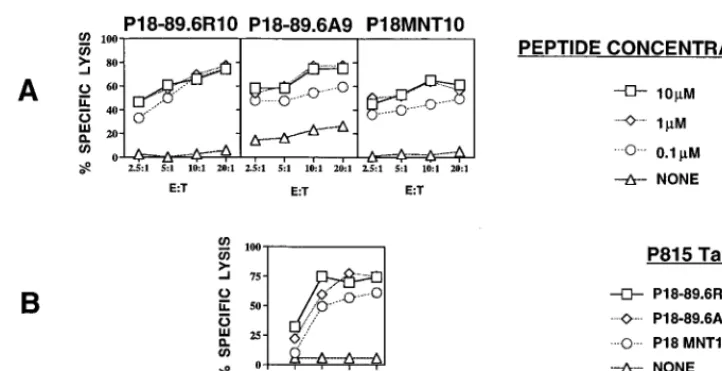
HIV MN V3 loop-specific CTLs cross-reactively recognize the corresponding epitope from HIV-1 89.6, whereas HIV IIIB-specific CTLs do not.To study CTL responses to the HIV-1 envelope protein expressed in MVA 89.6 and WR 89.6, we wanted to define a useful epitope to monitor the responses and, in particular, to determine whether the sequence of the 89.6 envelope protein homologous to the immunodominant epitope of HIV-1 IIIB and MN in the V3 loop region (called peptide 18-I10 or P18-I10 in strain IIIB [20, 28, 31, 35] or P18-MNT10 in strain MN [35]) was also a CTL epitope in this strain. We found that the 10-mer peptide sequence (IG-PGRAFYAR) from the V3 loop of the HIV-1 89.6 Env pro-tein is homologous to the 10-mer P18-MNT10 peptide se-quence (IGPGRAFYTT) from the V3 loop of HIV-1 MN envelope protein. We accordingly named the new 10-mer tide P18-89.6 R10. The differences between the 10-mer pep-tides from 89.6 envelope and MN envelope are two C-terminal residues, where the TT residues from the sequence of HIV-1 MN are replaced by AR in the 89.6 Env protein. We synthe-sized P18-89.6R10 and used it to determine whether the epitope can be cross-reactively recognized by a CTL line spe-cific for P18MN presented by H-2Dd. We found that the ability of the P18MN-specific CTL line to lyse the P815 target cells pulsed with P18-89.6R10 was similar to that for lysis of the target P815 cells pulsed with the original P18MN peptide (Fig. 1). Low concentrations of P18-89.6R10 peptide (0.1mM) were sufficient to sensitize target cells for lysis. For mapping of the minimal epitope from the 89.6 protein which can be recognized by the MN-specific CTL line, we used a 9-mer peptide (P18-89.6 A9 [IGPGRAFYA]) to pulse the target cells. The C-terminal Arg of P18-89.6R10 was not expected to be a good anchor for binding to H-2Dd(8), whereas the penultimate Ala at least had an aliphatic side chain, albeit not an optimal one. Surprisingly, the 9-mer P18-89.6A9 was not more active on a molar basis than the 10-mer for recognition by the MN-specific
CTL line, as might have been expected if the 10-mer required processing to remove the C-terminal Arg. The same results were found when BALB/c 3T3 fibroblasts were used as target cells (data not shown).
To determine whether HIV IIIB V3 loop-specific CTLs cross-reactively recognize the corresponding epitope from HIV-1 89.6, we studied the ability of the P18-I10-specific CTL line to lyse P815 target cells pulsed with 89.6R10 or P18-89.6A9. No cross-reactive recognition between the HIV IIIB V3 loop-specific CTL line and the corresponding epitope from HIV-1 89.6 was found, although the CTL killed positive con-trol targets pulsed with P18-I10 peptide of the IIIB strain (data not shown).
These results allowed us to use this epitope to measure the efficacy of recombinant vaccinia expressing the HIV-1 enve-lope protein of the 89.6 strain for induction of CTL.
The induction of long-lasting memory CTL responses in systemic and mucosal sites by i.r. immunization with MVA 89.6.Having defined a useful epitope for measurement of CTL activity specific for strain 89.6 envelope protein, we wished to study the induction of mucosal and systemic CTL responses by the replication-deficient strain of vaccinia virus MVA 89.6 ex-pressing the HIV-1 strain 89.6 envelope protein. A replication-competent WR strain of vaccinia virus (WR 89.6) that ex-presses the same HIV-1 89.6 env gene was used as a positive control. In vitro experiments indicated that the MVA recom-binant produced about twofold more Env protein, as measured by immunoprecipitation of [35S]methionine metabolically la-beled protein from infected cells and phosphoimager quanti-tation, than did the WR recombinant when a high multiplicity of virus sufficient to infect all cells was used. We anticipated, however, that the in vivo situation would be quite different since WR 89.6 can spread from cell to cell whereas the MVA 89.6 cannot. Thus, the ability to replicate might more than compensate for the slightly lower level of protein expression.
