0022-538X/94/$04.00+0
Copyright ©) 1994,American Society forMicrobiology
Complex Functional Interactions
at
the
Early
Enhancer of
the
PQ
Strain of BK Virus
ANNET.FERGUSONtANDSURESH SUBRAMANI*
DepartmentofBiology,
University
ofCalifomia, SanDiego, LaJolla, California92093-0322 Received1 November 1993/Accepted27 March 1994BKvirusis a humanpapovavirus that
latently
infectsamajority
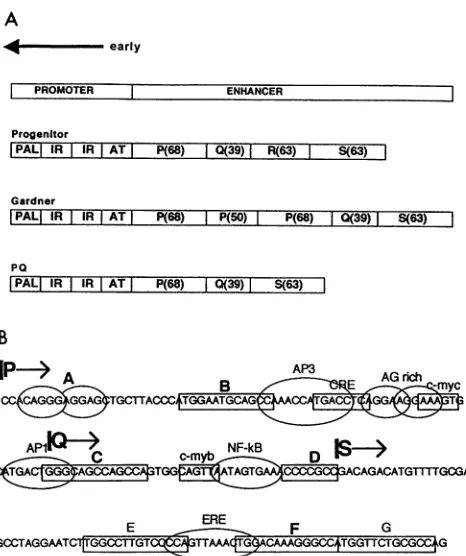
of the world'spopulation.Therearemore than 30 strains of thevirus,mostofwhich differin the structureof theearlyenhancerregion.
Theenhancer of theprogenitor strain,WW,from which the otherstrains canbe derived,consists of fourconserved DNA domains, P, Q, R, and S.Rearrangementofthe enhanceroccursupon passage in tissue culture and is thought tooccurduring virusreplication. The strain under study,PQ,wasselecteduponpassageoftheGardnerstrain (PPPQS)inthepermissive cellline, Vero. Mutationalanalysisoftheentire enhancerregiondemonstrates the importance of fivecis-acting sequences: DNA sites B, C, andF, which have homology to the NF-1 protein bindingsequence; onepurine-richmotifdesignated A;and siteD,which is similar toanSP-1proteinbinding site. Two sites, B and C, appear to have a negative influence on geneactivity.
To study the functional interactions in moredetail, promoter-enhancerconstructions that containdifferentcombinationsof the five DNAsiteslinkedtothechloramphenicolacetyltransferasegene weretestedforearly
geneactivity.
Theresults revealthat theproteinsbindingtotheenhancerfunctionally
cooperatewith each other. The effects ofmaking mutations attheDNAsitesarevery similartotheeffects ofusingexcessenhancerDNAsequences totitrate theproteins that bindtothecis-actingDNAsites (invivocompetition).Moreover,theeffects ofchangingthe spacing betweenthe DNA sitesalsodemonstrate thatthere arecooperativeinteractionsamongthe proteins thatbindtothePQ strainenhancer. DNAsitesB,C,and F areclearly protected from DNaseIdigestionby Vero cell nuclearproteins. In addition, mutation ofeach DNA site alters its sensitivity to DNase I in the presence of Vero cellproteins. Interestingly, mutation of siteBaffectsprotein bindingtosite Baswellasto sitesA, C, D,andF.These results suggestthatcooperative functionalandphysical interactionsoccur atthe early enhancer ofthePQ strain.Allstrains of the humanDNA tumorvirus BKvirus(BKV) haveverysimilarcoding regions. However, theDNA structure of the early enhancer, which controls expression of the viral earlygeneproducts, is different for each strain. The progenitor strain, WW (7), has an enhancer that consists of four con-served DNAsequenceblocksdesignatedP(68bp),Q(39bp),
R (63 bp), and S (63 bp) (29). The level of early gene expression controlled by the enhancer can directly and indi-rectly affect virusgrowth and tissuespecificityof its growth(5, 15, 47). In a permissive cell, the virus replicates, and its enhancerrearranges (41, 42). The virus strain thatgrowsthe best in a certain cell type has a selective advantage and eventuallypredominates in that cell(49).Tobetter understand the biology of BKV, we need more information about the functional interactions that occur at the enhancer. Enhancer elementsareduplicated inmoststrains,soitis easiesttostudy the simplest enhancer, that from the PQ strain. Hopefully, these resultscanbe usedtopredict earlygeneactivity of other virus strains aswell astheirgrowth potential.
