FORMULATION DEVELOPMENT AND VALIDATION OF
ANALYTICAL TECHNIQUE FOR NOVEL DRUG
Thesis submitted to
The TamilNadu Dr.M.G.R.Medical University Chennai-600032
As a partial fulfillment of the requirement for the award of Degree of
DOCTOR OF PHILOSOPHY (Faculty of Pharmacy)
Submitted by
I. SOWKAR BAIG, M.Pharm.
Under the Guidance of
Prof.Dr.T.VETRICHELVAN, M. Pharm., Ph.D. PRINCIPAL
ADHIPARASAKTHI COLLEGE OF PHARMACY MELMARUVATHUR-603319, TAMILNADU, INDIA.
CERTIFICATE
This is to certify that the thesis entitled “FORMULATION DEVELOPMENT AND VALIDATION OF ANALYTICAL TECHNIQUE FOR NOVEL DRUG” submitted to The TamilNadu Dr. M.G.R. Medical University, Guindy, Chennai – 600 032, TamilNadu, India as a partial requirements for the award of Degree of DOCTOR OF PHILOSOPHY (Faculty of Pharmacy) is a record of research work done by Mr. I. SOWKAR BAIG, M.Pharm., during 2008 - 2011 under my guidance and supervision at Adhiparasakthi College of Pharmacy, Melmaruvathur – 603 319, TamilNadu, India and that the thesis has not formed the basis for the award of any other degree, diploma, associateship, fellowship, or any other similar title to the candidate and the thesis represents independent work of the candidate.
Place : Melmaruvathur Prof.Dr.T.VETRICHELVAN, M.Pharm., Ph.D.,
Date : Head, Department of Pharmaceutical Analysis,
and Principal,
Adhiparasakthi College of Pharmacy, Melmaruvathur- 603 319,
DECLARATION
I hereby declare that the thesis entitled“FORMULATION DEVELOPMENT AND VALIDATION OF ANALYTICAL TECHNIQUE FOR NOVEL DRUG” submitted by me for the degree of DOCTOR OF PHILOSOPHY (Faculty of Pharmacy), is the record of
research work carried out by me during the period from 2008 to 2011, under the guidance of Prof.Dr. T. Vetrichelvan, M.Pharm., Ph.D.,Head, Department of Pharmaceutical Analysis,
and Principal, Adhiparasakthi College of Pharmacy, Melmaruvathur- 603 319, TamilNadu, India and has not formed the basis for the award of any degree, diploma, associateship, fellowship, title in this or any other university or similar institute of higher learning.
Place: Melmaruvathur (I. SOWKARBAIG)
Date:
ACKNOWLEDGEMENT
I wish to express my deep sense of gratitude to His Holiness “ARULTHIRU AMMA” for his ever-growing blessings in each step of the study. I am grateful to Thirumathi Lakshmi Bangaru Adigalar, Vice President, ACMEC Trust, Melmaruvathur, for having given me an opportunity and encouragement all the way in completing the study.
I extend my thanks to Dr.T.Ramesh, M.D., Managing Director, MAPIMS, Melmaruvathur and Mrs.B.Umadevi, M.Pharm., Correspondent for providing all the necessary facilities to carry out this work.
I feel immensely delighted expressing my copious sincere thanks from the core of my
heart and deep sense of my indelible gratitude to my guide Prof.Dr.T.Vetrichelvan, M.Pharm., Ph.D., Principal and Head, Department of
Pharmaceutical Analysis, Adhiparasakthi College of Pharmacy, Melmaruvathur, for the active guidance, innovative ideas, creative works, indulgement and enthusiastic guidance, valuable suggestions, a source of inspiration where the real treasure of my work.
I owe my sincere thanks with bounteous pleasure to my Doctoral Committee members Dr.C.Vijayaraghavan, M.Pharm., Ph.D., Vice Principal, PSG College of Pharmacy, Coimbatore and Prof.Dr.A.Shantha, M.Pharm., Ph.D., Principal, C.L.Baid Metha College of Pharmacy, Chennai, without their encouragement and moral support it would have been absolutely impossible to bring out the work in this manner.
I would like to express my sincere thanks to Dr.S.Kavimani, M.Pharm., Ph.D., Professor, Department of Pharmacology, MTPG & RIH, Puducherry for his valuable suggestion during the research work.
I am highly indebted to my friend K.Janaki Raman, M.Pharm., Ranbaxy Pvt.Ltd., for providing me Abacavir sulfate and Ritonavir gift sample.
I am deeply indebted to my friends Mr.P.Saravanan, M.Pharm., A.Harikrishnan, B.Pharm., and R. Iyappan, M.Pharm., for their valuable moral support
and immense help without that the work would be incomplete.
I have great pleasure in express my sincere heartfelt thanks to Dr.D.Nagavalli, M.Pharm., Ph.D., Professor, Department of Pharmaceutical Chemistry,
Mr.K.Sundaramoorthy, M.Pharm., Professor, Department of Pharmaceutics, Dr.S.Shanmugam, M.Pharm., Ph.D., Professor, Department of Pharmaceutics, Mr.A.ThirugnanaSambanthan, M.Pharm., Assistant Professor, Department of Pharmaceutical Chemistry, Mr.J.Samynathan, M.Pharm., Assistant Professor, Department of Pharmaceutical Analysis, Miss.G.Sankari, M.Pharm., Assistant Professor, for encouragement and support for the successful completion of this work.
My sincere thanks to our lab technicians Mrs.S.Karpagavalli, Mr.M.Vimalan, Mr.M.GomathiShankar, and Mrs.N.Thatchayani, for their kind help throughout this work.
I am indeed very much thankful to the librarian Mr.M.Suresh, M.L.I.S., for providing all reference books for the completion of this project.
Finally yet importantly, I gratefully forward my affectionate thanks to my parents, uncle and aunty, especially my wife for their frequent prayers, which has sustained me a lot in the successful completion of my project work.
Finally I take this opportunity to express my gratitude to all the people involved directly or indirectly in the successful completion of this dissertation work.
LIST OF ABBREVIATIONS
ABS - Abacavir Sulfate
AIDS - Acquired Immune Deficiency Syndrome
o
C - Degree Celsius
CPS - Centipoises
DSC - Differential Scanning calorimetry EE - Entrapment efficiency
F - Formulation
FTIR - Fourier Transform infra-red spectrum.
g - gram
HAART - Highly Active Anti Retroviral Therapy. ICH - International Conference on Hormonization IP - Indian Pharmacopoeia
KBr - Potassium bromide
λ - Lambda
LE - Loading efficiency
LC - Loading capacity
LOD - Loss on Drying
m - Slope, units of response
mg - milligram
µg - microgram
nm - Nanometer
NDDS - Novel Drug Delivery System
Np - Nanoparticle
PACA - Polyalkyl cyanoacrylate PBS - Phosphate Buffer saline
PCS - Photon Correlation spectroscopy
pH - Negative logarithm of Hydrogen ion concentration PDI - Poly Dispersity Index
PS - Particle size PVA - Polyvinyl alcohol
RES - Reticulo Endothelial system RH - Relative Humidity
% RSD - Percentage Relative Standard Deviation Rt or tR - Retention time
RP-HPLC - Reverse Phase High performance liquid chromatography
RT - Ritonavir
Rpm - Revolution per minute
SEM - Scanning Electron Microscope SD - Standard Deviation
Sol - Solution
t - Time
t1/2 - Biological half life
TEM - Transmission Electron Microscope USP - United State Pharmacopoeia UV - Ultraviolet visible spectroscopy
V/V - Volume/ Volume
CONTENTS Chapter
No. Title Page No.
1 Introduction 1-14
1.1. Novel drug delivery system 1
1.2. Concept of novel drug delivery 2
1.3. Nanotechnology 3
1.4. Methods for preparing Nanoparticles 4
1.5. Evaluation of preparing Nanoparticles 8
1.6. Application of Nanoparticles 8
1.7. Polymeric Nanoparticles 9
1.8. Classification of biodegradable polymers 12
1.9. Introduction to HIV/AIDS 13
2 Aim and objectives of the study 15
3 Review of literature 17-37
3.1. Drug Profile 17
3.2. Polymer Profile 22
3.3. Review of Nanoparticles 26
4 Scope and Plan of work 38
5 Materials and Methods 40-57
5.1. Materials used for the study 40
5.3. Preparation of Nanoparticles 42 Chapter
No. Title Page No.