BALB/c mice were immunized i.r. with 13107, 53107, or 13108infectious units of MVA 89.6 Env or WR 89.6 Env. Mice were studied either 2 weeks, 4 weeks, or 6 months later for memory CTL in the SP, PPs, or LP. We found that 2 weeks after a single i.r. immunization with doses of 13108and 53 107PFU, both MVA 89.6 and WR 89.6 elicited significant and
FIG. 1. (A) Recognition by the HIV-1 MN-specific CTL cell line of P815 target cells pulsed with different concentrations of P18MN peptides with substitutions. P815 targets were tested in the presence or absence of various concentrations of peptides as shown. (B) Killing of target cells pulsed with different concentrations of the peptides is compared with killing of unpulsed targets at an effector-to-target ratio (E:T) of 20:1. For both panels, standard errors of the means of triplicate cultures were all,5% of the mean.
on November 9, 2019 by guest
http://jvi.asm.org/
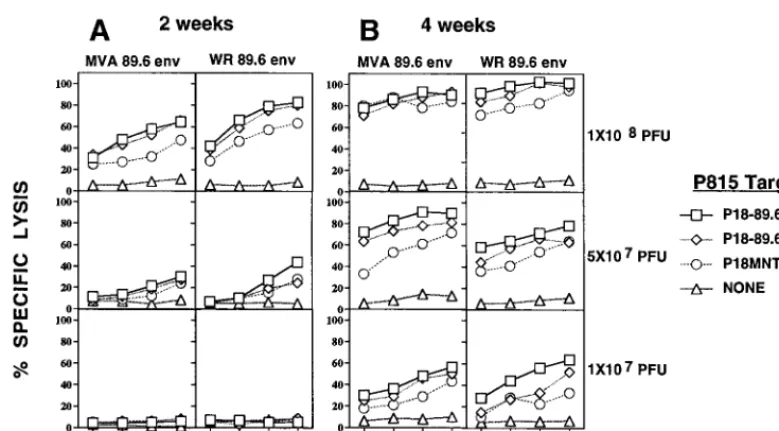
[image:3.612.117.478.70.255.2]comparable 89.6-specific CTL responses in the SP (Fig. 2A), whereas no CTLs were found 2 weeks after i.r. immunization with 13107PFU of MVA 89.6 Env or WR 89.6 Env (Fig. 2A). However, 4 weeks after i.r. immunization with MVA 89.6 or WR 89.6 virus, a significant increase in the level of the CTL responses in the SP at all three doses of the viruses was found (Fig. 2B). We found significant SP CTL memory 6 months after i.r. immunization with both MVA 89.6 and WR 89.6 virus (data not shown).
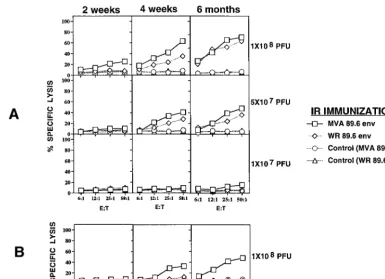
There was no direct parallel between induction of CTL re-sponses in mucosal sites and induction of CTL in the systemic immune system (SP). No significant CTL response in mucosal sites (either PP or LP cells) was found 2 weeks after i.r. im-munization with MVA 89.6 or WR 89.6 (Fig. 3). However, 4 weeks after i.r. immunization with MVA 89.6 or WR 89.6 at doses of 1 3 108 or 5 3 107 PFU, but not 1 3 107 PFU, significant CTL responses in PPs were found (Fig. 3A). In contrast, a significant LP CTL response occurred only 4 weeks after i.r. immunization with 13108PFU of MVA 89.6 (Fig. 3B), not with doses of 53107PFU (data not shown), and not with WR 89.6 virus even at doses of 13108PFU (Fig. 3B). PP CTL responses 6 months after i.r. immunization with MVA 89.6 were slightly higher than those after immunization with WR 89.6 virus (Fig. 3). Thus, replication-deficient MVA 89.6 virus was at least as effective for mucosal immunization as conventional recombinant vaccinia virus.
One month after i.r. immunization with MVA 89.6 or WR 89.6, several groups of mice were reimmunized with the same viruses (MVA 89.6 or WR 89.6) at the same dose. There was no significant increase in the level of mucosal or systemic CTL responses with MVA 89.6 or WR 89.6 after mucosal reimmu-nization (data not shown). One possible explanation for this is that recombinant vaccinia viruses induce strong anti-vaccinia virus mucosal immune responses (neutralizing antibody and CTL), which can lead to the neutralization of the viruses upon reimmunization.