The naturally occurring BKV strain studied in this work contains the enhancer structure PQS and was derived from passageofthe Gardner strain (PPPQS) inVero African green
monkeykidney cells (54) (Fig. 1A).We analyzed the role that cis-acting DNAsequences in the enhancer play in regulating earlygene expression inVero cells. We chose Vero cells for two reasons. First, Vero isa permissive primate cell line. In
*Correspondingauthor. Mailing address: Department of Biology,
UniversityofCalifornia,SanDiego, La Jolla, CA 92093-0322. Phone: (619)534-2327.Fax: (619)534-0053.
tPresent address:TheJohnsHopkins Oncology Center, Baltimore, MD21231.
addition, the PQ strain istranscriptionally active in Verocells and can replicate and lyse these cells efficiently, as PQ was selected forin thiscell type.
Previous work with otherstrains ofBKV in both semiper-missive and nonpermissive celltypeshasimplicated NF-1 and SP-1-like sites and factorsasimportant for early transcription (6, 15, 21, 29, 33). The work presented here reveals the importance oftwo newsites. OneisanNF-1-likesite found in the less studied S block. The second is an AG-rich motif, contained within the P block, which has homology to the binding motif for the Ets family oftranscription factors (26, 59).
We have also attempted to elucidate the details of the functionally and physically cooperativeinteractionsatthe early enhancer of PQ that occur among five DNA sites and the proteins that interact with them. Our analysis involves the generationofsingle,multiple,andspacermutations within the enhancer, use of in vivo competition experiments withexcess enhancer DNA sequence motifs, and DNase I footprinting. The results show that cooperative interactions among the enhancer-binding proteins contribute significantly to early promoteractivity.
MATERIALSANDMETHODS
Plasmid constructions used in the functional analysis of the PQ enhancer. (i) Single mutants.Previously,thewild-typePQ
early enhancer(pBK68; Fig. 1B) and linker-scan (LS) muta-tions spanningamajority of thePand Q regions ofthe early enhancer (LS 2 [A], LS 4 [B], LS 5 [C], LS 21 [cyclic AMP-responsive element], LS 31 [AP-1 binding site], LS 34 [C], LS 42[c-Mycbinding site], andLS 45[AP-3 binding site] 4274
on November 9, 2019 by guest
http://jvi.asm.org/
A
e learly
PROMOTER | ENHANCER
Progenitor
PALI IR IR ATI P(68) 0Q(39) R(63) S(63)
Gardner
PALI IR IR AT P(68) P(50) P(68) 0(39) 8(63)
Po
IPALI
IR IR AT P(68) 0(39) (63)B
AP,K
-- NF-kBAPlc c-myb
DNIS--=AGA GG 4 _ACA AGACTGTTTTGCNGA
E ERE F G
GCCTAGGMT GGTTCTGCGG
FIG. 1. (A)Structuresofthe noncodingregulatoryregions of three
BKV strains. The early promoter, which is the same in all strains, consists ofa palindrome (PAL), two inverted repeats (IR), and an
AT-rich regionof 20 bp. Theprogenitorstrain,WW,has theenhancer
structurethat contains single P (68-bp),Q (39-bp), R (63-bp), and S
(63-bp)blocks. The PQstrainis derived frompassageof theGardner
strain in Vero cells. Thewild-typePQenhancerwasusedtoconstruct the single-site mutations. (B) Putative transcription factor binding motifs in the BKVPQ enhancer. The boundaries of the P,Q,and S blocks are shown. Similarities to previously identified transcription factor bindingmotifsare labelled.Boxedsequences B, C, E, F, andG all have homologyto theNF-1 binding site, and siteDhas homology tothe SP-1 bindingmotif. Circled siteAisapurine-richsequencewith
similaritytothe binding motif for the Ets family of proteins. This is the early enhancerregion that is linkedtothe earlypromoterand theCAT andglobinreportergenes.
[15]) were made. (The site mutated by eachLS mutation is
indicated in brackets after the name of theLS mutant.) In
addition,deletion mutationsfrom theBamHI (late) end ofthe early enhancer (13) were made in the Gardner strain
back-ground. Three of the BamHI deletion mutations (AB3755, AB3720, and AB3699) were made, in the context of the PQ enhancer background, by digestion of these DNAs at their
unique Bsu36I sites, removal of the intervening sequence
containing the two P blocks, and religation of the large fragment. The resulting plasmids were named pAEFG-CAT,
which hasa deletionof the threeNF-1-like sites, E, F, and G;
pAFG-CAT, which has a deletion of sites F and G; and
pAG-CAT,which has adeletion of site G.
