0022-538X/00/$04.00⫹0
Copyright © 2000, American Society for Microbiology. All Rights Reserved.
The Carboxyl Terminus of v-Abl Protein Can Augment
SH2 Domain Function
DAVID WARREN,
1ANDREW J. HEILPERN,
2,3KENT BERG,
2ANDNAOMI ROSENBERG
1,2,3*
Department of Molecular Biology and Microbiology,
1Department of Pathology,
2and Graduate Program
in Immunology,
3Tufts University School of Medicine, Boston, Massachusetts 02111
Received 22 December 1999/Accepted 11 February 2000
Abelson murine leukemia virus (Ab-MLV) transforms NIH 3T3 and pre-B cells via expression of the v-Abl
tyrosine kinase. Although the enzymatic activity of this molecule is absolutely required for transformation,
other regions of the protein are also important for this response. Among these are the SH2 domain, involved
in phosphotyrosine-dependent protein-protein interactions, and the long carboxyl terminus, which plays an
important role in transformation of hematopoietic cells. Important signals are sent from each of these regions,
and transformation is most likely orchestrated by the concerted action of these different parts of the protein.
To explore this idea, we compared the ability of the v-Src SH2 domain to substitute for that of v-Abl in the
full-length P120 v-Abl protein and in P70 v-Abl, a protein that lacks the carboxyl terminus characteristic of Abl
family members. Ab-MLV strains expressing P70/S2 failed to transform NIH 3T3 cells and demonstrated a
greatly reduced capacity to mediate signaling events associated with the Ras-dependent mitogen-activated
protein (MAP) kinase pathway. In contrast, Ab-MLV strains expressing P120/S2 were indistinguishable from
P120 with respect to these features. Analyses of additional mutants demonstrated that the last 162 amino acids
of the carboxyl terminus were sufficient to restore transformation. These data demonstrate that an SH2 domain
with v-Abl substrate specificity is required for NIH 3T3 transformation in the absence of the carboxyl terminus
and suggest that cooperativity between the extreme carboxyl terminus and the SH2 domain facilitates the
transmission of transforming signals via the MAP kinase pathway.
Abelson murine leukemia virus (Ab-MLV) is a highly
onco-genic retrovirus that transforms NIH 3T3 cells and pre-B cells
in vitro and induces pre-B-cell lymphomas in vivo (60). The
virus arose via recombination between Moloney murine
leuke-mia virus (Mo-MLV) and the c-
abl
proto-oncogene gene and
encodes a single product, the v-Abl protein tyrosine kinase.
This protein contains amino-terminal determinants derived
from the Mo-MLV
gag
gene fused to
abl
-derived sequences
which specify the SH2 and catalytic domains and the long
carboxyl-terminal region characteristic of Abl protein family
members (18, 42). Although the protein tyrosine kinase activity
of v-Abl is absolutely required for all transforming functions
of the virus, other portions of the protein play important
roles. The SH2 domain facilitates
phosphotyrosine-mediat-ed protein-protein interactions (35), and the carboxyl terminus
is important for transformation of lymphoid cells (23, 40, 50,
58).
Mutations affecting different regions of the v-Abl protein
have shed light on their functions; however, the way these
re-gions may work together to orchestrate transformation is less
clear. For the modular SH2 domain, similar domains from
oth-er signaling molecules can partially substitute for its function in
transformation. For example, the SH2 domains of Crk and
several other proteins can substitute for endogenous SH2
se-quences in activated c-
abl
alleles (33, 38). However, the pattern
of tyrosine-phosphorylated cellular proteins is not identical (33),
and transformation usually occurs at a reduced frequency. In
contrast, chimeras in which the entire amino terminus of v-Src
has been fused to the v-Abl catalytic domain are fully
trans-forming for NIH 3T3 cells (22), suggesting that the presence of
an SH3 domain alters the response, perhaps by providing
dock-ing sites for cellular signaldock-ing molecules (34, 41).
The carboxyl terminus of Abl proteins is also involved in
protein-protein interactions (2, 8, 29, 36, 48, 65, 68), some of
which appear to involve cellular proteins which also interact
with other portions of the Abl protein (6, 64). In addition, the
carboxyl terminus also appears to signal to the Ras pathway
(15, 45, 54), an event that is critical for Ab-MLV
transforma-tion (62). Other studies have identified a Jak-interactive region
and shown that Jak activation is important for v-Abl-induced
factor-independent growth of hematopoietic cell lines (7, 8). In
addition to these functions, the carboxyl terminus contains a
DNA binding domain, an RNA polymerase II binding site,
multiple sites of serine phosphorylation, and regions that
in-teract with the cytoskeleton (2, 29, 36, 48, 68).
To more fully understand the ways in which the different
regions of the v-Abl protein work together to induce
transfor-mation, we have examined the ability of the v-Src SH2 domain
to substitute for that of v-Abl in the presence and absence of
the v-Abl carboxyl terminus. In contrast to results obtained
with chimeric proteins containing the entire amino-terminal
region of v-Src, our data demonstrate that the v-Src SH2
do-main can functionally substitute for the v-Abl SH2 dodo-main only
in the presence of a complete carboxyl terminus. Chimeric
proteins lacking the extreme carboxyl terminus fail to
trans-form NIH 3T3 cells in vitro, a feature that is correlated to
decreased activation of the mitogen-activated protein (MAP)
kinase pathway and diminished activation of the c-
fos
pro-moter. These data highlight a novel function of this region and
suggest that one role of these sequences is to facilitate
signal-ing through the MAP kinase pathway.
MATERIALS AND METHODS
Cells and viruses.NIH 3T3 cells, Ab-MLV-transformed NIH 3T3 cells, and
293T cells (12) were grown in Dulbecco’s modified Eagle’s medium (Life
Tech-* Corresponding author. Mailing address: SC-313, Tufts Medical
School, 136 Harrison Ave., Boston, MA 02111. Phone: (617) 636-2143.
Fax: (617) 636-0337. E-mail: [email protected].
4495
on November 9, 2019 by guest
http://jvi.asm.org/
nologies) supplemented with 10% fetal calf serum (Sigma) and 2 mML -glu-tamine (Gibco). Ab-MLV-transformed pre-B cells were grown in RPMI 1640 medium (Gibco) supplemented with 10% fetal calf serum, 2 mML-glutamine, and 50M 2-mercaptoethanol (Sigma). Helper-free viral stocks were prepared using Ab-MLV strains in the pSR␣MSVtkneo retroviral vector and the pSV-⌿ -E-MLV retroviral packaging plasmid (39). Briefly, 293T cells were plated at 4⫻ 106cells per 100-mm-diameter plate the day before transfection. The Ab-MLV plasmid and the packaging plasmid were precipitated, washed, and resuspended in sterile double-distilled water; CaCl2was added to a final concentration of 235 mM, and the DNA was added dropwise to the dish. Virus was harvested 36 to 72 h later, pooled on ice, filtered through a 0.45-m-pore-size filter, and stored at⫺70°C. To assess the amounts of infectious Ab-MLV in different viral stocks, 105NIH 3T3 cells were plated in 60-mm-diameter dishes and infected 24 h later with virus in the presence of Polybrene (8g/ml; Aldrich). After 48 h, the cells were lysed and the amount of v-Abl protein expressed by the cells was assessed by Western blotting (46). Transformation of NIH 3T3 cells was assessed by growth in soft agar. Cells were infected as described above, trypsinized 2 h later, and plated in an RPMI 1640-based medium containing 10% fetal calf serum and 0.3% agar (Difco) onto a 0.6% agar layer containing RPMI 1640 medium and 10% fetal calf serum. The plates were fed every 7 days, and macroscopic colonies were scored 3 to 4 weeks postinfection.