5.4. Characterization of Nanoparticles 46
5.5. Morphology Studies 47
5.6. Micromeritics studies 48
5.7. Invitro drug release study 51
5.8. Invitro release kinetics 51
5.9. Invivo studies 53
5.10. Pharmacokinetic Parameter Analysis 54
5.11. Stability Studies 54
5.12. Statistical Analysis 55
5.13. New analytical method development and the validation of best
formulated Nanoparticles 55
6 Results and Discussion 58-148
6.1. Interaction study 58
6.2. Calibration curve of Abacavir sulfate and Ritonavir 74
6.3. Prepared Nanoparticles 79
6.4. Morphology and Micromeritics properties of Nanoparticles 80
6.5. Entrapment efficiency of Nanoparticles 93
6.6 Invitro drug Release of Nanoparticles 94
6.7. Invivo study 132
6.9. New analytical method development and validation 137 Chapter
No. Title Page No.
7 Summary and Conclusion 149
8 Bibliography 152
LIST OF FIGURES
FigureNo. Contents Page No.
1 Schematic representation of the preparation of nanoparticles 45
2 Calibration curve of Abacavir sulfate in methanol 46
3 Calibration curve of Ritonavir in methanol 47
4 FT-IR spectra of a pure Abacavir sulfate powder 60
5 FT-IR spectra of Abacavir sulfate and chitosan polymer 61
6 FT-IR spectra of Abacavir sulfate and sodium alginate polymer 62
7 FT-IR spectra of formulation of Abacavir sulfate Nanoparticle 63
8 FT-IR spectra of a pure Ritonavir powder 63
9 FT-IR spectra of Ritonavir and chitosan polymer 64
10 FT-IR spectra of Ritonavir and sodium alginate polymer 65
11 FT-IR spectra of formulation of Ritonavir Nanoparticle 66
12 DSC curve of pure Abacavir sulfate powder 68
13 DSC curve of Abacavir sulfate and chitosan polymer 68
14 DSC curve of Abacavir sulfate and sodium alginate 69
15 DSC curve of formulation of Abacavir sulfate nanoparticles 69
16 DSC curve of pure Ritonavir powder 70
Figure
No. Contents Page No.
18 DSC curve of Ritonavir and sodium alginate 71
19 DSC curve of formulation of Ritonavir nanoparticles 71
20 x-ray diffraction pattern of pure Ritonavir powder 73
21 x-ray diffraction pattern of Ritonavir and sodium alginate polymer 73
22 x-ray diffraction pattern of Ritonavir nanoparticles 74
23 Calibration curve of Abacavir sulfate in 0.1 N HCl 75
24 Calibration curve of Ritonavir in 0.1 N HCl 76
25 Calibration Curve of Abacavir Sulfate in pH 7.4 Phosphate Buffer 78
26 Calibration Curve of Ritonavir in pH 7.4 Phosphate Buffer 79
27 PCS Particle size distribution of Abacavir sulfate nanoparticle AF4 82
28 PCS Particle size distribution of Abacavir sulfate nanoparticle AF5 82
29 PCS Particle size distribution of Abacavir sulfate nanoparticle AF6 83
30 PCS Particle size distribution of Ritonavir nanoparticle BF4 83
31 PCS Particle size distribution of Ritonavir nanoparticle BF5 84 32 PCS Particle size distribution of Ritonavir nanoparticle BF6 84
33 Zeta potencial of Abacavir sulfate nanoparticle AF4 85 34 Zeta potencial of Abacavir sulfate nanoparticle AF5 86
35 Zeta potencial of Abacavir sulfate nanoparticle AF6 87
Figure
No. Contents Page No.
37 Zeta potencial of Ritonavir nanoparticles BF5 89
38 Zeta potencial of Ritonavir nanoparticles BF6 90
39 TEM micrograph for formulation of Abacavir sulfate nanoparticle AF6 91
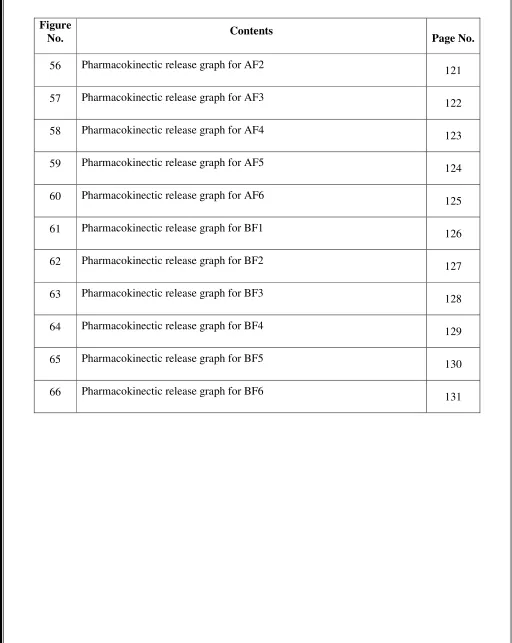
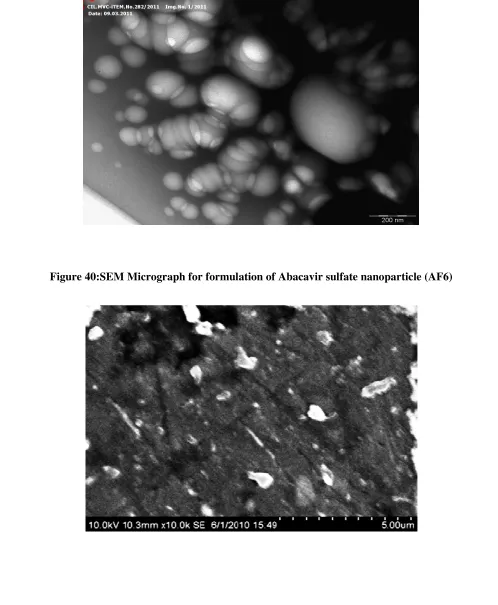
40 SEM micrograph for formulation of Abacavir sulfate nanoparticle AF6 91
41 TEM micrograph for formulation of Ritonavir nanoparticle BF6 92
42 SEM micrograph for formulation of Ritonavir nanoparticle BF6 92
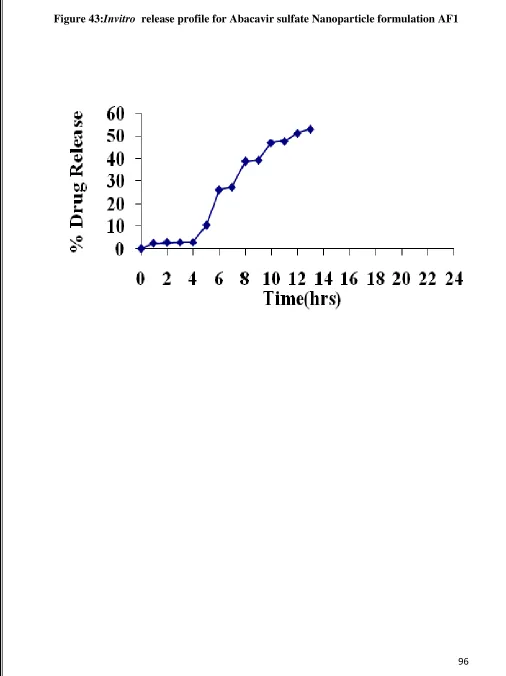
43 Invitro Release graph for Abacavir sulfate nanoparticle AF1 96
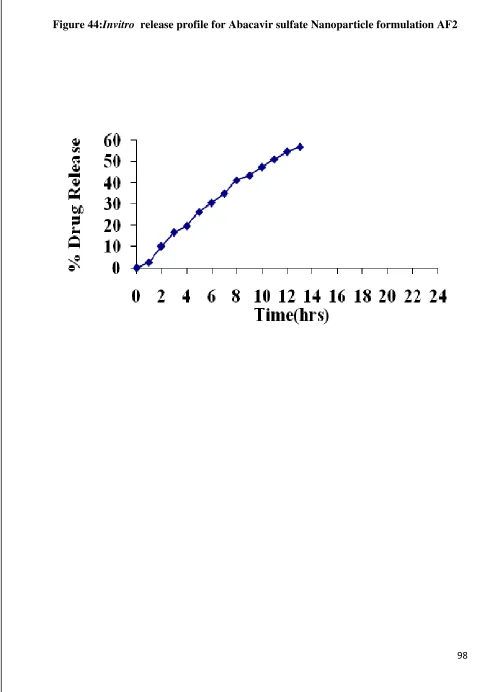
44 Invitro Release graph for Abacavir sulfate nanoparticle AF2 98
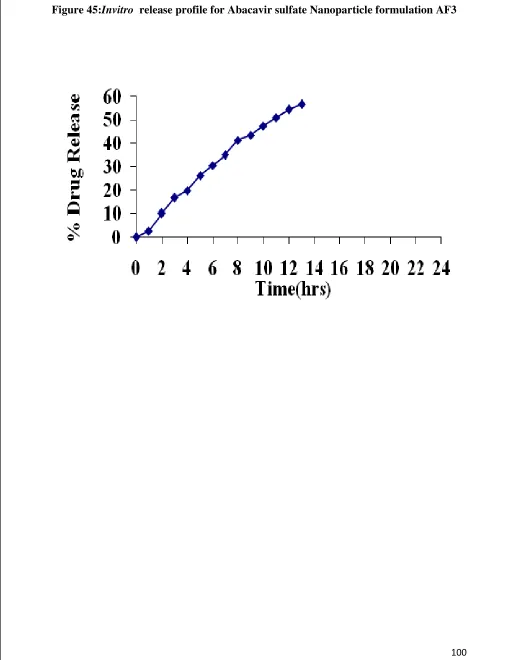
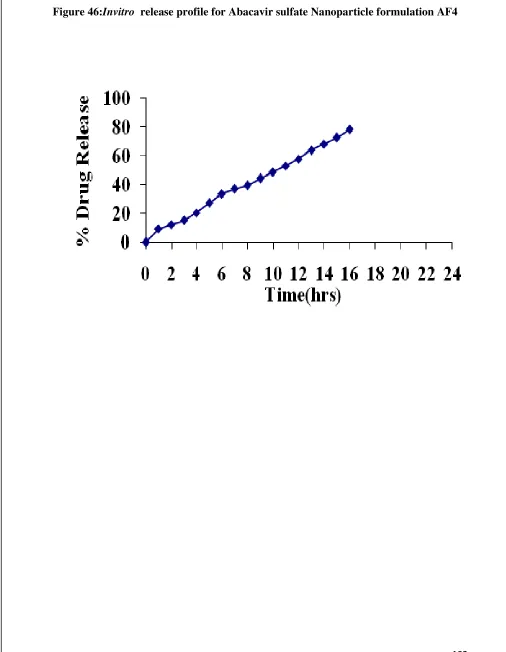
45 Invitro Release graph for Abacavir sulfate nanoparticle AF3 100 46 Invitro Release graph for Abacavir sulfate nanoparticle AF4 102