For comparison with mucosal immunization, we immunized
BALB/c mice i.p. with MVA 89.6 or WR 89.6 virus at doses of 108PFU. i.p. immunization with either virus induced high CTL responses in the SP but none in the LP. However, i.p. immu-nization with 108PFU of MVA 89.6, but not WR 89.6, was able to induce a CTL response in the PP (Fig. 4) that was modest compared to the response in the SP but was reproducible in three independent experiments with five mice each.
Because the P18-89.6R10, P18-89.6A9, or P18MN peptide was recognized in the above-mentioned studies on peptide-pulsed P815 target cells which do not express major histocom-patibility complex class II molecules, the data obtained imply that PP, LP, and SP CTL recognize peptide in the context of major histocompatibility complex class I molecules. In data not shown, by blocking response with CD8 monoclonal anti-body 2.43 (National Institutes of Health, Frederick, Md.) in the cultures, we also established that the CTL were CD81T
cells.
[image:4.612.104.494.70.285.2]Study of antigen presentation in mucosal inductive sites after i.r. immunization with MVA 89.6 and WR 89.6.The levels of mucosal CTL responses induced by i.r. immunization with MVA 89.6 and WR 89.6 could be dependent on antigen pre-sentation by cells in the mucosal site infected with the viruses. Therefore, we asked whether the APC, after in vivo immuni-zation of BALB/c mice with MVA 89.6 or WR 89.6, can present antigen in vitro to P18-89.6R10-specific CTL lines. To determine the antigen-presenting activity of cells exposed to virus in vivo, we immunized 10 BALB/c mice by both i.r. and i.p. routes with 2 3 108 PFU of MVA 89.6 or WR 89.6. Forty-eight hours after immunization the mice were killed and PPs and SPs were removed. Cells from 10 animals were pooled, and a positive-selection MACS sort for CD11c (N418) was performed for the isolation of APC from these organs. APC (23105) from PPs or SPs were cultivated with 23105cells of the P18-89.6R10-specific CTL line from PPs or SPs in 96-well U-bottom plates. As a control, we used APC from unimmu-nized animals which were cultivated with the same antigen-specific CTL cells. The antigen-presenting activity of the APC
FIG. 2. Induction of the 89.6-specific splenic CTL responses 2 and 4 weeks after i.r. immunization with replication-deficient MVA 89.6 and WR 89.6 expressing the HIV-1 envelope protein of HIV-1 strain 89.6. Induction of SP CTL responses 2 weeks (A) and 4 weeks (B) after i.r. immunization with the indicated recombinant viral vectors at the doses indicated to the right of panel B. Two weeks after i.r. immunization, SP cells were cultivated in vitro with 1mM concentrations of the indicated peptides. SP cells were stimulated in vitro for two 7-day culture periods before assay. Cytolytic activity of CTL lines was measured by a 4-h assay with51Cr-labeled P815 DBA/2 mastocytoma cell targets as described in Materials and Methods. E:T, effector-to-target ratio.
VOL. 72, 1998 REPLICATION-DEFICIENT VACCINIA VIRUS INDUCES MUCOSAL HIV CTL 8267
on November 9, 2019 by guest
http://jvi.asm.org/
was assessed by the concentration of IFN-g in the culture supernatant. We found that PP APC from mice infected in vivo with MVA 89.6 could significantly activate the PP P18-89.6-specific CTL line for the production of IFN-g (Fig. 5). In contrast, no significant production of IFN-gwas found after the stimulation of the P18-89.6-specific CTL line with APC from unimmunized animals (Fig. 5). As a specificity control, the same APC were tested for their ability to stimulate a P18IIIB-specific CTL line that does not cross-react with 89.6 envelope. No significant activation of the P18IIIB-specific CTL line was found. SP APC induced somewhat higher IFN-g pro-duction than did APC from the PPs (Fig. 5).
[image:5.612.107.492.73.352.2]Production of proinflammatory cytokines (IL-6 and TNF-a) in the mucosal site after i.r. immunization with MVA 89.6 and WR 89.6 viruses.Our hypothesis is that the induction of the mucosal CTL responses can be under the influence of the local inflammatory process and that proinflammatory cytokines can upregulate the inductive phase of the CTL response. In order to determine whether MVA 89.6 and WR 89.6 induced a significant inflammatory reaction, we studied proinflammatory cytokines produced by mononuclear cells from mucosal and systemic immune systems. We immunized 10 BALB/c mice each by the i.r. route with 23108PFU of MVA 89.6 or WR 89.6 (using a higher dose of inoculum to detect better cytokine
FIG. 3. Induction of PP and LP P18-89.6R10-specific CTL responses 2 weeks, 4 weeks, and 6 months after i.r. immunization with replication-deficient MVA 89.6 and WR 89.6. Induction of PP 89.6R10 (A)- and LP 89.6R10 (B)-specific CTL responses at the indicated times after i.r. immunization with MVA 89.6 and WR 89.6. Mice were immunized at doses as indicated to the right. The percent specific51Cr release was calculated as described in the legend to Fig. 2. E:T, effector-to-target ratio.