The mutation in site D (Fig. 2A) was made by PCR (43).
Briefly, a single-stranded primer containing an EcoRI site
(region underlined) in place of the GC box (5'GGGAA1T1C
GACAGACATGllFTTGCG3')
and pSK plasmid DNAcon-taining the wild-type PQ enhancer (pKS-BKV) were
hybrid-ized. Taqpolymerase (GibcoBRL)wasusedtosynthesizethe
mutant single-stranded DNA. Universal primer was used to
initiate thesynthesis of the other DNA strand. The amplified DNA was digested with EcoRI and BamHI, and the 83-bp DNAfragmentwasisolated. A plasmid containing the site D mutation, which lacked the3'-terminal 50 bp of the S block, was digested with these same restriction enzymes, and the large DNA fragment was isolated. pAD-CAT, which contains a mutation at site D, was made by ligation of these two EcoRI-BamHI-digested DNA fragments.
The site E mutant was also made by PCR. The BamHI primer (5'-GGGGATCCGCTGGCGCAGAACCATGGCCT TTGTCCAGTTTAACTGGGGACAAGGGTCGACTTCC TAGGCTCGC-3') containing the mutation as a newAccI site (region underlined) was used with pKS-BKV to synthesize the initial mutant DNA strand. M13 reverse primer (New England Biolabs) was used along with the BamHI primer to amplify the enhancer DNA fragment containing the site E mutation. This PCR-generated fragment was digested withBamHI and Hin-dIII. pBK68 was digested with BamHI and HindIII, and the large DNA fragment lacking the promoter-enhancer was iso-lated. The isolated DNA fragment and PCR-generated BamHI-HindIII DNA fragment were ligated, and theresulting plasmid with a mutation at site E was named pAE-CAT.
A mutation was created in the second AG-rich box by digestion of LS 39 (15) withXhoI, removal of the 5' overhang with 5 U of mung bean nuclease, and ligation of the blunt ends. This plasmid was named LS 39D. The site Fmutation (pAF-CAT) was made by partial digestion of pBK68 with NcoI, the use of mung bean nuclease to delete 4 bp from the NF-1 half-site, and ligation of the blunt ends. The DNAsequences of all of these mutations weredetermined by the method of Chen and Seeberg (8). All mutations weremade in the context of the chloramphenicol acetyltransferase (CAT) expression vector for analysis of CAT enzyme activity.
(ii) Multiple mutants.Mutations that delete sites C, D, and F and sites B, C, D, and F (AB3488 and AB3469) were previously made (13). The double mutation of sites A and B (pCDF-CAT, where CDF denotes the unmutated sites in the plasmid) was generated by site-directed mutagenesis (27). Briefly, a pTZ19U plasmid that contained the LS 2 (site A mutation) promoter-enhancer and a primer thatcontained the site B mutation
(5'GGTlh[GGCTGCATTCCCTCGGAGAA
GCAGC3') were used for the synthesis of the double mutant. pBK68 and its derivatives contain a unique Bsu36I restric-tion site at nucleotide 3502 (Fig. 2) between sites B and Cof the enhancer and the unique ApaI site in the simian virus 40 (SV40) polyadenylation sequence of the vector DNA. The mutations involving two sites, ACD, ACF, ADF, BCD, BCF, and BDF, were created by digestion of LS 2, LS 4, LS 34, pD-CAT, and pF-CAT withthese twoenzymes and ligation of the2-kbApaI-Bsu36I DNAfragment from LS4(ACDF) orLS 2 (BCDF) with the corresponding 3.4-kbBsu36I-ApaI DNA fragment from pF-CAT(ABCD), pD-CAT (ABCE), orLS 34 (ABDF) (underlined lettersdenote those sitesthatwerelinked together). The resulting plasmids were named pACD-CAT, pACF-CAT, pADF-CAT, pBCD-CAT, pBCF-CAT, and pBDF-CAT.
Similarly, the mutations involving three sites, CD, CF, and DF were made bydigestion of LS 34, pD-CAT,pF-CAT, and pCDF-CAT with ApaI and Bsu36I and ligation of the 2-kb ApaI-Bsu36I DNA fragment of pCDF-CAT, which contains the site A and site Bmutations, with the corresponding3.4-kb ApaI-BamHI DNA fragments from the single mutants pF-CAT (ABCD), pD-CAT (ABCE), and LS 34
(ABDF).