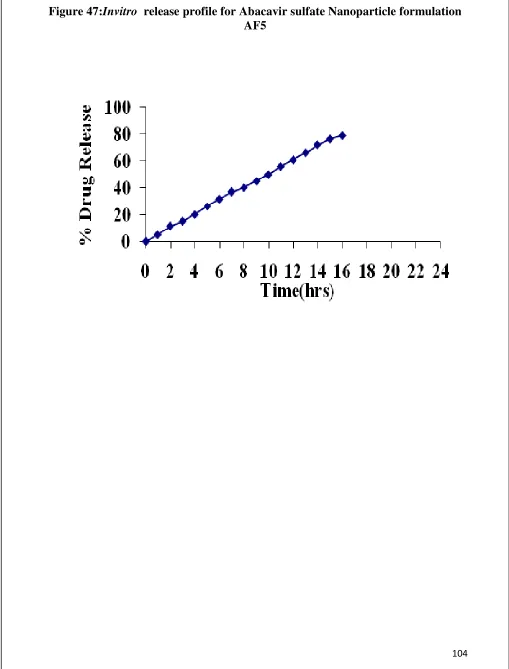
47 Invitro Release graph for Abacavir sulfate nanoparticle AF5 104
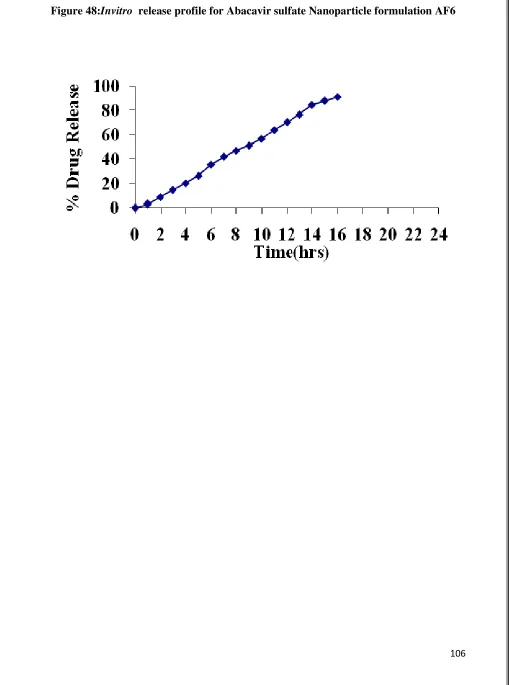
48 Invitro Release graph for Abacavir sulfate nanoparticle AF6 106 49 Invitro Release graph for Ritonavir nanoparticle BF1 108 50 Invitro Release graph for Ritonavir nanoparticle BF2
110 51 Invitro Release graph for Ritonavir nanoparticle BF3 112 52 Invitro Release graph for Ritonavir nanoparticle BF4 114 53 Invitro Release graph for Ritonavir nanoparticle BF5
116 54 Invitro Release graph for Ritonavir nanoparticle BF6
118
Figure
No. Contents Page No.
56 Pharmacokinectic release graph for AF2 121
57 Pharmacokinectic release graph for AF3 122
58 Pharmacokinectic release graph for AF4 123
59 Pharmacokinectic release graph for AF5
124 60 Pharmacokinectic release graph for AF6
125 61 Pharmacokinectic release graph for BF1
126 62 Pharmacokinectic release graph for BF2
127 63 Pharmacokinectic release graph for BF3
128 64 Pharmacokinectic release graph for BF4
129 65 Pharmacokinectic release graph for BF5
130 66 Pharmacokinectic release graph for BF6
LIST OF TABLES
Table No. Contents
Page No. 1 Composition of Abacavir sulfate and Ritonvir nanoparticle formulation 43
2 Data collection and absorbance of Abacavir sulfate in 0.1 N HCl 75
3 Data collection and absorbance of Ritonavir in 0.1 N HCl 76
4
Data collection and absorbance of Abacavir sulfate in pH 7.4 Phosphate
Buffer 77
5 Data collection and absorbance of Ritonavir in pH 7.4 Phosphate Buffer 78
6
Particle size, polydispersity and Zeta potencial studies of Abacavir
sulfate and Ritonavir nanoparticle 81
7 Entrapment efficiency of Abacavir sulfate and Ritonavir nanoparticles 93
8 In vitro release profile for Abacavir sulfate nanoparticle AF1 95
9 In vitro release profile for Abacavir sulfate nanoparticle AF2 97
10 In vitro release profile for Abacavir sulfate nanoparticle AF3 99
11 In vitro release profile for Abacavir sulfate nanoparticle AF4 101
12 In vitro release profile for Abacavir sulfate nanoparticle AF5 103
13 In vitro release profile for Abacavir sulfate nanoparticle AF6 105
14 In vitro release profile for Ritonavir nanoparticle BF1 107
15 In vitro release profile for Ritonavir nanoparticle BF2 109
16 In vitro release profile for Ritonavir nanoparticle BF3 111
Table No. Contents
Page No. 18 In vitro release profile for Ritonavir nanoparticle BF5 115
19 In vitro release profile for Ritonavir nanoparticle BF6 117
20
Pharmacokinetics parameter of Abacavir sulfate and Ritonavir
nanoparticles 132
21 Stability studies of Best formulation- Method AF6 and BF6 (Short term) 135
22 Stability studies of Best formulation- Method AF6 and BF6 (Long term) 136
23 Solubility profile of Abacavir sulfate in non polar and polar solvents 139
24 Optical characteristic of Abacavir sulfate by UV spectroscopic method 140
25 Quantification of formulation - Abacavir sulfate 141
26 Recovery studies for formulation- Abacavir sulfate 142
27 Interday and intraday analysis of formulation - Abacavir sulfate 143
28 Solubility profile of Ritonavir in non polar and polar solvents 144
29 Optical characteristic of Ritonavir by UV spectroscopic method 145
30 Quantification of formulation - Ritonavir 146
31 Recovery studies for formulation - Ritonavir 147
1
1. INTRODUCTION
1.1
Novel Drug delivery system
1For many decades, the treatment of an acute disease or a chronic illness has been mostly accomplished by the delivery of drugs to patients using various pharmaceutical dosage forms such as tablets, capsules, pills, suppositories, creams, ointments, liquids, aerosols and injectables. Even today, these conventional drug delivery systems are the primary pharmaceutical products commonly seen in prescriptions and which are available as over-the-counter medicines.