FIG. 4. Induction of systemic and mucosal CTL responses 4 weeks after i.p. immunization with replication-deficient MVA 89.6 and WR 89.6. Mice were immunized i.p. with 108PFU of either virus, and responses were measured 4 weeks later as described in the legends to Fig. 2 and 3. E:T, effector-to-target ratio.
on November 9, 2019 by guest
http://jvi.asm.org/
responses in vitro 2 days after immunization). Forty-eight hours after immunization, the mice were killed and PPs and SPs were removed and then mononuclear cells (MC) were separated by gradient centrifugation. MC were stimulated in vitro with bacterial LPS (10 ng/ml; Sigma) (Current Protocols in Immunology, Vol.3., Unit 14.4 and 14.5) for 4 days,
super-natants were collected, and IL-6 and TNF-awere assayed by ELISA. LPS-stimulated MC from the PPs of the mice immu-nized i.r. with MVA 89.6 produced the highest levels of IL-6 and TNF-a (Fig. 6). The concentration of IL-6 produced by LPS-stimulated MC from PPs was significantly higher than secretion of IL-6 by activated SP cells from the mice
[image:6.612.141.459.72.309.2]immu-FIG. 5. Ex vivo antigen presentation activity of cells from mucosal and systemic sites for mucosal and systemic CTL lines after i.r. and i.p. immunization with replication-deficient MVA 89.6 and WR 89.6. The same mice were immunized by both i.r. and i.p. routes with 23108PFU, and 48 h later APC were separated by MACS sorting for CD11c-positive cells from SP and PP. The PP (A) and SP (B) APC were cultivated in vitro in 96-well U-bottom plates with P18-89.6R10-specific or P18IIIB-specific CTL lines from SP and PP. The activation of the T-cell lines was studied by measuring IFN-gconcentration in culture supernatants. Similar results were obtained in three replicate experiments. The error bars represent the standard errors of the means.
FIG. 6. Proinflammatory cytokine (IL-6 and TNF-a) production by mucosal and systemic MC 48 h after i.r. immunization with replication-deficient MVA 89.6 and WR 89.6, measured after 4 days of in vitro stimulation with LPS (10 ng/ml). Cells were cultured at 250,000 per well in 96-well plates. Cytokines in the culture supernatants were measured by ELISA as described in Materials and Methods. The error bars represent the standard errors of the means.
VOL. 72, 1998 REPLICATION-DEFICIENT VACCINIA VIRUS INDUCES MUCOSAL HIV CTL 8269
on November 9, 2019 by guest
http://jvi.asm.org/
nized i.r. with MVA 89.6. In each case, the background levels in unstimulated cultures from MVA 89.6- and WR 89.6-immu-nized mice were not statistically different.
Because IL-6 can be produced not only by macrophages but also by antigen-specific T cells, we studied the production of IL-6 by PP and SP T cells 3 weeks after i.r. immunization with MVA 89.6 and WR 89.6 (to allow time for the development of antigen-specific T-cell response). PP and SP MC were stimu-lated in vitro with P18-89.9R10 for 4 days, and then the con-centration of IL-6 was determined. We found significant se-cretion of IL-6 by PP antigen-specific lymphocytes from MVA 89.6-immunized animals (Fig. 7). It is noteworthy that IL-6 production by PP MC was threefold higher than IL-6 secretion by SP MC. This active IL-6 production may be important for the regulation of IgA antibody responses in mucosal sites (2). One month after i.r. immunization with recombinant MVA 89.6, we found significant titers of P18-specific IgA and IgG antibodies in the rectal washes, but not after immunization with WR 89.6, as measured by ELISA on peptide P18-89.6R10-coated plates with goat anti-mouse IgA and IgG, as described in Materials and Methods (data not shown). How-ever, MVA 89.6 and WR 89.6 induced comparable serum IgG antibody responses (data not shown).