The resulting plasmids were named pCD-CAT,pCF-CAT,
and pDF-CAT.(iii) Mutants used in in vivo competitor assays. Some gross
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.62.295.77.355.2]A
_CAT geneexpression
EARLYPROMOTER ENHANCER
3362 3381 3408 3431 3455 p 3523 3562 s 3631
PAL IR IR AT A B D F
HindIII NCoo Bsu361
3475 3502
site seauence of site
sequenceof site after mutation
_~~~~~~~f _PS _ nwi IluaM l '7 *% wlvl
A AGGGAGGAGC AGGCCCTCGA 53.9/-2
B TGGAATGCAGCCA GGGAATGCAGCCA 222+1-9
C TGGGCAGCCAGCCA GGGGCAGCCAGCCA 304 -W-12
C TGGGCAGCCAGCCA TGGGCAQCTCGAGG 3374-13
D CCCGCC GAATTC 454-2
F TGGACAAAGGCCA TGGACAAAGGCQT 57+/-5
B
mutated site(s) intactsite(s) CATActivity
BCDF A 4+/-2
CDF AB
DF ABC 6+/-.4
F .ABCD 57+/-S
D ABCF 45+/-2
ABF CD 10+/-1
BF ACD 16+/-1
AF BCD 20+/-2
ABC :1DF 3+/-.2
BC ADF 50+/-1.5
AC BDF 43+/-1
AB CDF 43+/-3
B ACDF 222+/-9
C ABDF 337+/-13
A BCDF 53+/-2
ABD CF 38+/-2
BD ACF 69+/-2
AD BCf 62+/-2
ABCDf 100
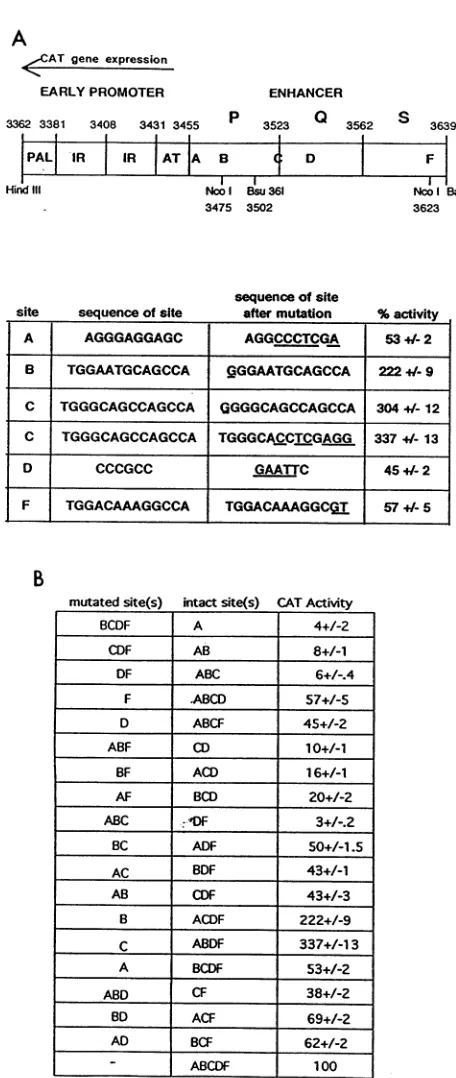
FIG. 2. (A)Transcription factor binding sitesfunctionally imp
tant for BKV early gene expression in Vero cells. The wild-tV sequences and corresponding mutations in the five functional si within the PQ enhancer are shown. Underlines highlight the spec nucleotide changes.Results of at least fivetransfections are indica
as means + standard errors (see Materials and Methods). Th( numbersarecorrectedfortransfection efficiency by using theratic CATactivity toluciferase activity. Site E was changed from TG( CTTGTCCCCAtoGACCCT(LGTCCCCA, andsite G was
chang
from TGGTTCTGCGCCA to TGGTTCTGCGCTC. Both of th mutationshad little effect on CAT activity (see Results). (B)Effects multiple mutations within the BKV early enhancer on CAT ge expression. CAT activity of the BKV enhancerless promoter-C,deletions within the enhancer of the Gardner strainweremade
previously (13).