Construction of viral strains.pSR␣MSVtkneo-Ab-MLV-P120 (pSR␣-P120)
and pSR␣MSVtkneo-Ab-MLV-P70 (pSR␣-P70) were constructed by replacing the sequence encoding the v-Abl carboxyl terminus in pSR␣MSVtkneo-P160 (39) with the corresponding 2,831-bpBstEII-BspEI fragment from pUC120 and pUC70 (14), respectively. P120⌬668-819 (Fig. 1A) was constructed by PCR using primers that amplified the sequences encoding amino acids 496 to 671 of P120 v-Abl. The 3⬘primer contained aSalI site and the amplified material contained theDraIII site at bp 2106 of the Ab-MLV-P120 genome. The amplified material was cloned into the TA vector and sequenced, and theDraIII-SalI fragment was
used to replace the sequence encoding amino acids 496 to 819 of P120v-Abl in pSR␣-P120. Ab-MLV-P90A has been described previously (40). To construct Ab-MLV strains in which the sequences encoding the v-Abl SH2 domain were replaced by those encoding the v-Src SH2 domain, pSR␣viruses and pSAK (22) were used. pSAK encodes a chimeric v-Abl–v-Src protein in which the v-Abl kinase domain is surrounded by v-Src flanking sequences (22). Specifically, the sequences encoding the v-Src SH1 domain (proviral bases 7901 to 8658 of the B77 strain of Rous sarcoma virus) have been replaced with those encoding the v-Abl SH1 domain (proviral bases 2052 to 2956 of Ab-MLV) by usingEcoRI linkers. To facilitate exchange of the SH2 domains between pSAK and the pSR␣ viruses, the intermediate vector TA/P70 was generated by cloning the 2,426-bp SacI fragment (proviral bases⫺36 to 2391) from pSR␣-P120 into the unique SacI site in the TA cloning vector (Invitrogen). The 1.3-kbHincII-EcoRI pSAK fragment (bases 7459 to 8658) encoding the v-Src SH2 domain linked to the v-Abl SH1 domain was used to replace the 1,100-bpHincII-EcoRI fragment of TA/P70, generating TA/P70S2. pSR␣-P120S2, pSR␣-P120S2⌬668-819, pSR␣ -P90S2, and pSR␣-P70S2 were subsequently generated by replacing the 1.4-kb BstEII-DraIII (bases 725 to 2106) fragment from pSR␣-P120, pSR␣-P120⌬ 668-819, pSR␣-P90, and pSR␣-P70 with the corresponding 1.6-kbBstEII-DraIII fragment from TA/P70S2 which contains the sequences encoding the v-Src SH2 domain linked to v-Abl sequences. The final constructs express chimeric v-Abl– v-Src proteins in which 88 amino acids of v-Abl (residues 241 to 328) have been replaced with 146 v-Src SH2 amino acids plus the four amino acids GINS. The P120⌬SH2 mutant in which the sequences encoding the SH2 domain are deleted was created by replacing the 1,386-bp BstEII-DraIII fragment in pSR␣-P120 vector with the corresponding fragment from pAM⌬SH2, a plasmid containing the first 708 bases of Mo-MLVgagfused to human c-abltype IV 15 bases into the c-ablcoding sequence; the c-ablsequences in this plasmid contain a deletion of c-abl bases which encode the SH2 domain (A. M. Pendergast, personal communication). The Myc-expressing retroviral vector, pSR␣-Myc, was created by inserting the 1.8-kb fragment encoding human c-Myc from pBS-myc-mut3 (19) into theEcoRI site of pSR␣MSVtkneo.
Protein analyses.Cell lysates were prepared as described previously (45).
Briefly, the cells were washed twice with phosphate-buffered saline (PBS), and the cell pellets were treated with lysis buffer (10 mM Tris [pH 7.4], 1% sodium dodecyl sulfate [SDS], 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride). The lysates were boiled immediately and sheared through a 25-gauge needle. The amount of protein in each lysate was quantitated using a bicincho-ninic acid protein assay kit (Pierce), and 50g of each sample was fractionated through an SDS-polyacrylamide gel. In some experiments, equivalent amounts of total cell protein were immunoprecipitated with 1g of purified antibody or serum on ice for 1 h. The immune complexes were recovered using protein A- or protein G-Sepharose beads (Pharmacia) and washed with buffer (10 mM sodium phosphate, 150 mM sodium chloride, 1% NP-40, 2 mM EDTA, 1 mM sodium vanadate, 1 mM phenylmethylsulfonyl fluoride). Bound proteins were eluted by heating the beads at 95°C in sample buffer (1% SDS, 50 mM Tris [pH 6.8], 10% glycerol, 0.03% bromophenol blue) for 5 min and analyzed by SDS-polyacryl-amide gel electrophoresis. The proteins were electrotransferred to polyvinyl-idene difluoride membranes (U.S. Biochemicals) which were blocked with PBS containing 0.2% I-block (Tropix) and 0.1% Tween 20 for at least 1 h. Blotting was performed according to the Western-Light kit protocol (Tropix), utilizing alkaline phosphatase-conjugated secondary antibodies with a CSPD substrate (Tropix). Blots were exposed to Kodak XAR-5 film and subsequently stripped by incubating in a pH 2.2 solution containing 0.2 M glycine and 1% Tween 20 for 3 h at 80°C. After stripping, blots were washed with PBS containing 0.1% Tween 20 and treated with blocking solution prior to reprobing. Proteins were analyzed using anti-Gag/v-Abl (H548) (5); antiphosphotyrosine (05-321; Upstate Biotech-nology); anti-Shc, anti-Grb2, and anti-Ras (S14630 or S14620, G16720, and R02120, respectively; Transduction Laboratories); anti-Myc (OP10L; Calbio-chem); anti-p42/44 MAP kinase, anti-phospho-p42/44 MAP kinase, anti-stress-activated (SAP) kinase/Jun N-terminal kinase (JNK), anti-phospho-SAP kinase/ JNK (9120, 9106, 9252, and 9251, respectively; New England Biochemical); and alkaline phosphatase-conjugated anti-mouse and anti-rabbit immunoglobulin G (S372B and S373B, respectively; Promega).
Ras assay.Levels of RasGTP were assessed using an assay in which RasGTP
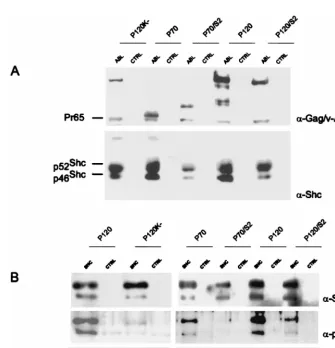
is recovered through its interaction with a glutathioneS-transferase (GST) fusion protein containing the Ras binding domain of Raf (RBD) (10, 21). To prepare the GST fusion proteins, log-phase Escherichia coli BL-21 cells containing pGEX-2T plasmids expressing either GST or GST-RBD were grown for 3 h in the presence of 0.1 mM isopropyl-1-thio--D-galactosidase and 12.5g of am-picillin per ml. The cells were pelleted, lysed in ice-cold lysis buffer (1⫻PBS, 1% Triton, 5 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM phenylmeth-ylsulfonyl fluoride, 10 g of leupeptin [Boehringer Mannheim] per ml) and subjected to three cycles of freeze-thawing. The extracts were centrifuged at 12,000 rpm for 30 min at 4°C, and the resulting supernatants were stored at ⫺20°C. The GST proteins were coupled to protein G-Sepharose beads (Phar-macia) according to the manufacturer’s protocol. The day before the experiment, 293T cells were plated at 106cells per 100-mm-diameter plate; the cells were fed with fresh medium 3 h before transfection and then transfected with 15g of a v-Abl expression plasmid in the pSR␣MSVtkneo vector and 15g of a c-Ha-Ras expression vector (62). For precipitation, 200 to 1,000g of cell lysate was incubated with 25l of packed beads prebound to either GST or GST-RBD and FIG. 1. Expression of enzymatically active chimeric viral proteins. (A)
Struc-tures of the P120, P120/S2, P70, P70/S2, and P120/K⫺v-Abl proteins. P120K⫺ encodes a v-Abl protein in which the aspartic acid at position 484 has been replaced by an asparagine (P120/D484N). (B) NIH 3T3 cells were infected with the Ab-MLV strains; lysates were prepared 48 h later and analyzed by Western blotting with the H548 anti-Gag/v-Abl monoclonal antibody (5). The blot was stripped and reprobed with antiphosphotyrosine antibody.