These types of dosage forms are known to provide prompt release of drugs. Therefore to achieve as well as to maintain the drug concentration within the therapeutically effective range needed for treatment, it is often necessary to take these dosage forms several times a day. This however results in a significant fluctuation in drug levels in the body. Recently, several technical advancements have resulted in the development of new techniques for drug release. These techniques are capable of controlling the rate of drug delivery, sustaining the duration of therapeutic activity and or targeting the delivery of drug to a tissue. These are referred to as novel drug delivery systems and they have revolutionized the method of medications, providing a number of therapeutic benefits.
The therapeutic benefits of novel drug delivery system over conventional dosage forms are,
• Improved therapy by increasing the duration of action and reducing the side effects.
• Potential prophylactic action.
• Increase patient compliance through decreased dosing frequency and convenient routes of administration.
2
1.2
Concept of Novel drug delivery
2Oral dosage forms are the most common pharmaceutical dosage forms due to their convenient of administration. However, the major drawback of these dosage forms is their erratic absorption from the gastro intestinal tract due to various gastric vagaries. Per oral conventional dosage forms are repeat action pharmaceuticals. These do not maintain constant drug concentration in blood. More over they get cannot by-pass the hepatic first pass system. A major quantity of the drug destroyed in the liver due to metabolic enzymes. The action of the drug is also delayed due to several such reasons. Through the injections, drugs are given directly to the blood circulation by passing the liver. This hepatic by pass mechanism results in quick high drug concentration in the blood and rapid therapeutic action.
However, injections are hazardous pharmaceutical dosage forms due to their manufacturing problems and other drawbacks associated with their administration. Scientists throughout the world are looking for pharmaceutical dosage forms, which will act injection but will not be an injection. Pharmaceutical scientist, have taken up a challenge for finding the solution of these problems.
This problem is further aggravated due to the reasons that next decade drugs will be small proteins and peptides derived by genetic engineering and biotechnology. Injections were earlier considered as the only dosage forms for these new drugs. However, several routes and devices are currently available for administration of these drugs. These formulations are already available in the pharmaceutical market and more will come in the near future.
3
1.3
Nanotechnology
3Nanoparticle have become an important area of research in the field of drug delivery, because they have the ability to deliver a wide range of drug to varying areas of the body for sustained periods of time.
Nanoparticles are said to be colloidal particles composed of natural, synthetic or semi synthetic polymers, with the size ranging from 1nm to 1000 nm. The drugs or other molecule may be dissolved, entrapped, encapsulated, absorbed or attached into the Nanoparticles.
Nanoparticles have further advantage over larger micro particles, because they are better suited for intravenous delivery and they have been highly exploited for controlled drug release and site specific drug targeting.
The advantages of using nanoparticles as a drug delivery system include the following:
• Particle size and surface characteristics of nanoparticles can be easily manipulated to achieve both passive and active drug targeting after oral administration.
• Nanoparticles posses a better stability as compared to liposome’s. This property may be very important for many modes of targeting.
• Controlled release and particle degradation characteristics can be readily modulated by the choice of matrix constituents. Drug loading is relatively high and drugs can be incorporated in to the systems without any chemical reactions. This is an important factor for preserving the drug activity.
• The method of preparation are simple, easier and reproducible.
• It can easily pass through syringe needle and exhibit good rheological properties.
Mechanism of Drug Release
44
• Osmatically driven burst mechanism
• Pore diffusion
• Erosion or degradation of polymer.
Methods of determination of drug release
The following methods for the determination of the in-vitro release have been used.
• Side by side diffusion cells with artificial or biological membranes.
• Dialysis bag diffusion techniques.
• Reverse dialysis sac techniques.
• Ultra centrifugation.
• Ultra filtration (centrifugal) technique.
1.4
Methods for preparing Nanoparticles
51) Solvent evaporation method
5 pilot production, alternative methods using low-energy emulsification are required. In this pursuit, following approaches have been attempted.
2) Spontaneous Emulsification/Solvent diffusion method
It is the modified version of the solvent evaporation method. In this water-soluble solvent like acetone or methanol along with the water insoluble organic solvents like dichloromethane or chloroform were used as an oilphase. Due to the spontaneous diffusion of water-soluble solvent (acetone or methanol), an interfacial turbulence is created between two phase loading to the formation of smaller particles. As the concentration of water-soluble solvent (acetone) increases, a considerable decrease in particle size can be achieved.
3)
Salting out / Emulsification – diffusion method
The method discussed above require the use of organic solvents, which are hazardous to the environment as well as to the physiological system. The USFDA has specified the residual amount of organic solvent injectable colloidal systems. In order to meet these requirements, Allemann and co-workers have developed two methods of preparing Nanoparticles. One is a salting out method while the second one is the emulsification solvent diffusion technique.
4)
Polymerization method
6 alkylcyanoacrylate at ambient temperature. Drug is dissolved in the polymerization medium either before the addition of the monomer or at the end of the polymerization reaction. The Nanoparticles suspension is then purified by ultracentrifugation or by resuspending the particles in an isotonic surfactant free medium.
5)
Coacervation / precipitation
6This method utilizes the physio chemical property of polymers, the polymers which are insoluble in alkaline pH medium, but precipitates/coacervates when it comes in contact with alkaline solution. Particles are produced by blowing polymer solution into an alkali solution like sodium hydroxide, sodium hydroxide-methanol or ethanediamine using a compressed air nozzle to form coacervate droplets. Separation and purification of particles was done by titration/ centrifugation followed by successive washing with hot and cold water. The method is schematically represented in figure, varying compressed air pressure or spray-nozzle diameter controlled the size of the particles and then using a cross linking agent to harden particles can control the drug release. In another technique, sodium sulfate solution was added drop wise to an aqueous acidic solution of polymer containing a surfactant under stirring and ultra sonication for 30 min. Nanoparticles were purified by centrifugation and re-suspended in demineralized water particles were cross linked with Glutaraldehyde. Particles produced by this method have better acid stability than observed by other methods.
6) Spray drying
7 to the formation of small droplets, from which solvent evaporates instantaneously leading to the formation of free flowing particles, as depicted in figure various process parameters are to be controlled to get the desired size of particles. Particle size depends upon the size of nozzle, spray flow rate, atomization pressure, inlet air temperature and extent of cross linking.
7)
Emulsion droplet coalescence method
The novel emulsion droplet coalescence method, which utilized the principles of both emulsion cross-linking and precipitation. However, in this method, instead of cross linking the stable droplets, precipitation is induced by allowing coalescence of polymer droplets with sodium hydroxide droplets. First, a stable emulsion containing aqueous solution of polymer along with drug is produced in liquid paraffin oil and then, another stable emulsion containing polymer aqueous solution of sodium hydroxide is produced in the same manner. When both emulsions are mixed under high speed stirring, droplets of each emulsion would collide at random and coalescence, there by precipitating polymer droplets to give small size particles.
8) Drug loading into Nanoparticles
Drug loading in nanoparticulate systems can be done by two methods, i.e.,
• During the preparation of particles (incorporation) and
• After the formation of particles (incubation)
8 mixed with polymer solution to form a homogenous mixture and then, particles can be produced by any of the method discussed before. For instance, Cisplastin was loaded during the formation of particles with encapsulation efficiency as high as 99%. The initial concentrated of cisplastin and volume of Glutaraldehyde had no effect on the encapsulation efficiency. Drug encapsulation increased as the concentration of polymer increased. Water-insoluble drugs and drugs that can precipitate in acidic pH solution can be loaded after the formation of particles by soaking the performed particles with the saturated solution of drug.
1.5
Evaluation of prepared Nanoparticles
7• Particle size determination.
• Polydispersity index.
• Particle shape and surface morphology.
• Determination of drug entrapment.
• Zetapotential.
• In-vitro drug release.
• Stability studies.
• Pharmacokinetic parameter.
1.6. Applications of Nanoparticles
Nanoparticles were first developed in the mid-seventies by Birrenbach and Speiser. Later on, the application for the design of drug delivery systems was made available by the use of biodegradable polymers that were considered to highly suitable for human applications. At that time, the research on colloidal carries was mainly focusing on liposomes, but no one was able to produce stable lipid vesicles suitable for clinical applications. In some cases, Nanoparticles have been shown to be more active than liposomes
9 (e.g, antibiotics, antiviral, antiparasitic drugs, cytostatics, protein and peptides) were associated to nanoparticles.