These included plasmids that contained anenhancerless BKVpromoter (AB3454), apromoterlinked to sites A and AB (AB3472and AB3516), and the promoterless enhancer DNA sequences that contained sites CDF and BCDF
(AH3641
andAH3581).TheAB3516plasmidcontained the AP-3 and c-Myc protein binding sites and the second AG-rich motif between unique Bsu36I andXhoI restriction BamHi sites. The DNA between these two sites was deleted from AB3516by
digestion
ofthe DNA tocompletion
withBsu36I
and
XhoI, polymerization
of the staggered ends of DNAby
using
theKlenowfragment
of DNApolymeraseI, andligation
of the two blunt ends of DNA. This plasmid was named
pComp-AB-CAT.
A mutation was made in the F site of AH3641 and AH3581byusing the NcoI siteatnucleotide 3623 (Fig. 2A)oftheenhancerasdescribed above. These plasmids were named pNP-CD-CAT andpNP-BCD-CAT.
BglI isa unique restriction site within thepBR322
1-lacta-mase gene of all of the BKV promoter-enhancer
plasmids.
There isaunique XhoI restriction site at nucleotide 3455(Fig. 2A) ofplasmid AB3454. pNP-CD-CAT and pNP-BCD-CAT haveuniqueXhoI restriction sitesatthe5' ends of sites C and B, respectively. Two plasmids that contained the BKV pro-moterlinked to the enhancer DNA sites C andD and sitesB, C,and Dweremade.AB3454,pNP-CD-CAT, and pNP-BCD-CATweredigestedwithBglIandXhoI. Onesetofcompetitor plasmids wasmade by ligation of the 3.4-kb BglI-XhoI DNA fragmentfromAB3454 with the corresponding 2-kbXhoI-BglI DNAfragment of pNP-CD-CAT and pNP-BCD-CAT. These plasmids were named pComp-CD-CAT and pComp-BCD-CAT. The finalcompetitorplasmidwascreatedby ligation of the 3.4-kb BglI-XhoI DNA fragment from AB3472 with the 2-kbXhoI-BglI DNA fragment of pNP-CD-CAT. Thisplasmid was named pComp-ACD-CAT. A frameshift mutation was made in the CAT gene of each of these six competitors (a wild-type construct was included) by partial digestion within the CATgene withEcoRI and polymerization of thestaggered end of DNA by using the Klenow fragment of DNA
poly-merase I, which created a new AseI site. The competitor plasmids were the same as the test plasmid except that they hadonly segments of enhancer DNA and aframeshift muta-tion in their CAT genes(CAT-). Thecompetitor plasmid that contained the enhancerless promoter was named pComp-prom-CAT-, and the competitor plasmid that contained the wild-type promoter-enhancer was namedpComp-WT-CAT-. Competitor plasmids that contained sites A, AB, CD, ACD, and BCD were namedpComp-A-CAT-, pComp-AB-CAT-, pComp-CD-CAT-, pComp-ACD-CAT-, and pComp-BCD-CAT-, respectively.
(iv) Mutants withDNA insertions between sites B and C. Previously,5bpof DNA wasinsertedbetweensites B and C of the enhancer (LS21) (15). Insertions of 11 bp (pBCMX.11) and 15 bp (pBCMX.15) of DNA between sites B and C were made by digestion of pBK68 with the restriction en-or- zymeBsu36I, polymerization of thestaggered end of DNA by ype using the Klenowfragment of DNApolymeraseI, which adds tes 3 bp of DNA, and blunt ligation of an 8- or 12-bp double--ific strandedoligonucleotide that contains anXhoI restriction site. ted pBCMX.48 (insertion of 48 bp) andpBCMX.81 (insertion of
ese 81bp)weremadebythemethod used formaking pBCMX.15
G°of except that after a12-bpdouble-stranded oligonucleotide was
ged
ese
sof ene ,AT
plasmid (AB3454[11]) is 1%. Allconstructions have intactEand G sites, but these sitesplaynosignificant role in activity.
9%activitv
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.65.293.67.606.2]ligated, another one or two 33-bp XhoI DNA fragments derived from the multiple cloning site of pKS- (Bluescript) were also added. These XhoI insertions were introduced in both orientations. The sequences from pKS- that were used forthe insertion mutations were inspected for consensus DNA sequencesrepresentingbinding sites for over 10 common and cell-type-specific transcription factors, and none were found (22, 23).