on November 9, 2019 by guest
http://jvi.asm.org/
rocked at 4°C for 90 min. The beads were washed three times with wash buffer (50 mM Tris [pH 7.5], 20 mM NaCl, 10 mM MgCl2, 0.5% NP-40), and the bound proteins were eluted with SDS sample buffer, resolved by electrophoresis, and analyzed by Western blotting.
Transfection assays.The day before the experiment, 293T cells were plated at
2.5⫻105cells per 60-mm-diameter plate and fed with fresh medium 3 h before transfection. For experiments using luciferase expression vectors, the cells were transfected with 1g of pSVOA⌬5⬘, which contains a 379-bp murine c-fos promoter upstream of the firefly luciferase gene (20), 4g of a v-Abl expression plasmid in the pSR␣MSVtkneo vector, and 0.7 g of pRL-TK, an internal control reporter plasmid expressing renilla luciferase (Promega). In some exper-iments, pSVOA⌬5⬘was replaced with plasmids that contained mutations in three of thecis-acting elements of the murine c-fospromoter as described previously (28) (see Results). Other experiments included 4g of a fourth DNA which encoded either dominant-negative (DN) Ras (Ras; Ras/S17N) (62) or DN-Akt (DN-Akt K179M) (13). The transfections were performed as described earlier. The medium was changed 8 to 12 h posttransfection, and the cells were fed with 5 ml serum-free medium 24 h posttransfection. Luciferase activity was measured using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer’s protocol. Results were normalized to the expression of renilla luciferase. These transfections were carried out in triplicate. In other experi-ments, 293T cells were transfected with 4g of pJ3M-Erk, expressing Myc-tagged Erk from the simian virus 40 promoter (L. Feig, personal communica-tion), or pcDNA, expressing Flag-Jnk1 (17), and 4 g of a v-Abl expression plasmid in the pSR␣MSVtkneo vector. Cells were serum starved, and lysates were prepared 24 h later. The samples were analyzed by Western blotting.
RESULTS
P70/S2 fails to transform NIH 3T3 cells.
Previous studies
had demonstrated that the v-Src amino-terminal region,
com-prising the SH3 and SH2 domains, can function in concert with
the v-Abl kinase domain and mediate transformation of NIH
[image:3.612.90.507.72.410.2]3T3 cells (22). To test the ability of the v-Src SH2 domain to
supply functions normally provided by the v-Abl SH2 domain,
chimeric Ab-MLV strains in which the v-Abl SH2 domain was
replaced with the SH2 domain of v-Src were constructed using
both the P120 wild-type background and that of the P70
car-boxyl-terminal truncation mutant (Fig. 1A). The v-Abl protein
encoded by this latter strain is similar to v-Src in that it
termi-nates at the end of the kinase domain and lacks the
carboxyl-terminal region characteristic of Abl proteins (14). To confirm
that all of the viruses expressed proteins of the expected size,
NIH 3T3 cells were infected and 48 h postinfection cell lysates
were analyzed by Western blotting. All of the samples
con-tained proteins of the expected size and the levels of expression
were similar (Fig. 1B). In addition, total cellular
phosphoty-rosine levels were elevated in all infected cells except those
infected with P120/D484N, a control virus expressing a
kinase-negative v-Abl protein (52). These data suggest the kinase
ac-tivity of the chimeric proteins is similar to that of v-Abl protein.
Although both the P120/S2- and P70/S2-infected cells
dis-played elevated levels of cellular phosphotyrosine, only those
infected with the P120/S2 strain displayed the transformed
morphology characteristic of cells infected with Ab-MLV-P120
or P70 (Fig. 2). Cells infected with the P70/S2 strain failed to
form foci of rounded up cells characteristic of
Ab-MLV-trans-formed cells. However, the cells assumed a more fusiform
appearance and grew in a more irregular pattern compared to
uninfected cells or cells infected with the P120/D484N strain
FIG. 2. NIH 3T3 cells infected with the P70/S2 are not morphologically transformed. NIH 3T3 cells were infected with the Ab-MLV strains and photographed 48 h later.on November 9, 2019 by guest
http://jvi.asm.org/
(Fig. 2). To investigate this difference further, virus stocks were
standardized by assessing levels of Abl proteins expressed in
NIH 3T3 cells after a 48 h infection and tested for the ability
to induce transformation in soft agar. Consistent with the
ap-pearance of the infected cells, similar numbers of colonies
were obtained from cultures infected with the P120, P120/S2,
and P70 strains (Table 1); in contrast, no colonies were evident
in cells infected with the P70/S2 strain. These results suggest
that the v-Abl SH2 domain plays a vital role in v-Abl-mediated
fibroblast transformation and that the carboxyl terminus is able
to complement this function in the P120/S2 strain.
Shc phosphorylation and Grb-2 association are decreased in
NIH 3T3 cells infected with the P70/S2 strain.
v-Abl
associa-tion with Shc and the formaassocia-tion of a Shc-Grb2 complex has
been implicated as one pathway by which the v-Abl protein
activates Ras and mediates transformation (44, 52). To
exam-ine whether the chimeric viral proteins were associated with
Shc, lysates were prepared from infected NIH 3T3 cells.
West-ern analysis of samples immunoprecipitated with anti-Abl
an-tibody and probed with anti-Shc anan-tibody revealed that Shc
associated with all of the v-Abl proteins, including P70/S2 (Fig.
3A). To examine whether Shc was tyrosine phosphorylated and
associated with Grb2, NIH 3T3 cells were infected and lysates
were prepared 48 h later. Western analysis of samples
immu-noprecipitated with anti-Shc antibody and probed with
an-tiphosphotyrosine and anti-Grb2 antibodies revealed that Shc
was tyrosine phosphorylated and associated with Grb2 in cells
that had been infected with the P120, P120/S2, and P70 strains
but not in cells that had been infected with the P70/S2 strain
(Fig. 3B). Thus, the presence of a full COOH terminus
en-hances the ability of a v-Abl protein containing the Src SH2
domain to interact with Shc. These results raise the possibility
[image:4.612.53.294.84.164.2]FIG. 3. P70/S2 does not mediate Shc phosphorylation and Grb2 association. NIH 3T3 cells were infected and lysed. (A) The v-Abl proteins were immunoprecipi-tated with the anti-Gag/v-Abl monoclonal antibody H548 (5) and analyzed by Western blotting with the H548 antibody; the blot was then stripped and reprobed with an anti-Shc antibody. (B) The lysates were immunoprecipitated with anti-Shc antibodies and analyzed by Western blotting. The upper portion of the blot was probed with anti-Shc antibodies; the blot was stripped and reprobed with an antiphosphotyrosine (␣-pTyr) antibody. The lower portion of the blot was probed with anti-Grb2 antibody. CTRL, control.