DISCUSSION
Several experimental vaccination strategies have been devel-oped to prevent primary infection with HIV-1 and as immu-notherapeutics for infected individuals. Many of the putative vaccines have been tested in animal models to determine their safety and efficacy and to delineate immune correlates of pro-tection. One of the candidates for the development of HIV recombinant vaccines is the highly attenuated, replication-de-ficient vaccinia virus MVA. To date, however, our knowledge about the efficacy and the immunogenicity of recombinant MVA is very limited. It was shown that mice immunized in-tramuscularly with MVA containing the hemagglutinin and
nucleoprotein genes of H1N1 influenza virus developed IgG and CTL responses and were completely protected against lethal lower respiratory infections (4, 30). In addition, intra-gastric immunization induced anti-IgA to titers that protected against intranasal challenge with influenza virus (30). Cotton rats inoculated intramuscularly or intranasally with MVA ex-pressing parainfluenza virus 3 membrane proteins were pro-tected against both upper and lower respiratory tract infections (38). In addition, rhesus macaques immunized with MVA ex-pressing SIV Env and Gag/Pol proteins were partially pro-tected against intravenous challenge with SIV (18). Surpris-ingly, in both the influenza virus and SIV challenge studies, nonreplicating recombinant MVA appeared more potent than similar recombinant viruses derived from the replication-com-petent Wyeth or WR strains of vaccinia virus. Blanchard et al. (5) suggested that the loss of several host immune evasion genes by MVA might be responsible for the good immunoge-nicity. However, although a recombinant adenovirus vector expressing glycoprotein B (gB) of herpes simplex virus had been reported to induce mucosal CTL against this herpesvirus protein (14), the ability of MVA recombinants to elicit a mu-cosal CTL response had not been reported previously. Indeed, we know of no publications that describe the generation of mucosal HIV-specific CTL responses after mucosal immuni-zation with recombinant viral vectors. It might have been ex-pected that the lack of viral replication of the MVA recombi-nant and therefore presumed lower level of inflammatory response induced would together have resulted in a lower mucosal CTL response. Therefore, we undertook to test the ability of a recombinant MVA expressing HIV-1 envelope pro-tein to elicit a mucosal CTL response.
As a vaccine prototype, we developed a recombinant MVA expressing the HIV-1 89.6 envelope protein. HIV-1 89.6 is a primary isolate with both T-cell tropic and macrophage-tropic properties. In this study we characterized the immunogenicity of MVA 89.6 following a single i.r. immunization of BALB/c mice, focusing on the induction of mucosal CTLs specific for the dominant epitope of the HIV 89.6 envelope protein.
To do so, we identified a dominant CTL epitope P18-89.6R10 of the 89.6 strain of HIV-1 envelope protein which allowed us to measure the efficacy of recombinant vaccinia virus expressing the HIV-1 envelope protein of the 89.6 strain for induction of CTLs in mucosal and systemic lymphoid tis-sues. This epitope corresponds to a sequence of the V3 loop homologous to a CTL epitope we have previously defined in HIV-1 IIIB (31) and MN (34). We found that HIV-MN V3 loop-specific CTLs cross-reactively recognize the correspond-ing epitope from HIV-1 89.6, whereas HIV IIIB-specific CTLs do not.
Our finding that MVA 89.6 Env was more immunogenic than WR 89.6 Env is consistent with comparisons of other recombinant viruses (18, 30) and may reflect intrinsic proper-ties of the vectors. However, differences in the levels of expres-sion of the 89.6 Env protein may also be important. We found by immunoprecipitation that when NIH 3T3 cells were in-fected with MVA 89.6 and WR 89.6 at multiplicities of infec-tion that were sufficient to infect all cells, the MVA expressed approximately two times more HIV-1 envelope protein. This was a surprising result, because the synthetic early-late pro-moter used to regulate HIV envelope expression in the WR vector is stronger than the modified H5 promoter used in the MVA construct (38). In vivo, however, the WR virus should compensate for this by replicating and infecting more cells. Factors other than the level of expression would also affect CTL induction, in particular the induction of local inflamma-tion and the level of antigen presentainflamma-tion. Mucosal cell
popu-FIG. 7. IL-6 production by MC 3 weeks after immunization with replication-deficient MVA 89.6 and WR 89.6, measured after 4 days of in vitro stimulation with P18-89.6R10 Env peptide (1mM). Cells were cultured at 250,000 per well in 96-well plates. Cytokines in the culture supernatants were measured by ELISA as described in Materials and Methods. The error bars represent the standard errors of the means.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.51.290.71.281.2]lations enriched for APC from mice infected mucosally with MVA 89.6 effectively presented antigen ex vivo to HIV enve-lope-specific CTLs in the absence of any additional source of antigen. Also, we found a significant production of proinflam-matory cytokines by MC after i.r. immunization with MVA 89.6, surprisingly more so than after immunization with WR 89.6. Both factors, local mucosal inflammation and antigen presentation in the inductive mucosal site, can lead to the recruitment of a significant number of 89.6-specific CTLs in the regional lymphoid system. Antigen-specific CTL from the inductive mucosal site can easily repopulate the effector sites both in mucosal and systemic immune tissues. However, it is possible that some MVA 89.6 virus particles can reach the systemic tissues by blood or lymphoid circulation and result in antigen presentation in the systemic lymphoid tissues. Probably infection of the local mucosal DC with MVA 89.6 can signif-icantly increase the migration of these cells into the organized regional lymphoid tissue (inductive site of the mucosal immune system) and enhance antigen presentation broadly throughout the intestinal mucosa. Also, it is possible that the proinflam-matory cytokines activate and mobilize DC from their tissues of origin (pararectal lymphoid nodes) and augment the dis-semination of these cells widely into the intestinal mucosa. It was shown that adhesion of Langerhans cells (LC) to keratin-ocytes is mediated by E-cadherin (19, 36). IL-1, TNF-a, and LPS all reduce E-cadherin expression and thereby mobilize LC from the epidermis and presumably attenuate LC-keratinocyte adhesion (19, 36). All of these features of immunization with MVA 89.6 could thus contribute to its surprisingly greater immunogenicity, despite the fact that it is nonreplicating.