• Intravenous Administration
• Application to the treatment of HIV/AIDS
• Subcutaneous\Intramuscular Administration
• Oral route
• Ocular Delivery
1.7 Polymeric Nanoparticles
8Polymeric Nanoparticles are particles of less than 1µm diameter that are prepared from natural or synthetic polymers. Nanoparticles have become an important area of research in the field of drug delivery because they have the ability to deliver wide range of drugs to varying areas of the body for sustained period of time.
Polymeric Nanoparticles have many advantage such as:
• Easy to fabricate and characterize
• Process is inexpensive so the final cost of formulation is reduced.
• Polymers are biocompatible, biodegradable, non-toxic and non-immunogenic that are suitable for preparing different formulations.
• Mostly polymers are water soluble such as chitosan and sodium alginate.
• Applicable to a broad category of small molecule drugs, genes, proteins and polynucleotides.
• Can be lyophilized and are stable after reconstitution with medium for application.
10
Polymers in the preparation of Nanoparticles
A wide variety of delivery system has been developed for the purpose of prolonging the release and ultimately bioavailability of drugs to the body. Examples include the transdermal patch, oral dosage forms such as osmotic pumps and swellable hydrophilic polymer matrices, and various types of polymer-based parenteral formula Depot sustained release formulations can overcome limitations associated with oral or transdermal administration routes.
Several of these limitations include poor drug stability in the GI track, low drug permeability through the GI mucosa or stratum cornea, rapid clearance from first-pass metabolism, and the need for delivery for more than a few days. Many interesting and successful parenteral depot systems for sustained release applications have been developed. Such system can be distinguished into degradable and non-degradable delivery systems based on the properties of the polymers.
Mechanism of Biodegradable polymers
The use of biodegradable polymers precludes the need for retrieval at the conclusion of the dosing regimen, thereby avoiding the potential complications associated with the use of non–degradable systems. Degradation may take place by variety of mechanisms, although it generally relies on either erosion or chemical changes to the polymer. Degradation by erosion normally take place in devices that are prepared from soluble polymers. In such instances, the devices erode as water is absorbed into the system causing the polymer chain to hydrate, swell and ultimately dissolve away from the dosage.
11 describes that characterize low chemical degradation of the polymer or of polymer-drug conjugates can be utilized to achieve drug release.
The most widely studied biodegradable polymers include those which undergo chemical degradation through random hydrolysis of the covalent bond constituting the backbone of the polymer chains. Random chain scission results in reduction in the molecular weight of the polymer. As this process continues over time, polymer chain become progressively shorter resulting, at some point in time, in the loss of mechanical integrity in the dosage form. Ultimately, the degradation process proceeds until polymers fragments are degraded to soluble oligomers or individual monomers. As a necessity due to their process, bio degradable polymers and their degradation products must be biologically compatible and non-toxic.
12
1.8 Classification of Biodegradable polymers.
4 & 9Type Class Examples
Synthetic Polymer Polyamide Polyamino acids Polypeptides Polyesters Poly (glycolide) Poly(D,L-Lactide) Poly(D,L-lactide-co-glycollde) Poly(E-Caprolactone) Poly(dioxanone) Poly(hydroxybutyrate) Poly anhydride Poly orthoester Poly phosphazene Poly phosphoester Poly phosphate Poly phsophonate Poly phosphite Naturally Occurring Polymers Polysaccharides Chitosan Alginate Dextron Starch Hyaluronic acid Polypeptides, Proteins Collagen Gelatin
13
1.9. Introduction to HIV/AIDS
10Human Immune Deficiency Virus (HIV) infection and acquired immune deficiency syndrome (AIDS), commonly referred to as HIV/AIDS, constitute one of the most serious infectious disease challenges to public health globally, and has had a crippling effect in certain parts of the world especially in sub-saharan Africa. There are currently 33.2 million people living with HIV/AIDS globally of this total number, an over whelming 22.5 million people are HIV positive in sub-saharan Africa specifically, representing 67.8% of the global number. Intraventions such as AIDS counselling educational tools and antiretroviral drug therapy have contributed to transforming HIV infection from a fatal to manageable chronic infections disease.
Despite the availability of these measures, the above Statistics indicate that much remain to be accomplished as the number of newly reported. HIV infections still remains unacceptably high.
There are currently two known species of HIV, viz., HIV-1 and HIV-2, with their respective sub species. HIV-1 is the globally common infection while HIV-2 is more prevalent in west Africa, and takes a longer time to develop into immunodeficiency from infection than HIV-1.
HIV infection in the human body results mainly from integration of its viral genome into the host cell for the purpose of cell replication and AIDS is the advanced stage of the disease caused by HIV infection. The virus infects the host cell by binding the viral SP 120 Protein to two transmembrane receptors, i.e., CD4+ and either of the two chemokine
receptors, CCRS and CXCR4.
14
Nanotechnology for HIV/AIDS treatment
11Nanotechnology-based platforms for systemic delivery of antiretroviral could have similar advantages, sustained-release delivery systems can enhance their half-lives, keeping them in circulation at therapeutic concentrations for longer period of time. This could have major implication in improving adherence to the drugs. Nanoscale delivery systems also enhance and modulate the distribution of hydrophobic and hydrophilic drugs into within different tissue due to item small size.
This particular feature of nanoscale delivery system appears to hold the most promise of their use in clinical treatment and prevention of HIV. Specifically, target delivery of antiretroviral drugs to CD4+ cells and macrophages as well as delivery to the brain and other organ system could ensure that drug reach latent reservoirs.
15
2.
AIM AND OBJECTIVES OF THE STUDY
Novel drug delivery system present an opportunity for formulation to overcome the many challenges associated with Antiretroviral (ARV) drug therapy thereby improving the management of patients with HIV/AIDS.
Many promising antiviral agents against HIV unfortunately have disadvantages in physiochemical properties which lead to a poor bio-distribution and insufficient cellular up take. One presupposition for a therapeutic approach is to maintain adequate drug level at the sites viral replication over extended period of time. The well-known adverse reaction and side effects of antiviral treatment are often related to the accumulation of the drug at inappropriate site.
Symptomatic HIV/AIDS patients have to take their drugs by 3 times during the day in order to maintain the drug level in their bodies. In order to overcome this challenging problems mentioned above, the research in nanotechnology has been initiated to investigated this work and describes some promising strategies of using nanoparticles to improve in HIV/AIDS drug formulations.
16 1) Reduce the dosing frequently. 2) Increase the bioavailability and decrease the degradation /metabolism in the gastro–intestinal tract 3) Deliver them to the target cells selectively with minimal side effects.
This work provides a antiretroviral novel drug delivery system for achieving sustained drug release kinetics, and for addressing formulation difficulties such as poor solubility, stability and drug entrapment.
This aim highlights the significant potential that novel drug delivery system have for the future effective treatment of HIV/AIDS patients on Antiretroviral drug therapy.
The objective of the present investigation was to describe the formulation and characterization of Abacavir sulfate and Ritonavir nanoparticles specifically, the following themes were examined.
Effect of polymer on nanoparticle preparation and selection of the best polymer. Preparation of biodegradable nanoparticles (using selected polymer) by two
different methods.
Comparison of various drug, polymer ratio in case of biodegradable nanoparticle by evaluating the release characteristics.
Evaluation of in-vitro release kinetics, pharmacokinetic parameter of nanoparticles prepared with the best drug polymer ratio.
17
3. REVIEW OF LITERATURE
3.1. Drug profile
12Abacavir sulfate
Official status – Official in I.P., Merck Index. Molecular formula – (C14H18N6O)2, H2SO4 Molecular weight – 670.8
Structure
Chemical Name - (1S, 4R) –a-{2-Amino-6(cyclo propylamino)-9H-purin-9-yl} -2- cyclopentene -1- methanol hemisulfate.
Description – A white or almost white crystalline powder. Solubility – Freely soluble in water and methanol. Melting point – 165oC
Storage – Stored at a temperature not exceeding 30oC
Half life – 1.54 ± 0.63 h Protein binding – 50%
18
Pharmacology
13• Abacavir sulfate is a nucleoside reverse transcriptase inhibitor (NRTI) with activity against Human immunodeficiency virus type 1 (HIV-1).
• Abacavir sulfate is phosphorylated to active metabolites that compete for incorporation into viral DNA. They inhibit the HIV reverse transcriptase enzyme competitively and act as a chain terminator of DNA synthesis.
• The lack of a 3' -OH group in the incorporated nucleoside analogue presents the
formation of the 5' to 3'phosphodiester linkage essential for DNA chain elongation, and therefore, the viral DNA growth is terminated.