TABLE 1. P70/S2 fails to transform NIH 3T3 cells
aVirus Colonies/10
5cells⫾SEM
Expt 1 Expt 2
P120
121
⫾
16
86.5
⫾
6.9
P120/S2
226.5
⫾
12
124.8
⫾
4.3
P70
103.5
⫾
13
69.5
⫾
4.3
P70/S2
⬍1
⬍1
P120K⫺
⬍1
⬍1
aCells were infected with virus stocks of matched titer and plated in agar
following virus adsorption. Macroscopic colonies were counted 4 weeks after the cells were plated. Values given as⬍1 indicate that no colonies were observed and reflect the minimum number of colonies that could have been detected.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.134.469.336.684.2]that the absence of the Shc/Grb2 signal contributes to the
absence of transformation in cells expressing P70/S2.
P70/S2 can stimulate increased RasGTP levels.
Expression
of DN-Ras inhibits transformation of NIH 3T3 and bone
mar-row cells by the virus (62). To determine if expression of
P70/S2 is capable of activating Ras, 293T cells were transfected
with DNAs encoding the different v-Abl proteins and a
c-Ha-Ras expression plasmid and serum starved for 24 h. Cell lysates
were prepared, and the levels of activated Ras were assessed
using a GST-RBD fusion protein (21). This protein contains
the RBD of Raf, a region which binds preferentially to
Ras-GTP. Western analysis of the GST-RBD complexes revealed
that all of the kinase-active v-Abl proteins tested, when
ex-pressed at comparable levels (Fig. 4), stimulated Ras activity
above the background level observed with the P120/D484N
mutant. Although this assay does not monitor Ras activation in
NIH 3T3 cells, these results suggest that differences in Ras
activation do not explain the different biological phenotypes
associated with the different chimeric proteins.
P70/S2 is compromised in activation of ERK but not JNK.
Activated Ras transmits signals which activate both the MAP
and SAP kinase pathways (11, 37). Both of these pathways are
activated by v-Abl (53, 55). To test the ability of the chimeric
v-Abl proteins to activate ERK and JNK, 293T cells were
cotransfected with DNAs encoding the viral proteins and
ex-pression plasmids encoding either ERK or JNK. Analysis of
cell extracts prepared from serum-starved cells revealed that
cells expressing P70, P120, and P120/S2 contained elevated
levels of phosphorylated ERK. However, levels of
phosphory-lated ERK recovered from cells expressing P70/S2 were similar
to those recovered from cells expressing P120/D484N (Fig.
5A). In contrast to these results, levels of phosphorylated JNK
were recovered from all cells expressing kinase active v-Abl
proteins (Fig. 5B). These results suggest P70/S2 is
compro-mised in its ability to activate ERK but not JNK and that the
carboxyl terminus is able to complement this function. These
results also suggest that activation of the JNK pathway alone is
not sufficient to induce NIH 3T3 cell transformation.
P70/S2 fails to transactivate the c-
fos
promoter.
c-
fos
is one
important gene stimulated by signals from the MAP and SAP
kinase pathways (27, 66). To test the ability of the chimeric
proteins to activate transcription from the c-
fos
promoter,
293T cells were transfected with DNAs encoding the viral
proteins and a plasmid expressing firefly luciferase under the
control of the c-
fos
promoter. The pRL-TK vector encoding
renilla luciferase was included as a transfection control. The
cells were serum starved, and luciferase activity was analyzed
using the Dual-Luciferase reporter assay system (Promega). As
expected, the P120 protein stimulated luciferase activity (55),
as did expression of P120/S2 and P70 (Fig. 6A). However, in
these latter instances, levels of luciferase activity were twofold
lower than those recovered from cells expressing the P120
protein. In contrast to these results, expression of P70/S2 failed
to stimulate luciferase expression above the background levels
obtained with P120/D484N. These data suggest that P70/S2 is
compromised in its ability to transactivate the c-
fos
promoter.
The ability of P120/S2 to stimulate the c-
fos
promoter suggests
that the carboxyl terminus is able to complement functions
normally provided by the v-Abl SH2 domain with respect to
c-
fos
activation.
P120/S2 and P70 mediate c-
fos
activation via the
cis
-acting
SRE element and Ras.
The promoter region of the c-
fos
gene
FIG. 4. P70/S2 stimulates increased RasGTP levels. 293T cells weretrans-fected with 15g of plasmids expressing the different v-Abl forms and 15g of a c-Ha-Ras expression vector. The cells were serum starved for 24 h, and extracts were prepared 48 h after transfection. (A) Portions of the lysate were analyzed by Western blotting with the H548 monoclonal antibody (5). (B) Portions of the lysates were incubated with GST or GST-RBD, and the affinity precipitates were analyzed by Western blotting with anti-Ras antibody (10, 21). The signals ob-tained were analyzed by densitometry, and the RasGTP levels were normalized to the levels of viral protein. The value for the P120 sample was set as 1.
FIG. 5. P70/S2 activates JNK but not ERK. 293T cells were transfected with 4g of plasmids expressing the v-Abl proteins and 4g of either pJ3M-ERK (A) (L. Feig, personal communication) or pcDNA expressing Flag-Jnk1 (17) (B). The cells were serum starved for 24 h, and extracts were prepared 48 h after transfection. Equivalent amounts of extract were analyzed by Western blotting. The blots were probed with the anti-Gag/v-Abl H548 monoclonal antibody (5) and anti-ERK or anti-JNK antibodies and reprobed with either anti-phospho-ERK or anti-phospho-JNK antibodies. The pattern of degradation observed for the v-Abl proteins in panel A is not typical.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.54.292.72.279.2]contains various
cis
-acting elements including a cyclic
AMP-responsive element (CRE), an AP-1-AMP-responsive element, a
c-sis
–platelet-derived growth factor-inducible element (SIE),
and a serum-responsive element (SRE) (3, 61, 69). The SRE
contains binding sites for both ternary complex factors (TCFs)
and serum response factors (SRFs) (43, 63, 67). The TCFs bind
to the SRE only in the presence of SRF or its core subdomain
(63), while the SRFs can activate c-
fos
independently of the
TCFs (24). The SRE is the major site at which Ras-dependent
MAP kinase signals are integrated at the c-
fos
promoter (9, 27,
37), and others have demonstrated the ability of v-Abl to
trans-activate promoters containing SREs (55, 56).
To determine which
cis
-acting elements in the c-
fos
moter are required for transactivation by the P70 v-Abl
pro-tein, 293T cells were transfected with DNAs encoding v-Abl
proteins and luciferase reporter plasmids under the control of
c-
fos
promoters containing mutations in the various upstream
elements. Mutation of the SIE had no effect on luciferase
expression, and mutation of either the AP-1 or CRE site had
minimal effects. In contrast, mutation of the SRF binding site
within the SRE ablated transaction of the c-
fos
promoter
com-pletely (Fig. 6B). These results indicate that the SRE is the
principal site at which signals from v-Abl proteins integrate at
the c-
fos
promoter. Mutation of the TCF binding site within
this region also reduced transactivation significantly (Fig. 6C),
suggesting that an SRF/TCF-dependent mechanism is involved
in v-Abl-mediated transactivation of the c-
fos
promoter.
Con-sistent with the role of Ras in transducing signals to SREs (58),
expression of DN-Ras but not DN-Akt ablated induction of
the c-
fos
promoter (Fig. 6D). This result suggests that signals
transmitted via Ras play a critical role in the response.