Our data show that the systemic immunization (i.p.) with MVA 89.6 virus can induce reproducible 89.6-specific CTL responses in the PP. No CTL responses were found in the inductive site of the mucosal immune system after i.p. immu-nization with WR 89.6. Since, the wild-type virus WR 89.6 would be expected to disseminate more widely than MVA 89.6, this result suggests that it is not dissemination of virus that produces PP CTL after i.p. infection with MVA 89.6, but rather dissemination of immune cells. Probably, some common lymphoid circulation exists between the inductive sites of the systemic lymphoid tissues and the organized mucosal lymphoid tissues (PPs).
Our previous study (3) showed that a synthetic, multideter-minant HIV-1 IIIB peptide construct vaccine plus cholera toxin adjuvant induced long-lasting, antigen-specific CTL memory in both the inductive (PP) and effector (LP) mucosal sites, as well as in systemic sites (SP), whereas systemic immu-nization induced specific CTL only in the SP. We found that i.r. immunization with the synthetic HIV peptide vaccine pro-tected mice against infection via mucosal challenge with a recombinant vaccinia virus expressing HIV IIIB gp160 (3). It was shown that this protection was CD8 dependent (unpub-lished data). Unfortunately, we are not able to study the pro-tective immunity after mucosal immunization with recombi-nant replication-deficient virus expressing 89.6 protein, because the cross-reactive vaccinia virus-specific immunity in-duced by MVA virus would recognize any other recombinant vaccinia viruses that could be used to challenge.
Probably immunogenicity and protective immunity induced after i.r. immunization with recombinant MVA 89.6 viruses and synthetic HIV peptide vaccine are different and depend on multiple factors. For example, induction of mucosal memory CTL by using the HIV peptide vaccine construct required several reimmunizations, whereas long-lasting mucosal CTL responses can be induced by one mucosal immunization with vaccinia virus, probably because of the longer persistence of
the virus. Another important consideration is that the anti-HIV immunogenicity of MVA 89.6 viruses and other recom-binant vaccinia viruses in the humans already exposed to vac-cinia virus (vacvac-cinia small pox immunization) may be low, because of preexisting immunity to vaccinia virus antigens. However, a combined regime whereby the animals were first primed with the DNA vaccine and then given boosters with MVA was the most potent protocol for the induction of both IFN-g-producing and cytolytic T cells against two CTL epitopes simultaneously (15). The mucosal inductive sites may still be naive to the previous immunization with vaccinia virus and may develop a vigorous immune response to the recom-binant HIV protein.
Thus, this study makes the novel observation that MVA can serve as a highly efficient vector for the expression of HIV protein in the mucosal immune system, despite an inability to replicate in mammalian cells. Therefore, for safety reasons, we suggest that MVA replace less-attenuated strains of vaccinia virus for production of recombinant proteins, because for mu-cosal immunization, MVA 89.6 is at least as effective as con-ventional recombinant vaccinia virus.
ACKNOWLEDGMENTS
We thank Michael A. Derby and Vanessa Hirsch for critical reading of the manuscript and helpful suggestions.
REFERENCES
1. Alexander-Miller, M. A., G. R. Leggatt, A. Sarin, and J. A. Berzofsky. 1996. Role of antigen, CD8, and CTL avidity in high dose antigen induction of apoptosis of effector CTL. J. Exp. Med. 184:485–492.
2. Beagley, K. W., and C. O. Elson. 1992. Cells and cytokines in mucosal immunity and inflammation. Gastroenterol. Clin. North Am. 21:347–366. 3. Belyakov, I. M., M. A. Derby, J. D. Ahlers, B. L. Kelsall, P. Earl, B. Moss, W.
Strober, and J. A. Berzofsky.1998. Mucosal immunization with HIV-1 pep-tide vaccine induces mucosal and systemic cytotoxic T lymphocytes and protective immunity in mice against intrarectal recombinant HIV-vaccinia challenge. Proc. Natl. Acad. Sci. USA 95:1709–1714.
4. Bender, B. S., C. A. Rowe, S. F. Taylor, L. S. Wyatt, B. Moss, and P. A. Small, Jr.1996. Oral immunization with a replication-deficient recombinant vac-cinia virus protects mice against influenza. J. Virol. 70:6418–6424. 5. Blanchard, T. J., A. Alcami, P. Andrea, and G. L. Smith. 1998. Modified
vaccinia Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vac-cine. J. Gen. Virol. 79:1159–1167.