Mechanism of Action
• Abacavir is a carbocyclic synthetic nucleoside analogue. Intra cellularly, Abacavir is converted by cellular enzymes to the active metabolite carbovir triphosphate, an analogue of deoxy guanosine -5'-triphosphate (dGTP)
• Carbovir triphosphate inhibits the activity of HIV-1 reverse transcriptase (RT) both by competing with the natural substrate dGTP and by its incorporation into viral DNA.
Pharmacokinetic parameter
• The pharmacokinetic properties of Abacavir were independent of dose over the range of 300 to 1,200 mg once day.
• Abacavir was rapidly and extensively absorbed after oral administration. The geometric mean absolute bioavailability of the tablet was 83%. After oral administration of 300 mg twice daily.
19
• Abacavir is not significantly metabolized by cytochrome P450 enzymes. The primary routes of elimination of Abacavir are metabolism by alcohol dehydrogenase to form the 51-carboxylic acid and glucuronyl transferase to form the 51-glucuronide. The metabolites do not have antiviral activity.
Distribution
• The apparent volume of distribution after IV administration of Abacavir was 0.86±0.15 L/Kg, suggestion that Abacavir distribution into extravascular space.
• In 3 subjects, the CSF AUC (0-6 h) to plasma Abacavir AUC (0-6 h) ratio ranged from 27% to 33%.
Side effects
Abdominal pain, cough, diarrhea, fatigue, fever, headache, insomnia, joint pain, loss of appetites, mouth ulcers, muscle aches, nausea, rash, severe blisters in the mouth and eyes, shortness of breath sleep disorders, sore throat, swelling, tiredness and vomiting.
Dose of Abacavir sulfate for adults
The recommended Abacavir sulfate dose for treating adults (17 Years old and older) with HIV or AIDS is Abacavir sulfate 300 mg twice daily or 600 mg once daily. One study suggested that people who took Abacavir sulfate 600 mg once daily (Instead of Abacavir 300 mg twice daily) had a higher risk of developing dangerous allergic reactions. Abacavir sulfate should be used in combination with other HIV medications, as part of an HIV “Cocktail” (this is known as Highly Active Anti-Retroviral Therapy, or HAART).
Abacavir sulfate Dosages for children
20 Ritonavir14
Official status – Official in I.P., Merck Index. Molecular Formula – C37H48N6O5S2
Structure
Chemical Name - 10-Hydroxy-2-methyl-5-(1-methylethyl)-1-[2-(1-methyl ethyl)-4- [thiazolyl]-3,6,-dioxo-8,11 bis (Phenyl methyl)-2,4,7,12- Tetra azatrudecan-13-oicacid, 5-thiazolymethyl ester, [5S (5R*,8R*,10R*,11R*)]
Category – Antiretroviral (Protease inhibitor) Description – A white or almost white powder
Solubility – Practically insoluble in water, freely soluble in methanol, sparingly soluble in acetone and very slightly soluble in acetonitrile.
Storage – Ritonavir should be kept in a well closed container and protected from light.
Half life – 3 to 5 h Bioavailability – 63%
21
Mechanism of action
13Ritonavir is a selective competitive reversible inhibitor of HIV protease interferes with the formation of essential protein and enzyme. After which the formation of immature and non-infectious viruses follows. It also interferes with the production of infectious HIV and limits further infection spread of the virus.
Ritonavir produces most common adverse effects are dose dumping, and include G.I.T complaint such as nausea, diarrhoea, anorexia, abdominal pain and taste prevention.
Pharmacokinetic parameter
Ritonavir is one of the most essential protease inhibitor (PI) for AIDS treatment with its oral bioavailability over 63% the pharmacokinetic properties of Ritonavir were independent of dose over the range of 600 to 1200 mg/day, and it was rapidly and extensively absorbed after oral administration.
Side effects
Fatigue or a general ill feeling (Malaise), Loss of appetite, Nausea, Yellow eyes or skin (Jaundice), Tenderness near the liver (the upper-right abdomen), pale stools, high blood sugar, Anemia, symptoms of inflammation of the pancreas such as: tender or swollen abdomen, nausea and vomiting, fever, a rapid heart rate (tachycardia) and Rapid breathing.
Ritonavir Dosing for Adults
22
3. 2. Polymer Profile
15-16Chitosan
Chitosan is a natural polysaccharide comprising co polymers of glucosamine and N-acetyl glucosamine. It can be derived by partial deN-acetylation of chitin from crustacean shells. Although the discovery dates from nineteenth century, it has recently received lot interest for drug delivery as well as biomedical applications.
Chitosan have a positive charge when compared with many other natural polymers and is muco adhesive.Hence it is widely used in drug delivery system. Chitosan is a week base and is insoluble in water and organic solvents, however, it is soluble in dilute aqueous and acidic solution (pH 6.5).
Commercially chitosan is available in the form of dry flakes, solution and fine powder properties such as biodegradability, low toxicity and good compatibility make it suitable for use in biomedical and pharmaceutical formulations.
Physical properties
23
Pharmaceutical Applications
Properties such as biodegradability, low toxicity and good biocompatability make it suitable for use in biomedical and pharmaceutical formulations, e.g. it is used for hydro billirubinaemic and hypochloestrolemic effects, antacid and antiulcer activities, wound and burn healing properties, immobilization of enzymes and living cell and in ophthalmology. Since chitosan has a capacity of forming film it has been suggested as a biopolymer of choice for the development of contact lens (Soft and hard contact lenses).
24 Sodium Alginate17-19
Alginates are linear, unbranched polysaccharides composed of random chains of glucuronic and mannuronic acid. In aqueous media, the sodium ions from salts of these anionic, hetero polymers exchange, divalent cation, such as calcium to form water in soluble gels.
Alginates are non-Immunogenic and available in wide range of molecular weight as characterized by their inherent viscosity.Alginate Nanoparticles are prepared by extruding on aqueous sodium alginate solution through a narrow-bore needle into an aqueous solution of a cationic agent, such as calcium ions, chitosan, or poly-1-lysine. The cation cross link the glucuronic and mannuronic acid to form an egg box structure that forms the core of the gel matrix.
Properties of Alginates
Solubility
25
Viscosity
Various grades of sodium alginates are available, yielding aqueous solutions of varying viscosity within a range of 20-400 centipoises.
Pharmaceutical applications
Alginates have been widely used as tablet disintegrator, binding agent, viscosity modifying agent, as a stabilizer in disperse system in the production of suspension and emulsion and also thickening agent in pharmaceutical industries. The most important advantage of using alginate as a matrix for sustained release formulations is its biodegradability because it is degraded and is absorbed by the body during and or a after drug release without any toxic effects.
The following properties of alginates have enabled it to be used as a most for sustained drug delivery,
• It is readily available and is relatively inexpensive.
• It is non – toxic when taken orally and also has a protective effect processing and hence stability, toxicological and environmental problems associated with solvents can be minimized.
26
3.3. Review of Nanoparticles
Benoy Brata Bhowmik et al.,20 have developed slow release Testosterone-loaded nano capsules in alginate, biodegradable hydropolymer were prepared by insitu nanoemulsion-polymer cross linking approach. Different formulation varying in the drug loading solvent phase were prepared. Four different drug-loading solvents were assayed and the food grade hexane provided nanocapsules testosterone load of 30%. Testosterone loading was confirmed by FT-IR, DSC and Quantitated by HPLC. Prepared nanocapsules appeared spherical with a dense drug core in Transmission Electron Microscopy studies. The nanocapsulation technique developed can be a good choice for the development of different sustained steroid hormonal drug carriers.
Kavitha S.Rao et al.,21 have reported a steady increase in the drug parenchyma capillary ratio over time without disrupting the BBB integrity suggests that TAT-conjugated NPs are first immobilized in the brain vascularate prior to their transport into parenchyma. Localization of NPs in the brain parenchyma was further confirmed with histological analysis of the brain section. The brain drug level with conjugated NPs was 800-fold higher than that with drug in solution at two weeks. Drug clearance was seen within four weeks.
27 Lavanya Varatharajan et al.,23 have reported a better appreciation of the transporters present at the brain barriers will prove a valuable millestone in understanding the limits brain penetration of anti-HIV drugs and drug combinations, with enhanced efficiency in the CNS. This review aims to summarise current knowledge on the transport of anti-HIV drugs across the blood-brain barriers and the chorid plexus as well as provide recommendations for future research.
Xiaowei Dong et al.,24 have reported these novel paclitaxel nanoparticles were stable at 4oC over five months and PBS at 37oC over 102 h as measured by Physical stability. Release of paclitaxel was slow and sustained without initial burst release. Cytotoxicity studies in MDA-MB-231 cancer cells showed that both nanoparticles have similar anticancer activities compared to Taxol.