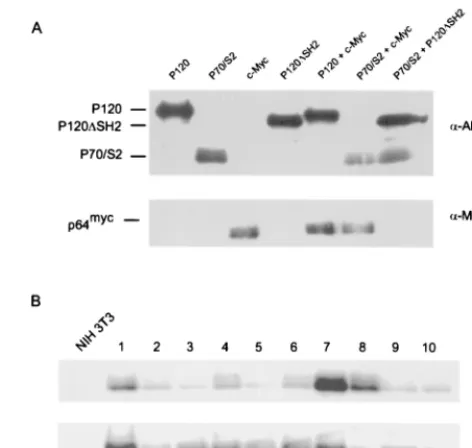
[image:6.612.58.540.67.371.2]Complementation by the carboxyl terminus maps to the
extreme COOH terminus.
The ability of P120/S2, but not P70/
S2, to transform NIH 3T3 cells and stimulate c-
fos
expression
demonstrates that functions contributed by the carboxyl
termi-nus of v-Abl can complement functions normally provided by
the v-Abl SH2 domain. To define the region within the
car-boxyl terminus required for SH2 complementation, the P90/S2
and P120/S2
⌬
668-819 Ab-MLV strains were constructed. The
P90/S2 protein contains the v-Src SH2 domain and the first 144
amino acids of the carboxyl terminus present in P120 v-Abl; the
P120/S2
⌬
668-819 protein contains the v-Src SH2 domain but
lacks the carboxyl-terminal amino acids 668 to 819 as a
conse-quence of an in-frame coding seconse-quence deletion (Fig. 7). The
Ab-MLV strains expressing these proteins were tested for the
ability to induce NIH 3T3 transformation in soft agar assays.
As expected, colonies were readily detected following infection
with Ab-MLV-P90 (59). However, the P90/S2 strain was
sim-ilar to the P70/S2 strain and did not induce colony formation
(Table 2). In contrast, P120/S2 and P120/S2
⌬
668-819 induced
similar numbers of colonies. Consistent with these data, both
P120/S2 and P120/S2
⌬
668-819 activated expression of the c-
fos
FIG. 6. v-Abl transactivates the c-fospromoter via Ras and the SRE. 293T cells were transfected with 4g of a pSR␣v-Abl expression plasmid, 1g of different reporter plasmids, and 0.7g of pRL-TK, an internal control reporter plasmid. The cells were serum starved for 24 h, and extracts were prepared 48 h after transfection. Luciferase activity was measured using the Dual-Luciferase reporter assay system (Promega). Each transfection was done in triplicate. For each replicate, firefly luciferase activity was normalized to renilla luciferase activity over three or more serial dilutions and averaged for each sample. The error bars reflect standard deviations. (A) Cells were transfected as described above with the pSVOA⌬5⬘reporter plasmid, which contains a 379-bp murine c-fospromoter upstream of the firefly luciferase gene (20). (B and C) Cells were transfected with plasmids that contained mutations in variouscis-acting elements (28): mSIE, mutant SIE; mCRE, mutant CRE; mAP-1, mutant AP-1 binding site; mSRE, mutant SRE; mTCF, mutant TCF binding site. WT, wild type. (D) Cells were transfected with pSVOA⌬5⬘and plasmids encoding DN-Ras (62) or DN-Akt (13).
on November 9, 2019 by guest
http://jvi.asm.org/
promoter, while P90/S2 failed to stimulate activity above the
background levels obtained with the kinase-inactive P120/D484N
protein (data not shown). Thus, sequences within the last 162
carboxyl-terminal amino acids are required for SH2 domain
complementation, and at least one function mediated by these
sequences leads to activation of c-
fos
expression.
Carboxyl-terminal sequences are required in
cis.
To
deter-mine whether the carboxyl-terminal v-Abl sequences were
re-quired in
cis
or
trans
for complementation of SH2 function, the
ability of the transformation-defective P120/
⌬
SH2 strain to
restore transforming function to P70/S2 was tested. The P120/
⌬
SH2 strain expresses a v-Abl protein that contains a complete
carboxyl terminus but from which sequences encoding the SH2
domain have been deleted (G. Raffel, personal
communica-tion). NIH 3T3 cells were also infected with pSR
␣
-Myc, a
retrovirus which expresses the c-Myc protein, since
overexpres-sion of c-Myc has been shown to complement the
transforma-tion-defective phenotype of a variety of
abl
alleles (1). NIH
3T3 cells were infected, either singularly or in combination,
with P70/S2, P120/
⌬
SH2, P120, and pSR
␣
-Myc and plated in
soft agar (Fig. 8A). Colonies were scored 3 to 4 weeks later.
When used individually, the P70/S2, P120/
⌬
SH2, and pSR
␣
-Myc strains all failed to transform NIH 3T3 cells. Coexpression
of both P120/
⌬
SH2 and P70/S2 failed to induce transformation
in the agar assay, while coexpression of c-Myc weakly
comple-mented P70/S2 in the agar transformation assay (Table 3).
Colonies obtained from cells coinfected with c-Myc and P70/S2
were screened via Western analysis to confirm the expression
of both proteins (Fig. 8B). Although some samples contained
lower amounts of both proteins, probably reflecting smaller
colony size, all expressed both proteins. These data
demon-strate that carboxyl-terminally mediated complementation of
v-Abl SH2 function requires the carboxyl terminus in
cis
. In
addition, overexpression of c-Myc only partially complemented
P70/S2, suggesting that defective signaling to c-Myc may be
partly, but not wholly, responsible for the transformation
de-fect of P70/S2.
DISCUSSION
Our analysis of chimeric v-Abl/v-Src proteins demonstrates
that sequences within the carboxyl terminus of the v-Abl
pro-tein can complement functions normally supplied by the v-Abl
SH2 domain. As shown previously for chimeras in which the
v-Src SH2 domain was inserted into other full-length,
trans-forming Abl proteins (33, 35), the v-Src SH2 region functions
well in the context of the P120 strain. However, even though
Ab-MLV strains encoding carboxyl-terminally truncated v-Abl
proteins transform NIH 3T3 cells efficiently (14, 23, 50, 58),
substitution of the v-Src SH2 domain in this context abolishes
transformation competency. Although the carboxyl terminus
has long been appreciated as playing an important role in
lymphoid transformation (23, 40, 50, 58), this is the first
dem-FIG. 7. Structures of the P90, P90/S2, P120⌬668-819, P120/S2⌬668-819, andP120 viral proteins.
[image:7.612.52.309.72.175.2]FIG. 8. P70/S2 is not complemented by P120⌬SH2. NIH 3T3 cells were infected, either singularly or in combination, with retroviruses viruses expressing different v-Abl proteins or c-Myc protein. (A) Samples of infected cells were lysed 48 h postinfection, and levels of the different v-Abl proteins were assessed by Western blotting with the anti-Gag/v-Abl monoclonal antibody H548 (5). (B) Colonies obtained from doubly infected populations were expanded and ana-lyzed by Western blotting with the anti-Gag/v-Abl monoclonal antibody H548 and an anti-Myc antibody.
TABLE 2. Carboxyl-terminal sequences are required in
cis
aVirus Colonies/10
5cells⫾SEM
Expt 1 Expt 2
P120
130.8
⫾
9.2
229.6
⫾
6.81
P70/S2
⬍0.1
⬍0.1
P120/⌬SH2
⬍0.1
⬍0.1
c-Myc
⬍0.1
⬍0.1
P120
⫹
c-Myc
367.2
⫾
29.32
468.4
⫾
19.46
P70/S2
⫹
c-Myc
10.4
⫾
1.78
2.1
⫾
0.5
P120/⌬SH2
⫹
P70/S2
⬍0.1
⬍0.1
aCells were infected with virus stocks of matched titer and plated in agar
fol-lowing virus adsorption. Macroscopic colonies were counted 4 to 5 weeks after the cells were plated. Values given as⬍0.1 indicate that no colonies were observed and reflect the minimum number of colonies that could have been detected.