Transfection of Vero cells. Prior to transfections, the DNA sequenceof each of the mutationswasverified by the method of Chen andSeeberg (8).VeroAfrican green monkey kidney cells were grown in Dulbecco's modified Eagle medium sup-plemented with 10% bovine calf serum (Gibco BRL Labora-tories). Cells were transferred at approximately 25 to 50% confluencyto 100-mm-diameterplates the day before transfec-tion. Transfection was performed by a modification of the calcium phosphate method (25, 37) that included a 2-min glycerol shock 4 h after the
DNA-Ca2"
precipitate was added to the cells. In the assay of thesingle, multiple, and insertion mutation plasmids, 8 pug of reporter CAT plasmid was used with 2 ,ug of the reference plasmid, pRSV-luciferase (11). Luciferase activity was used to correct for the efficiency of transfection. The Rous sarcoma virus (RSV) promoter does not compete with the BKV early promoter for limitingtran-scriptionfactors under the assay conditions used(45).For the experiments done underreplicating conditions, 5
jig
of plas-midcontaining the3-globin
gene underthe control of thePQ earlypromoter-enhancer was usedwith 5 ,ug of pRSV-BKV T-antigen expression plasmid. For the competition experi-ments,4,ugofpBK68 (testplasmid),2,IgofpRSV-luciferase (referenceplasmid),and4jig
(1:1),8 ,ug(2:1),or14,ug(3.5:1) of competitor plasmid were used. When necessary, pComp-prom-CAT-wasusedas asupplementsothatatotal of 20 ,ug ofDNA was usedfor each transfection.CAT and luciferase assays.Cell lysateswere made approx-imately48 hposttransfection by freeze-thawingthesamplesin 0.1Mpotassium phosphate(pH 7.8) (3, 11). CAT assayswere
performedso that the positivecontrol, pRSV-CAT, achieved 25 to 95% conversion of chloramphenicol to its acetylated form. Luciferase activities for samples, as measured by the number of light units, were at least 100-fold higher for DNA-transfected cells than for mock-transfected cells. Each set of experiments consisted of at least five
independent
transfections with the mutant
plasmid
and two transfections with thewild-type plasmid, pBK68. The CAT activity gener-atedbyeachset ofplasmidswasfirst normalized forefficiency
oftransfectionbyusingtheratio of CAT
activity
toluciferase activity.The results forpBK68wereaveragedandset as100%. The results of all five normalized transfections for each of the mutant plasmids were then averaged, compared with the average for pBK68, and thenexpressed
as a percentage of wild-typeactivity.The standarderrorsfor eachmutantplasmid
werealso calculated from the data from all five transfections. The percent activity in the
competition
analysis
represents gene activity of the testplasmid
as the percentage of theactivity
ofpBK68,
using
the enhancerless promoterplasmid,
pComp-prom-CAT-, asthe
competitor.
Statistical analysis. An
exponential
regression
fit the data for the gene activities of themultiple
mutations the best because the datahadalarge
rangeofvalues,
andexponential
regressions fit on the basis of percentage errors. In other words, the data fit an
equation
that representscooperative
rather than
independent
interactions among theproteins
that bindtothe fivecis-acting
sites.Theequation
usedtomake the predictionsis Z= eeO+ 0BB + ODD+ OFF+OABAB+OACA C +OADAD+(AFAF+OBCBC+OCDCD+OABCABC+ OBDFBDF+OCDFCDF+OBCDFBCDF
+ 0
ABCFABCF.
Thedependent variable,Z,is the gene activityor datum. The coefficients are denoted by 0. The independent variables,which representthecis-acting sites,aredenoted byA toF.Specificnumerical values for each of these independent variables were obtained from the data. In this model, each independent variable is a dummy variable, which can have either of twovalues, 1 or0. Thedummyvariables represent whether or not the DNA site, which is represented by the capital letter,is wild typeormutated.Avalue of1denotes that the DNA site is wild type, and a value of 0 denotes that the DNAsite is mutated. Forinstance,if allof thecis-actingsites are mutated orA = B = C = D = F =0,
thenZ =e'O.
In anotherexample,theimpactof thetwocis-acting sites,Aand B,canbe assessedby changingthedummyvariables:A =B=1 and C = D = F = 0. The predicted value forZ or gene activityisZ =
e'O
+ 6BB +OABAB,
and therefore the additionalimpactofDNAsites AandB onthe activityofacompletely mutated enhancerise08B + OAAB
Construction ofplasmids forRNA analysis. Plasmids that contained the rabbit
3-globin
gene under the control of the BKV early regulatory region (both wild-type and mutantenhancers)
weremadebyreplacement
of the CAT gene from plasmids pBK68,LS2,LS4,LS34,pAD-CAT,
pAF-CAT,and pBK3362 (13) (which expresses the CAT gene under the controlof thepromoter-enhancer
of the Gardnerstrain)
withthe
3-globin
gene to formpWT-Globin,
pAA-Globin,
pAB-Globin,
pAC-Globin, pAD-Globin, pAF-Globin,
andpGard-ner-Globin. These plasmid DNAs were then digested with ApaI (the site is within SV40 VP1
coding region),
and the staggered end wasmade into ablunt endwith the use of the KlenowfragmentofDNApolymeraseI. Plasmid RSV-NeoK, whichhas the RSV promoter upstream of theneomycingene(a gift
ofFereydoun
G.Sajjadi-Gensia Pharmaceuticals,
Inc.),
was digested with BamHI (the site is at the RSV promoter
end).