28 Jason R. Mc Carthy et al.,26 have studied magnetic nanoparticles have become important tools for the imaging of prevalent diseases, such as cancer, atherosclerosis, diabetes and others. While first generation nanoparticles were fairly nonspecific, newer generations have been targeted to specific cell types and molecules targets via affinity ligands. Commonly, these ligands emerge from phage or small molecule screens.
Nicolas Anton et al.,27 have studied the formulation of nanoparticles drug carriers, based on nano emulsion formulation, appears at first to require adaptation to the therapeutic aims and specificity of the drug to encapsulate. That is to say, both high and low energy nanoemulsion formulation methods whether or not they include the use of organic solvents, have to be adapted according to the active molecule properties. i.e Sensitivity to temperature, high-shear devices, contact with organic solvents etc.
Zonghua Li et al.,28 have studied Natural polysaccharides, due to their out standing merits, have received more and more attention in the field of drug delivery systems. In particular, polysaccharides seem to be the most promising materials in the preparation of nanometric carriers. This work relates to the newest developments in the preparation of polysaccharides-based nanoparticles. In this work, four mechanism are introduced to prepare polysaccharides – based nanoparticles, that is covalent cross linking ionic cross linking, polyelectrolyte complex and the self-assembly of hydrophobically modified polysaccharides.
29 degradation through exposure of chitosan to lysosyme contained in the mucus. However, the authors speculate that chitosan may ultimately depleted the mucus layers of intestinal cells through enhanced secretion of globlet cell mucus, thereby allowing remaining unbound chitosan to interact directly with cell surfaces.
Lakkireddy et al.,30 have developed Etopside-incorporated tripalmitin nanoparticles with negative (ETN) and positive (ETP) were prepared by melt emulsification and high pressure homogenization techniques. Spray drying of nanoparticles led to free flowing powder with excellent redispersibility. The nanoparticles were characterized by size analysis, Zeta Potential Instruments and Scanning Electron Microscope. The mean diameter of ETN and ETP nanoparticles was 391 nm and 362 nm respectively and the entrapment efficiency was more than 96%.
30 Guowei Qu et al., 32 have reported chitosan derivatives with hydrophobic moieties of octyl and hydrophilic moieties of sulfates and polyethylene glycol mono methyl ether (mPEG) groups were synthesized. The values of critical micelle concentration (CMC) were found to be 0.011 – 0.079 mg/ml, and the log CMC was linearly relative to four structure parameters, degree of substitution (DS) of chitosan unit, sulfate group, PEG unit and octyl group by mole per kilogram. Paclitaxel (PTX)- loaded micelles was designed and conducted, including interaction of the drug loaded micelles with protein and the kinetics of PTX-loaded micelles below CMC.
Petras Juzenas et al.,33 have developed Gold nanoparticles with Silica shell have recently been described as bio compatible contrast agents possessing increased absorption of X-rays. Metallic nanoparticles may increase therapeutic efficacy of radiotherapy by such selective scattering and or absorption of ionizing radiation and thus allowing to decrease radiation dose and minimizing side effects. Nanoparticles containing antioxidants have also been shown as effective adjuvant for radio therapy. Nanoparticles are being investigated as light source for PDT for deep cancer treatment.
31 David C. Bibby et al.,35 have reported the biodistribution and pharmacokinetics of a cyclic doxorubicin-nanoparticles, formulation in tumor-bearing mice. The NPs core was composed of insulin multi-methacrylate with a targeting peptide, cyclic RGD, covalently attached to the NPs via PEG-400. 72% of the doxorubicin was attached to the NPs matrix via an amide bond; 28% of doxorubicin was entrapped as unconjugated drug. The Pharmacokinetics of total, unconjugated and metabolized doxorubicin was examined for 5 days following intravenous (iv) administration of the NPs formulation (250 µg doxorubicin equiv.,) revealing a bi-exponential fix with a terminal half-life of 5.99 h.
Mayank D. Bhavsar et al.,36 have prepared by further protecting the DNA-loaded nanoparticles in a poly (epsilon-caprolactone) (PCL) matrix to form microsphere of less than 5.0 µm in diameter. Inorder to evaluate the biodistribution following oral administration, radio labeled gelatin nanoparticles and NiMOs were administered orally to fasted wistar rats. The results of biodistribution studies showed that, while gelatin nanoparticles traversed through the GI tract fairly quickly with more than 85% of the administered dose per gram localizing in the large intestine within the first hour, NiMOs resided in the stomach and small intestine for relatively longer duration.
Jingduan Chi et al.,37 have reported a sensitive and rapid Liquid Chromatography tandem Mass spectrometry (LC-MS) method has been developed to measure the level of five
HIV protease inhibitor Nelfinavir (NFV) Indinavir (IDV), Ritonavir (RTV), Saquinavir (SQU) and Amprenavir (APV) in human plasma. The analytes and internal
32 Vaibhav Saini et al.,38 have studied viruses are well known for that ability to cause disease, but their beneficial usefulness as vectors for gene therapy have been noted as well. As an extension of their use in a gene therapy context, their combination with nanotechnology is starting to benefit many areas of sciences and medicine. These include nanofabrication and medical diagnostics, here defined as the combination of viral biology with nanotechnology to create new therapeutics avenues to treat disease.
Partha Gosh et al.,39 have developed Gold nanoparticles AUNPs provide non-toxic carriers for drug and gene delivery application with these system, the gold core imparts stability to the assembly, while the monolayer allows tuning of surface properties such as charge and hydrophobicity. An additional attractive feature of AUNPs is their interaction with thiols, providing an effective and selective means of controlled intracellular release.
33 Samuel K. Lai et al.,41 have reported the PCI- PMMA nanoparticles internalized and released carried plasmid DNA into HeLa cells very efficiently. DNA molecules carried by the nanoparticles were specifically delivered into the nucleus of the target cells. The PEI-PMMA nanoparticles maintaining the stable spherical structure, the monodispersed size distribution, together structure and the proper net positive surface charge after forming with plasmid DNA did not obviously disorder the cultured cells observed by confocal laser scanning microscopy.
Takami Akagi et al.,42 have developed novel PGA-graft-LPAE copolymers formed nanoparticles with a mean diameter around 200 nm and a monodispersed size distribution when prepared by a precipitation and dialysis method. The PGA nanoparticles showed a highly negative zeta potential in PBS. This could attribute to the presence of ionized carboxyl group from the PGA on the nanoparticles. Therefore, these PGA nanoparticles may be considered promising biodegradable and bio compatible protein carriers for modulated biodistribution and site and one or cell- specific drug delivery systems.
Elias Fattal et al.,43 have studied oligonucleotides, associated with biodegradable polyalkylcyanoacrylate nanoparticles through the formation of ion pairs between the negatively charged oligonucleotides bound to these nanoparticles were found to be protected from nuclease attack in cell cultural media and their cellular uptake was increased as the results of the capture of nanoparticle.
34 degradation velocity. Use of the sterically stabilizing polaxamer 188 could prevent the invitro degradation of well-degradable Dynasan 114 particles.
Archana Swami et al.,45 have developed the nanoparticles and were characterized with respect to DNA interaction, hydrodynamic diameter, zeta potential, invitro cytotoxicity and transfection efficiency on model cell lines. One nanoparticles formed a loose complex with DNA compared to the corresponding native PEI, leading to more efficient unpackaging of DNA. The maximum level of reporter gene expression was mediated by IPPL nanoparticles in both the series. The cytotoxicity, another pertinent problem with cationic polymers, was also negligible in case of IPP nanoparticles.
Surendra Nimesh et al.,46 have studied a nanoplex gene delivery system employing PAA and DS with Zinc sulfate as a stabilizer and has been developed in an attempt to enhance PAAs transfection efficacy along with increase cell viability. These nanoplexes are found to have enhanced transfection activity within the reduction in the intrinsic toxicity of the PAA component. The size and surface charge of nanoparticles are two essential parameter that control their uptake by cells. The mean size of particles prepared using above methodology were found to be dependent on the PAA to DS ratio. The particle sizes reduced from 274 nm observed at a polymer ratio of 1:1 to 143 nm at a polymer ratio of 3:1. A zeta potential above +22 mv would be expected to inhibit aggregation because of charge repulsion.
35 polymer. A 207 nm mean diameter were obtained, which remained stabilized over 120 days at 4oC and the penicillin encapsulation ratio in the nanocapsules was 35%.