TABLE 3. P90/S2 fails to transform NIH 3T3 cells
aVirus Colonies/10
5cells⫾SEM
Expt 1 Expt 2
P90
171.5
⫾
16.0
97.5
⫾
7.6
P90/S2
⬍1
⫾
0.1
⬍1
⫾
0.1
P120⌬668-819
112.0
⫾
19.0
94.3
⫾
9.0
P120/S2⌬668-819
190.0
⫾
12.0
98.8
⫾
5.8
P120K⫺
⬍1
⫾
0.1
⬍1
⫾
0.1
aCells were infected with virus stocks of matched titer and plated in agar
following virus adsorption. Macroscopic colonies were counted 4 to 5 weeks after the cells were plated. Values given as⬍1 indicate that no colonies were observed and reflect the minimum number of colonies that could have been detected.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.312.548.74.298.2] [image:7.612.53.293.227.327.2] [image:7.612.311.551.614.695.2]onstration that the region can influence transformation of
im-mortalized fibroblast cell lines.
The inability of the P70/S2 strain to transform NIH 3T3 cells
demonstrates that the v-Abl and v-Src SH2 domains are not
functionally equivalent. Earlier work (22), using different
chi-meras, reached the opposite conclusion. However, in these
experiments, the entire amino terminus of v-Src was appended
to a truncated v-Abl protein similar to P70. Consequently,
these chimeras also contained the v-Src SH3 domain. Several
Src SH3-binding proteins are phosphorylated in v-Src
trans-formed cells but not in cells transtrans-formed by a variant v-Src
allele lacking the SH3 domain (70). Thus, the Src SH3 domain
plays a role in v-Src-mediated tyrosine phosphorylation of
sub-strates, and its presence may influence transformation
poten-tial in NIH 3T3 cells.
Consistent with the inability of P70/S2 to transform cells,
several downstream signals associated with Ab-MLV
transfor-mation are missing in cells expressing this protein. For
exam-ple, tyrosine phosphorylation of the Shc adapter protein and
association with Grb-2 does not occur. This interaction is
be-lieved to be one way in which Abl proteins activate Ras (16, 44,
52). The p42 and p44 ERK proteins, elements that function
downstream of Ras and are critical components of the MAP
kinase cascade, are not phosphorylated in cells expressing P70/
S2. Consistent with this, P70/S2 is compromised in its ability to
activate transcription from the c-
fos
promoter. All of these
events are thought to be important for v-Abl-mediated
trans-formation (45, 62). These data contrast with the ability of
P70/S2 to stimulate Ras activation. Because Ras activation was
examined using a transfection system, the results may not
re-flect events occurring in the NIH 3T3 target cells.
Alterna-tively, Ras activation can be achieved in many ways, raising the
more intriguing possibility that the way in which Ras is
acti-vated influences transmission of downstream signals.
SH2 domains contribute significantly to the specificity with
which tyrosine kinases activate downstream effectors (31, 66),
and the SH2 domains of v-Src and v-Abl have been placed in
different subgroups based on their ability to interact with
ty-rosine phosphorylated peptides (66). The ability of the v-Abl
SH2 domain but not the Src SH2 domain to interact with the
Shc adapter protein (31, 44, 52) may reflect this property. The
inability of P70/S2 to mediate phosphorylation of Shc suggests
that one pathway by which v-Abl normally signals to Ras is not
functional in these cells, a feature that may contribute to the
transformation defective nature of this strain.
Although Shc phosphorylation and assembly of the
Shc-Grb2-Sos complex is classically associated with Ras activation,
these proteins may mediate other types of responses. For
ex-ample, complex formation does not always correlate with MAP
kinase activation (44, 49), and injection of Grb2 antibodies
inhibits membrane ruffing and cell growth in response to
epi-dermal growth factor, even though microinjection of anti-Ras
antibodies affects only growth (32, 57). In addition, dShc, the
Drosophila
homologue, lacks the residue analogous to the
mammalian Grb-2 interaction site, and no complex between
dShc and Drk, the
Drosophila
Grb-2 homologue, can be
de-tected (30). Considered in combination with the finding that
Ras can be activated by P70/S2, this information raises the
possibility that the absence of Shc-Grb2-Sos complexes in cells
expressing P70/S2 reflects the loss of other functions important
for v-Abl-mediated transformation.
P70/S2 was unable to activate transcription from a c-
fos
promoter, a response that involves interactions at both the
SRF and TCF sites within this element. SRF activation does
not appear to require the JNK and ERK MAP kinases; TCF
can be activated by both of these kinases (4). The inability of
P70/S2 to activate ERK may influence the TCF activity and
contribute to the transformation-defective phenotype of the
P70/S2 strain. If this is indeed the case, interactions with SAP-1
might be involved. This protein appears to be important in
murine cell lines such as NIH 3T3 cells and is activated in a
Ras-independent manner with minimal influence of JNK (25).
The final 162 amino acids of the v-Abl carboxyl terminus are
sufficient to complement the transformation defect in P70/S2
and to restore Shc phosphorylation. Analyses of other mutants
suggest that the carboxyl terminus does not bind Shc (52);
because the region appears to be required in
cis
, it may simply
stabilize the interaction between v-Abl and Shc. Some
pro-teins, including Bcr/Abl, can recruit Grb2-Sos complexes
di-rectly (47, 51), bypassing a need for Shc interaction. However,
Bcr/Abl-Grb2 interaction is mediated by residues in the Bcr
portion of the protein (47, 51), and sequences within the Abl
carboxyl terminus have not been shown to interact with Grb2.
Although the effects on Shc and Grb2 are striking, they may
not be the critical feature involved. The extreme carboxyl
ter-minus contains motifs that mediate interactions with the
cy-toskeleton (36, 68). Other studies have shown that this region
binds RNA polymerase II, facilitating phosphorylation of this
protein by c-Abl (2). While this nuclear event is probably not
involved in Ab-MLV-mediated transformation, it does
high-light a role for this region in protein-protein interaction.
Un-raveling the mechanism by which these sequences affect
trans-formation should shed light on the function of the v-Abl
carboxyl terminus. Because the chimeric v-Abl proteins studied
here display strong phenotypic differences in a readily
manip-ulable cell type, they provide an excellent model to uncover the
mechanism underlying the function of the carboxyl-terminal
residues.
ACKNOWLEDGMENTS
We thank Steve Goff, Brent Cochran, Anne Marie Pendergast,
Charles Sawyers, and Larry Feig for supplying reagents and Tony
Baughn and Jonah Rainey for assistance with some of the experiments.
K.B. was supported by grant T35 HL07785, and the experiments
were supported by grant CA22440 from the National Cancer Institute.
REFERENCES
1.Afar, D. E. H., A. Goga, J. McLaughlin, O. N. Witte, and C. L. Sawyers.1994.
Differential complementation of Bcr-Abl point mutants with c-Myc. Science
264:424–426.
2.Baskaran, R., M. E. Dahmus, and J. Y. Wang.1993. Tyrosine
phosphoryla-tion of mammalian RNA polymerase II carboxyl-terminal domain. Proc. Natl. Acad. Sci. USA90:11167–11171.
3.Berkowitz, L. A., K. T. Riabowol, and M. Z. Gilman.1989. Multiple sequence
elements of a single functional class are required for cyclic AMP respon-siveness of the mouse c-fospromoter. Mol. Cell. Biol.9:4272–4281.
4.Cahil, M. A., R. Janknecht, and A. Nordheim.1996. Signalling pathways:
Jack of all cascades. Curr. Biol.6:16–19.
5.Chesebro, B., K. Wehrly, M. Cloyd, W. Britt, J. Portis, J. Collins, and J.
Nisho.1981. Characterization of mouse monoclonal antibodies specific for
Friend murine leukemia virus-induced erythroleukemia cells: Friend-specific and FMR-specific antigens. Virology112:131–144.