6. Blauvelt, A., H. Asada, M. W. Saville, V. Klaus-Kovtun, D. J. Altman, R. Yarchoan, and S. I. Katz.1997. Productive infection of dendritic cells by HIV-1 and their ability to capture virus are mediated through separate pathways. J. Clin. Invest. 100:2043–2053.
7. Chakrabarti, S., J. R. Sisler, and B. Moss. 1997. Compact, synthetic, vaccinia virus early/late promoter for protein expression. BioTechniques 23:1094– 1097.
8. Corr, M., L. F. Boyd, E. A. Padlan, and D. H. Margulies. 1993. H-2Dd exploits a four residue peptide binding motif. J. Exp. Med. 178:1877–1892. 9. Cranage, M. P., A. M. Whatmore, S. A. Sharpe, N. Cook, N. Polyanskaya, S. Leech, J. D. Smith, E. W. Rud, M. J. Dennis, and G. A. Hall.1997. Macaques infected with live attenuated SIVmac are protected against superinfection via the rectal mucosa. Virology 229:143–154.
10. Doranz, B. J., J. Rucker, Y. Yi, R. J. Smyth, M. Samson, S. C. Peiper, M. Parmentier, R. G. Collman, and R. W. Doms.1996. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell 85:1149–1158.
11. Earl, P., N. Cooper, and B. Moss. 1991. Preparation of cell cultures and virus stocks, p. 16.16.1–16.16.7. Greene Publishing Associates/Wiley Interscience, New York, N.Y.
12. Earl, P. L., A. W. Hugin, and B. Moss. 1990. Removal of cryptic poxvirus transcription termination signals from the human immunodeficiency virus type 1 envelope gene enhances expression and immunogenicity of a recom-binant vaccinia virus. J. Virol. 64:2448–2451.
13. Fenner, F. 1989. Risks and benefits of vaccinia vaccine use in the worldwide smallpox eradication campaign. Res. Virol. 140:465–466.
14. Gallichan, W. S., and K. L. Rosenthal. 1996. Long-lived cytotoxic T lympho-cyte memory in mucosal tissues after mucosal but not systemic immuniza-tion. J. Exp. Med. 184:1879–1890.
15. Hanke, T., T. J. Blanchard, J. Schneider, C. M. Hannan, M. Becker, S. C. Gilbert, A. V. S. Hill, G. L. Smith, and A. McMichael.1998. Enhancement of
VOL. 72, 1998 REPLICATION-DEFICIENT VACCINIA VIRUS INDUCES MUCOSAL HIV CTL 8271
on November 9, 2019 by guest
http://jvi.asm.org/
MHC class I-restricted peptide-specific T cell induction by a DNA prime/ MVA boost vaccination regime. Vaccine 16:439–445.
16. Hanke, T., T. J. Blanchard, J. Schneider, G. S. Ogg, R. Tan, M. Becker, S. C. Gilbert, A. V. Hill, G. L. Smith, and A. McMichael.1998. Immunogenicities of intravenous and intramuscular administrations of modified vaccinia virus Ankara-based multi-CTL epitope vaccine for human immunodeficiency virus type 1 in mice. J. Gen. Virol. 79:83–90.
17. Hanke, T., J. Schneider, S. C. Gilbert, A. V. S. Hill, and A. McMichael. 1998. DNA multi-CTL epitope vaccines for HIV and Plasmodium falciparum: immunogenicity in mice. Vaccine 16:426–435.
18. Hirsch, V. M., T. R. Fuerst, G. Sutter, M. W. Carroll, L. C. Yang, S. Goldstein, M. Piatak, Jr., W. R. Elkins, W. G. Alvord, D. C. Montefiori, B. Moss, and J. D. Lifson.1996. Patterns of viral replication correlate with outcome in simian immunodeficiency virus (SIV)-infected macaques: effect of prior immunization with a trivalent SIV vaccine in modified vaccinia virus Ankara. J. Virol. 70:3741–3752.
19. Jakob, T., and M. C. Udey. 1998. Regulation of E-cadherin-mediated adhe-sion in Langerhans cell-like dentritic cells by inflammatory mediators that mobilize Langerhans cells in vivo. J. Immunol. 160:1–7.
20. Kozlowski, S., M. Corr, T. Takeshita, L. F. Boyd, C. D. Pendleton, R. N. Germain, J. A. Berzofsky, and D. H. Margulies.1992. Serum angiotensin-1 converting enzyme activity processes an HIV 1 gp160 peptide for presenta-tion by MHC class I molecules. J. Exp. Med. 175:1417–1422.