Ying-Ying Kuo et al.,48 have developed the loading efficiency of stavudine a human immunodeficiency antiretroviral agent, on the external surface of polybutyl cyano acrylate were prepared. The experiment result indicate that the larger the polymeric nanoparticles, the smaller loading efficiency of stavudine on the two kinds of biomaterials. Freeze-drying of the two nanoparticles, however, yields an increase in particle size and an increase in loading efficiency of stavudine. In general, stability studies at 4oC over 6 weeks leads to an increase in particle size and a decrease in loading efficiency of stavudine. In general, stability studies at 4oC over 6 weeks leads to an increase in particle size and a decrease in loading efficiency of stavudine.
Nikhil. K Sachan et al.,49 have reported the potential of sodium alginate as a biopolymer in the formulation development and its allied applications. The sodium alginate is a versatile functional biomaterial for viscosity enhancement, stabilizer, matrixing agent, encapsulation polymer, bioadhesive and film former in transdermal and transmucosal drug delivery.
36 Faiyaz Shakeel et al.,51 have reported the present investigation was to compare pharmacokinetic profile (bioavailability) of Acelofenac by transdermal and oral application, Nanoemulsion, nanoemulsion gel and marketed tablet (Aceclofar) were subjected to pharmacokinetic (bioavailability) studies on wistar male rats. Several pharmacokinetic parameters like Cmax, Tmax, AUCo t, AUCo α, Ke and T1/2 were determined for each
formulation.
Appala Raju et al.,52 have reported two simple, accurate, rapid and sensitive method (A and B) which have been developed for the estimation of Abacavir sulfate in its pharmaceutical dosage form. The method A and B are based on the formation of chloroform extractable complex of Abacavir sulfate with Bromophenolblue (method A) and Bromocresol green (Method B), which shows absorbance maxima at 460 nm and 469 nm respectively. The absorbance-concentration plot is linear over the range of 1-10µg/ml for method A and B respectively.
Pawan K, Saini et al.,53 have reported the developed HPLC method was found suitable for the analysis of Abacavir sulphate and Lamivudine in marketed tablet dosage form. Statistical analysis proves that the method is repeatable and selective for the analysis of Abacavir sulphate and Lamivudine simultaneously. This method might be employed for quality control analysis. This method has the advantage of being specific for both drugs without the need for additional sample preparation, such as extraction of the active constituents.
37 adjusted dose regimens of Lopinavir-Ritonavir were tested in combination with rifampin, thirty-two healthy subjects participated in a randomized two arm, open label, multiple dose, within subject controlled study.
Amir Dustgani et al., 55 have investigated the synthesis and characterization of novel biodegradable nanoparticles based on Chitosan for encapsulation of dexamethasone sodium phosphate. To achieve this objective, ionic gelation method were used. Drug containing nanoparticles were prepared with different amount of drug. The mean size and size distribution of nanoparticles were measured by dynamic laser light scattering. The mean particle size, varied in the range of 250-350 nm.
Zahoor Ahmed et al., 56 have reported high drug encapsulation efficiency was achieved in alignate nanoparticles, ranging from 70% - 90%. A single oral dose resulted in therapeutic drug concentrations in the plasma drug concentration in the plasma drug concentration in the plasma for 7-11 days and in the organs (Lungs, Liver and Spleen) for 15 days. In comparison to free drug which are cleared from Plasma / Organs within 12.24 h, there was significant enhancement in the relative bioavailability of encapsulated drug.
Joseph N.M et al.,57 have reported the purpose of the present study was to prepare modified gelatin nanoparticles by desolvation method. Polyethylene glycol was used to modify the gelatin nanoparticles. Particle characteristics size, zeta potential, surface morphology, encapsulation efficiency, invitro drug release were investigated. The prepared gelatin nanoparticle size ranged between 281 nm – 889 nm.
38
4
.SCOPE AND PLAN OF WORK
The overall scope of this research work is to develop effective bidegradable loaded nanoparticle with Abacavir sulfate and Ritonavir. The efficacy and effectiveness of the orally administrated Abacavir sulfate and Ritonavir nanoparticles are studied by various invitro and invivo methods.
• Preparation of polymeric nanoparticles nanoemulsion by polymeric cross linking method and solvent evaporation method by using biodegradable polymers like Sodium alginate and chitosan using various polymer ratio (1:1 – 1:3).
• Interaction study
1. FT-IR studies
2. DSC analysis
3. X-ray diffraction analysis
• Calibration of Standard curve
• Design of Biodegradable nanoparticles
1. Formulation of nanoparticles
2. Characterization of nanoparticles
2.1.Percentage of Drug entrapment efficiency
2.2.Particle size distribution
2.3.Polydispersity index
39 3. Morphology studies
3.1Scanning Electron Microscope (SEM)
3.2Transmission Electron Microscope (TEM)
• Invitro release studies
• Invivo study of best formulated nanoparticles
• Stability analysis
40
5. MATERIALS AND METHODS
5.1. Materials used for the study
a) Drugs and Excipients
Abacavir sulfate and Ritonavir was collected from Ranbaxy Pvt. Ltd., Guargon, India. Sodium alginate (250 Cps) was purchased from Sigma Aldrich, Bangalore. Chitosan (11 Cps) was kindly gifted from India sea foods, Cochin, Kerala, India. Dialysis bag was purchased from Himedia laboratories, Mumbai, India. Propylene glycol was obtained from NR Chemicals, Mumbai. Polyvinyl alcohol was obtained from Grifton Laboratories Pvt. Ltd., Mumbai, India.
b) Chemicals / Reagents
Chemicals Suppliers
Methanol, Glacial acetic acid, Sodium Chloride, Sodium Hydroxide,
Potassium Bromide, Disodium hydrogen Phosphate, Potassium dihydrogen Phosphate, Calcium Chloride and Glycerol.
Qualigens Fine Chemicals, Mumbai.
Hydrochloric acid Sd Fine Chemical, Ltd., Mumbai. Capsule Shell Sunil Health care Pvt. Ltd., Mumbai.
c) Equipments
41 and Rotary shaker (Malvern Instrument, U.K), Transmission Electron Microscope (Bio Twin Netherland, FEI Tech 12) and Zetapotential (Zeta sizer, Malvern U.K) were employed for the studies.
d) Software used
1. PCP – Disso v – 2.08 (BUDU Pune College of Pharmacy) 2. Chemdraw (For Drug Structure)
3. The Statistical Analysis were performed for the experimental results using MS office- Word, Excel in 2007 Version.
4. Mini Tab 2002 – V 13.20 (For Factorial Design) and pk solution (For pharmacokinetic studies).
5.2. Interaction study
59-62Before formulation of drug Substances into a dosage form, it is essential that the drug and polymer should be chemically and physically characterized. Interaction studies give the information needed to define the nature of the drug substances and provide a frame work for the combination with pharmaceutical excipients in the fabrication of dosage form.
a) FT-IR
42 b) DSC
Thermal analysis was preformed for all the multiparticles as well as the raw materials used for the study. Samples were accurately weighed (200 ± 0.5 mg) and placed into ventilated aluminum pan and thermograms were obtained at a scanning rate of 20oC/min over a range of temperature of 25oC – 400oC in a standard differential calorimeter. The DSC was performed and compared for Pure drug, polymer, and physical mixture of drug with polymer.
c) X-ray Diffraction
X-ray diffraction is a proven tool to study crystal lattice arrangements and yields very useful information in degree of sample crystallinity. X-ray diffraction pattern of pure drug, physical mixture and formulation were obtained and compared which revealed marked difference in the molecular state of Ritonavir nanoparticles.
5.3. Preparation of nanoparticles
43 Table 1:Composition of Abacavir sulfate and Ritonavir in nanoparticles preparation
Formulation Abacavir
Sulfate Ritonavir
SodiumAlginate (0.1%) Chitosan solution (1%) Tween 80 CaCl2 solution (2M) Polyvinyl Alcohol 2%
AF1 10 mg - - 10 ml - - 25 ml
AF2 10 mg - - 20 ml - - 25 ml
AF3 10 mg - - 30 ml - - 25 ml
AF4 10 mg - 10 ml - 0.09 mg 2.5 ml -
AF5 10 mg - 20 ml - 0.09 mg 2.5 ml -
AF6 10 mg - 30 ml - 0.09 mg 2.5 ml -
BF1 - 10 mg - 10 ml - - 25 ml
BF2 - 10 mg - 20 ml - - 25 ml
BF3 - 10 mg - 30 ml - - 25 ml
BF4 - 10 mg 10 ml - 0.09 mg 2.5 ml -
BF5 - 10 mg 20 ml - 0.09 mg 2.5 ml -
44
a)
Nano emulsion- polymer cross linking method
17Abacavir sulfate and Ritonavir