6.Dai, Z., and A. M. Pendergast.1995. Abi-2, a novel SH3-containing protein
interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity. Genes Dev.9:2569–2582.
7.Danial, N. N., J. A. Losman, T. Lu, N. Yip, K. Krishnan, J. Krolewski, S. P.
Goff, J. Y. Wang, and P. B. Rothman.1998. Direct interaction of Jak1 and
v-Abl is required for v-Abl-induced activation of STATs and proliferation. Mol. Cell. Biol.18:6795–6804.
8.Danial, N. N., A. Pernis, and P. B. Rothman.1995. Jak/STAT signaling
induced by the v-Abl oncogene. Science269:1875–1877.
9.Davis, R. J.1999. Signal transduction by the c-Jun N-terminal kinase.
Bio-chem. Soc. Symp.64:1–12.
10. de Rooij, J., and J. L. Bos.1997. Minimal Ras-binding domain of Raf1 can
be used as an activation-specific probe for Ras. Oncogene14:623–625.
11. de Vries-Smits, A., B. Burgering, S. Leevers, C. Marshall, and J. Bos.1992.
Involvement of p21rasin activation of extracellular regulated kinase 2.
Na-ture (London)357:602–604.
on November 9, 2019 by guest
http://jvi.asm.org/
12.Dubridge, R. B., P. Tang, C. Hsia, P. M. Leong, J. H. Miller, and M. P. Calos.
1987. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol.7:379–387.
13.Dudek, H., S. R. Datta, T. F. Franke, M. J. Birnbaum, R. Yao, G. M. Cooper,
R. A. Segal, D. R. Kaplan, and M. E. Greenberg. 1997. Regulation of
neuronal survival by the serine-threonine protein kinase Akt. Science275:
661–664.
14.Engelman, A., and N. Rosenberg.1987. Isolation of temperature-sensitive
Abelson virus mutants by site-directed mutagenesis. Proc. Natl. Acad. Sci. USA84:8021–8025.
15. Feller, S. M., B. Knudsen, and H. Hanafusa.1994. c-Abl kinase regulates the
protein binding activity of c-Crk. EMBO J.13:2341–2351.
16. Goga, A., J. McLaughlin, D. E. H. Afar, D. C. Saffran, and O. N. Witte.1995.
Alternative signals to Ras for hematopoietic transformation by the Bcr-Abl oncogene. Cell82:981–988.
17. Gupta, S., H. Yan, L. H. Wong, S. Ralph, J. Krolewski, and C. Schindler.
1996. The SH2 domains of Stat1 and Stat2 mediate multiple interactions in the signal transduction of IFN-␣signals. EMBO J.15:1075–1084.
18. Hanks, S. K., A. M. Quinn, and T. Hunter.1988. The protein kinase family:
conserved features and deduced phylogeny of the catalytic domains. Science
241:42–51.
19. Hann, R. S., M. W. King, D. L. Bentley, C. W. Anderson, and R. N.
Eisen-man.1988. A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lym-phomas. Cell52:185–195.
20. Harvat, B. L., and W. Wharton.1995. Serum response element and flanking
sequences mediate the synergistic transcriptional activation of c-fos by 12-O-tetradecanoylphorbol-13-acetate and cholera toxin in AKR-2B cells. Cell Growth Differ.6:955–964.
21. Herrmann, C., G. A. Martin, and A. Wittinghofer.1995. Quantitative
anal-ysis of the complex between p21rasand the Ras-binding domain of the human Raf-1 protein kinase. J. Biol. Chem.270:2901–2905.
22. Hevezi, P., K. Alin, and S. P. Goff.1993. Transforming activity and tissue
tropism of hybrid retroviral genomes containing portions of the v-abl and v-src oncogenes. Oncogene8:2413–2423.
23. Hevezi, P., K. Alin, R. Rees-Jones, and S. P. Goff.1992. Bone
marrow-transforming activity of linker insertion mutants of Abelson murine leukemia virus. Oncogene7:2323–2328.
24. Hill, C. S., J. Wynne, and R. Treisman.1995. The Rho family GTPases
RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell
81:1159–1170.
25. Hipskind, R. A., D. Buscher, A. Nordheim, and M. Baccarini.1994. Ras/
MAP kinase-dependent and -independent signaling pathways target distinct ternary complex factors. Genes Dev.8:1803–1816.
27. Hipskind, R. A., V. N. Rao, C. G. F. Mueller, E. S. P. Reddy, and A.
Nordheim.1991. Ets-related protein Elk-1 is homologous to the c-fos
regu-latory factor p62TCF. Nature (London)354:531–534.
28. Karin, M.1996. The regulation of AP-1 activity by mitogen-activated protein
kinases. Philos. Trans. Roy. Soc. Lond. B351:127–134.
29. Kim, D. W., V. Cheriyath, A. L. Roy, and B. H. Cochran.1998. TFII-I
enhances activation of the c-fos promoter through interactions with up-stream elements. Mol. Cell. Biol.18:3310–3320.
30. Kipreos, E. T., and J. Y. J. Wang.1992. Cell cycle-regulated binding of c-Abl
tyrosine kinase to DNA. Science256:382–385.
31. Lai, K.-M. V., J. P. Olivier, G. D. Gish, M. Henkemeyer, J. McGlade, and T.
Pawson.1995. ADrosophila shcgene product is implicated in signaling by the
DER receptor tyrosine kinase. Mol. Cell. Biol.15:4810–4818.
32. Marengere, L. E. M., Z. Songyang, G. D. Gish, M. D. Schaller, J. T. Parsons,
M. J. Stearn, L. C. Cantley, and T. Pawson.1994. SH2 domain specificity and
activity modified by a single residue. Nature (London)369:502–505.
33. Matuoka, K., F. Shibasaki, M. Shibata, and T. Takenawa.1993. Ash/Grb-2,
a SH2/SH3-containing protein, couples to signaling for mitogenesis and cytoskeletal reorganization by EGF and PDGF. EMBO J.12:3467–3473.
34. Mayer, B. J., and D. Baltimore.1994. Mutagenic analysis of the roles of SH2
and SH3 domains in regulation of the Abl tyrosine kinase. Mol. Cell. Biol.
14:2883–2894.
34. Mayer, B. J., and D. Baltimore.1993. Signalling through SH2 and SH3
domains. Trends Cell Biol.3:8–13.
35. Mayer, B. J., P. K. Jackson, R. A. van Etten, and D. Baltimore.1992. Point
mutations in the Abl SH2 domain coordinately impair phosphotyrosine bind-ing in vitro and transformbind-ing activity in vivo. Mol. Cell. Biol.12:609–618.
36. McWhirter, J. R., and J. Y. J. Wang.1991. Activation of tyrosine kinase and
microfilament-binding functions of c-Abl by Bcr sequences in Bcr/Abl fusion proteins. Mol. Cell. Biol.11:1553–1565.
37. Minden, A., A. Lin, M. McMahon, C. Lange-Carter, B. Derijard, R. J.
Davies, G. L. Johnson, and M. Karin.1994. Differential activation of ERK
and JNK mitogen-activated protein kinases by Raf-1 and MEKK. Science
266:1719–1723.
38. Muller, A. J., A.-M. Pendergast, K. Parmar, M. H. Pavlik, N. Rosenberg, and
O. N. Witte.1993. En bloc substitution of the Src homology region 2 domain
activates the transforming potential of the c-Abl protein tyrosine kinase. Proc. Natl. Acad. Sci. USA90:3457–3461.