Thisstaggered
endwas made intoablunt end with the use of the Klenowfragment
ofDNApolymerase
I. Then the DNA was digested with SmaI (the site is downstream of the neomycingene)
andtreated with calf intestinal alkalinephos-phatase.
The 1.4-kbfragment
containing
theneomycin
tran-scription
unitwasisolated andligated
into theBKV promoter-enhancer-globin vectors. Theplasmid
was made to allow expressionof both theneomycinandglobin
reporter genesso that both G418 resistance and globin geneexpression
can be monitored. Theplasmids containing
thewild-type
and five enhancer mutations in this context are namedpWT-Neo-Globin,
pAA-Neo-Globin, pAB-Neo-Globin,
pAC-Neo-Glo-bin, pAD-Neo-Globin,and
pAF-Neo-Globin.
Creation of stable cell lines.
Twenty
100-mm-diameter platesof 25% confluentVerocellsweretransfected with 20 ,ug ofpWT-Neo-Globin,
pAA-Neo-Globin,
pAB-Neo-Globin,
pAC-Neo-Globin, pAD-Neo-Globin, and pAF-Neo-Globin
DNA. At 48 h
posttransfection,
the cells on eachplate
weresplit
intotwo150-mm-diameterplates,
andG418-resistant cells wereselected forby
using
medium thatcontained 400 jig of Geneticin(G418 sulfate;
GibcoBRL)
perml.After 1 month ofselection,
clonalpopulations
ofcells,
whichwere resistant toG418, were isolated.
Eighteen
clones of cells derived from transfection withpWT-Neo-Globin,
16clones of cells derived from transfection withpAA-Neo-Globin,
26 clones of cells derived fromtransfection withpAB-Neo-Globin,
13clones of cells derived fromtransfection withpAC-Neo-Globin,
8clones of cells derived from transfection withpAD-Neo-Globin,
and 19 clones of cells derived from transfection withpAF-Neo-Globin were grown
independently
until cellswereconfluent. The clones of cells derived from each transfection wereon November 9, 2019 by guest
http://jvi.asm.org/
TABLE 1. Oligonucleotidesusedforgel shift assays
Sequence'
Site
Wild-typeoligonucleotide Mutantoligonucleotide
B 5'-GCTTACCCATGGAATGCAGCCAAAC-3' 5'-GCCTCGAGGGAATGCAGCCAAAC-3'
C 5'-GACTGGGCAGCCAGCCAGTG-3' 5'-GACTGGGCACCTQGAGGGC-3'
F 5'-CGATGGACAAAGGCCATCC-3' 5'-CGATGGACAAAGGCGTTCc-3'
aTranscriptionfactor consensusmotifs(10,40)areitalicized,sequences thataremutatedareunderlined.
combined andpassaged in the presence of 200 ,ug of Geneticin perml.
mRNA isolation. Isolation of poly(A) RNA was accom-plished with the Fast TrackmRNAisolation kitas instructed by the manufacturer (Invitrogen Corp., San Diego, Calif.).