21. Lehner, T., Y. Wang, M. Cranage, L. A. Bergmeier, E. Mitchell, L. Tao, G. Hall, M. Dennis, N. Cook, R. Brookes, L. Klavinskis, I. Jones, C. Doyle, and R. Ward.1996. Protective mucosal immunity elicited by targeted iliac lymph node immunization with a subunit SIV envelope and core vaccine in ma-caques. Nat. Med. 2:767–775.
22. Mayr, A., V. Hochstein-Mintzel, and H. Stickl. 1975. Abstammung, Eigen-schaften und Verwendung des attenuierten Vaccinia-Stammes MVA. Infec-tion 3:6–14.
23. Mayr, A., H. Stickl, H. K. Muller, K. Danner, and H. Singer. 1978. The smallpox vaccination strain MVA: marker, genetic structure, experience gained with the parenteral vaccination and behavior in organisms with a debilitated defence mechanism. Zentbl. Bakteriol. B 167:375–390. (In Ger-man.)
24. Meyer, H., G. Sutter, and A. Mayr. 1991. Mapping of deletions in the genome of the highly attenuated vaccinia virus MVA and their influence on virulence. J. Gen. Virol. 72:1031–1038.
25. Moss, B. 1991. Vaccinia virus: a tool for research and vaccine development. Science 252:1662–1667.
26. Myers, G., S. F. Josephs, J. A. Berzofsky, A. B. Rabson, T. F. Smith, and F. Wong-Staal.1989. Human retroviruses and AIDS 1989. Los Alamos Na-tional Laboratory, Los Alamos, N.Mex.
27. Redfield, R. R., D. C. Wright, W. D. James, T. S. Jones, C. Brown, and D. S. Burke.1987. Disseminated vaccinia in a military recruit with human immu-nodeficiency virus (HIV) disease. N. Engl. J. Med. 316:673–676. 28. Shirai, M., C. D. Pendleton, and J. A. Berzofsky. 1992. Broad recognition of
cytotoxic T-cell epitopes from the HIV-1 envelope protein with multiple class I histocompatibility molecules. J. Immunol. 148:1657–1667. 29. Sutter, G., and B. Moss. 1992. Nonreplicating vaccinia vector efficiently
expresses recombinant genes. Proc. Natl. Acad. Sci. USA 89:10847–10851. 30. Sutter, G., L. S. Wyatt, P. L. Foley, J. R. Bennink, and B. Moss. 1994. A
recombinant vector derived from the host range-restricted and highly atten-uated MVA strain of vaccinia virus stimulates protective immunity in mice to influenza virus. Vaccine 12:1032–1040.
31. Takahashi, H., J. Cohen, A. Hosmalin, K. B. Cease, R. Houghten, J. Cor-nette, C. DeLisi, B. Moss, R. N. Germain, and J. A. Berzofsky.1988. An immunodominant epitope of the HIV gp160 envelope glycoprotein recog-nized by class I MHC molecule-restricted murine cytotoxic T lymphocytes. Proc. Natl. Acad. Sci. USA 85:3105–3109.
32. Takahashi, H., R. N. Germain, B. Moss, and J. A. Berzofsky. 1990. An immunodominant class I-restricted cytotoxic T lymphocyte determinant of human immunodeficiency virus type 1 induces CD4 class II-restricted help for itself. J. Exp. Med. 171:571–576.
33. Takahashi, H., R. Houghten, S. D. Putney, D. H. Margulies, B. Moss, R. N. Germain, and J. A. Berzofsky. 1989. Structural requirements for class-I MHC molecule-mediated antigen presentation and cytotoxic T-cell recogni-tion of an immunodominant determinant of the HIV envelope protein. J. Exp. Med. 170:2023–2035.
34. Takahashi, H., S. Merli, S. D. Putney, R. Houghten, B. Moss, R. N. Germain, and J. A. Berzofsky.1989. A single amino acid interchange yields reciprocal CTL specificities for HIV gp160. Science 246:118–121.
35. Takeshita, T., H. Takahashi, S. Kozlowski, J. D. Ahlers, C. D. Pendleton, R. L. Moore, Y. Nakagawa, K. Yokomuro, B. S. Fox, D. H. Margulies, and J. A. Berzofsky.1995. Molecular analysis of the same HIV peptide function-ally binding to both a class I and a class II MHC molecule. J. Immunol. 154:1973–1986.
36. Tang, A., M. Amagai, L. G. Granger, J. R. Stanley, and M. C. Udey. 1993. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature 361:82–85.
37. Wittmann, M. M., A. Wittmann, and D. H. Wittmann. 1996. AIDS, emer-gency operations, and infection control. Infect. Control Hosp. Epidemiol. 17:532–538.
38. Wyatt, L. S., S. T. Shors, B. R. Murphy, and B. Moss. 1996. Development of a replication-deficient recombinant vaccinia virus vaccine effective against parainfluenza virus 3 infection in an animal model. Vaccine 14:1451–1458. 39. Wyatt, L. S., and B. Moss. Unpublished data.