39. Muller, A. J., J. C. Young, A.-M. Pendergast, M. Pondel, N. R. Landau, D. R.
Littman, and O. N. Witte.1991. BCR first exon sequences specifically
acti-vate theBCR/ABLtyrosine kinase oncogene of Philadelphia chromosome-positive human leukemias. Mol. Cell. Biol.11:1785–1792.
40. Murtagh, K., G. Skladany, J. Hoag, and N. Rosenberg.1986. Abelson
mu-rine leukemia virus variants with increased oncogenic potential. J. Virol.60:
599–606.
41. Musacchio, A., T. Gibson, V. P. Lehto, and M. Saraste. 1992. SH3- an
abundant protein domain in search of a function. FEBS Lett.307:55–61.
42. Mysliwiec, T., R. Perego, and G. D. Kruh.1996. Analysis of chimeric
Gag-Arg/Abl molecules indicates a distinct negative regulatory role for the Arg C-terminal domain. Oncogene12:631–640.
43. Norman, C., M. Runswick, R. Pollock, and R. Treisman.1988. Isolation and
properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fosserum response element. Cell55:989–1003.
44. Owen-Lynch, P. J., A. K. Y. Wong, and A. D. Whetton.1995. v-abl-mediated
apoptotic suppression is associated with shc phosphorylation without con-commitant mitogen-activated protein kinase activation. J. Biol. Chem.270:
5956–5962.
45. Parmar, K., and N. Rosenberg.1996. Ras complements the carboxyl
termi-nus of v-Abl protein in lymphoid transformation. J. Virol.70:1009–1015.
46. Pendergast, A. M., M. L. Gishizky, M. H. Havlik, and O. N. Witte.1993. SH1
domain autophosphorylation of p210 BCR/ABL is required for transforma-tion but not growth factor independence. Mol. Cell. Biol.13:1728–1736.
47. Pendergast, A. M., L. A. Quilliam, L. D. Cripe, C. H. Bassing, Z. Dai, N. Li,
A. Batzer, K. M. Rabun, C. J. Der, J. Schlessinger, M. L. Gishizky.1993.
BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell75:175–185.
48. Pendergast, A. M., J. A. Traugh, and O. N. Witte.1987. Normal cellular and
transformation-associated Abl proteins share common sites for protein ki-nase C phosphorylation. Mol. Cell. Biol.7:4280–4289.
49. Pruett, W., Y. Yuan, E. Rose, A. G. Batzer, N. Harada, and E. Y. Skolnik.
1995. Association between Grb2/Sos and insulin receptor substrate 1 is not sufficient for activation of extracellular signal-regulated kinases by interleu-kin-4: implications forrasactivation by insulin. Mol. Cell. Biol.15:1778– 1785.
50. Prywes, R., J. G. Foulkes, N. Rosenberg, and D. Baltimore.1983. Sequences
of the A-MuLV protein needed for fibroblast and lymphoid cell transfor-mation. Cell34:569–579.
51. Puil, L., J. Liu, G. Gish, G. Mbamalu, D. Bowtell, P. G. Pelicci, R.
Arling-haus, and T. Pawson.1994. Bcr-Abl oncoproteins bind directly to activators
of the Ras signalling pathway. EMBO J.13:764–773.
52. Raffel, G., K. Parmar, and N. Rosenberg.1996.In vivoassociation of v-Abl
with Shc mediated by a non-phosphotyrosine-dependent SH2 interaction. J. Biol. Chem.271:4640–4645.
53. Raitano, A. B., J. R. Halpern, T. M. Hambuch, and C. L. Sawyers.1995. The
bcr-ablleukemia oncogene activates Jun kinase and requires Jun for trans-formation. Proc. Natl. Acad. Sci. USA92:11746–11750.
54. Ren, R., Z.-S. Ye, and D. Baltimore.1994. Abl protein-tyrosine kinase selects
the crk adapter as a substrate using SH3-binding sites. Genes Dev.8:783– 795.
55. Renshaw, M. W., E. Lea-Chou, and Y. J. Wang.1996. Rac is required for
v-Abl tyrosine kinase to activate mitogenesis. Curr. Biol.6:76–83.
56. Renshaw, M. W., J. R. McWhirter, and J. Y. J. Wang.1995. The human
leukemia oncogenebcr-ablabrogates the anchorage requirement but not the growth factor requirement for proliferation. Mol. Cell. Biol.15:1286–1293.
57. Ridley, A. J., H. F. Paterson, C. L. Johnston, D. Diekmann, and A. Hall.
1992. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell70:401–410.
58. Rosenberg, N., D. R. Clark, and O. N. Witte.1980. Abelson murine leukemia
virus mutants deficient in kinase activity and lymphoid transformation. J. Vi-rol.36:766–774.
59. Rosenberg, N., and O. N. Witte.1980. Abelson murine leukemia virus
(A-MuLV) mutants with alterations in the A-MuLV-specific P120 molecule. J. Virol.33:340–348.
60. Rosenberg, N., and O. N. Witte.1988. The viral and cellular forms of the
Abelson (abl) oncogene. Adv. Virus Res.35:39–81.
61. Sadowski, H. B., K. Shuai, J. E. Darnell, Jr., and M. Z. Gilman.1993. A
common nuclear signal transduction pathway activated by growth factor and cytokine receptors. Science263:1739–1744.
62. Sawyers, C. L., J. McLaughlin, and O. N. Witte.1995. Genetic requirement
for ras in the transformation of fibroblasts and hematopoietic cells by the bcr-abl oncogene. J. Exp. Med.181:307–313.
63. Shaw, P. E., H. Schroter, and A. Nordheim.1989. The ability of a ternary
complex to form over the serum response element correlates with serum inducibility of the human c-fos promoter. Cell.56:563–572.
64. Shi, Y., K. Alin, and S. P. Goff.1995. Abl-interactor-1, a novel SH3 protein
binding to the carboxy-terminal portion of the Abl protein, suppresses v-Abl transforming activity. Genes Dev.9:2583–2597.
65. Smith, J. M., S. Katz, and B. J. Mayer.1999. Activation of the Abl tyrosine
kinase in vivo by Src homology 3 domains from the Src homology 2/Src homology 3 adaptor Nck. J. Biol. Chem.274:27956–27962.
on November 9, 2019 by guest
http://jvi.asm.org/
66.Songyang, Z., S. E. Shoelson, M. Chadhuri, G. Gish, T. Pawson, W. G. Haser, F. King, T. Roberts, S. Ratnofsky, R. J. Lechleider, B. G. Neel, R. B. Birge, J. E. Fajardo, M. M. Chou, H. Hanafusa, B. Schaffhausen, and L. C.
Cantley.1993. SH2 domains recognize specific phosphopeptide sequences.
Cell72:767–778.
67.Treisman, T.1990. The SRE: a growth factor responsive transcriptional
regulator. Semin. Cancer Biol.1:47–58.
68.Van Etten, R. A., P. K. Jackson, D. Baltimore, M. C. Sanders, P. T.
Matsu-daira, and P. A. Janmey.1994. The COOH terminus of the c-abl tyrosine
kinase contains distinct F- and G-actin binding domains with bundling ac-tivity. J. Cell Biol.124:325–340.
69. Wagner, B. J., T. H. Hayes, C. J. Hoban, and B. H. Cochran.1990. The SIF
binding element confers sis/PDGF inducibility onto the c-fos promoter. EMBO J.9:4477–4484.
70. Zhigang, W., S. M. Thomas, R. J. Rickles, J. A. Taylor, A. W. Brauer, C.
Seidel-Dugan, W. M. Michael, G. Dreyfuss, and J. S. Brugge.1994.
Identi-fication of Src, Fyn, and Lyn SH3-binding proteins: implications for a func-tion of SH3 domains. Mol. Cell. Biol.14:4509–4521.
on November 9, 2019 by guest
http://jvi.asm.org/