RNase protection assays. The pKS--Globin plasmid con-tained the BKVpromoter-enhancer-globingeneon a BamHI-AccIDNAfragmentthatwasderived frompWT-Globin. The pKS--Actin plasmid contained the actin gene on an EcoRI DNA fragment. Both globin (495-bp) and actin (265-bp) antisense RNA probeswere synthesized invitro byusing the InvitroScript RNAtranscription system(Invitrogen). Approx-imately 3 x 105 cpm ofprobe was incubated with 20 ,ug of tRNAandapproximately 1 ,ugofmRNA(notincluded in the negative control [lane T] ofFig.3) inhybridizationbuffer[40
mM
piperazine-N,N'-bis(2-ethanesulfonic
acid (PIPES; pH 6.4), 0.4M NaCl, 1 mM EDTA] at85°Cfor 10 min and then overnightat45°C. The mixturewastreatedwith 5 ,ug of RNase Apermlfor 45 min at30°C, treated with 250 ,ug of proteinase Kper ml for 30 min, extracted with phenol-chloroform, and precipitated with ethanol. The products were subjected toelectrophoresisin a4% denaturing polyacrylamide gel. Vero cell nuclear extracts. Vero cells were grown as de-scribed for transfection experiments. Cells were grown in twenty
1,050-cm2
roller bottles and supplemented with 5% CO2.Cellswerefed and boostedwith5%CO2every 3daysfor 1 week,atwhich point they were 70% confluent. For harvest-ing, the cells were washed twice withTD(140mMNaCl,5mMKCl, 0.7 mM Na2HPO4) and then treated with trypsin. The detached cells were pelleted bycentrifugationandwashed with phosphate-buffered saline (140mM NaCl, 3 mM KCl, 6 mM
Na2HPO4, 1.5 mM KH2PO4) supplemented with 1 mg of MgCl2perml.Nuclear extracts were made from the cellsby a modification of the procedures described by Dignametal.(16) and Briggset al. (4).
DNase I footprinting. The 277-bp BamHI-HindIII DNA fragment containing the BKV early promoter and enhancer (wildtype ormutant) wasradioactively labelled at theHindlll
(coding
strand) orBamHI (noncoding strand) end, using T4 DNA kinase. Approximately 2 x 105 cpm of this probe was usedper DNase Ifootprinting reaction. The DNA probe was incubated with nonspecific competitor DNA (sonicated calf thymus DNA), polyvinyl alcohol at a final concentration of 0.4%,and110jig of Vero cell nuclear protein in TM reaction buffer (final concentration of 50 mMTris-HCl
[pH 7.9], 100 mM KCl, 8jiM
ZnSO4, 10mMMgCl2,
20%glycerol, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, and 1 mMsodiummetabisulfite). The mixture was incubated for 15 min on ice, atwhich point 5 mMCaCl2-10 mM MgCl2
was added,and DNase I digestion was begun. Digestion products were subjected to electrophoresis in a 5% denaturing poly-acrylamide gel. The DNA sequence of the wild-type DNA probe was determined by the method of Maxam and Gilbert (30).Oligonucleotidesused forgelshiftassays.Sequencesfor the
oligonucleotides
usedare shown in Table 1.Mobility shift analysis. DNAprobes for the mobility shift assaysincludeddouble-strandedoligonucleotidesfor the wild-type and mutant sites B, C, and F mentioned above. These oli onucleotides were phosphorylated by using 10 jiCi of [ry- 2P]ATP and 1 U of T4 polynucleotide kinase.
Radioac-tively
labelledoligonucleotideswereisolatedon a 1% agarosegel.
Then theoligonucleotidesweredilutedto5,000 cpm/,ul.A total of10,000cpm of DNAprobewasusedper25-,ul reaction mixture. The DNA probe andVero cell proteins were incu-bated in thesame0.1 M TM buffer used for DNase Ifootprint
analysisfor 15minonicebeforesubjectionto
electrophoresis
ina5%nondenaturing polyacrylamide gel
(38).
RESULTS
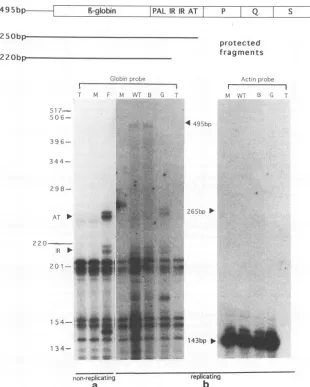
Determination of the transcriptional initiation sites for early geneexpression.Any changes in CATactivity observed with mutations in the PQ enhancer could be due to either altered concentration of thetranscript (transcriptionalcontrol)
oraltered efficiency oftranslation of the transcript (posttran-scriptionalcontrol). If control is strictly transcriptional, the 5' termini of wild-type and mutant enhancer mRNAs are ex-pectedtobe thesame.Wedetermined the transcriptionalstart sites by using RNase protection analysis (Fig. 3). Promoter-enhancerconstructs were linkedto the rabbit 3-globin gene, since it is knownthat CAT mRNA is unstable. Forexperiments done underreplicatingconditions, these constructs were tran-siently transfected with the pRSV-BK T-antigen expression plasmid.The Tantigen is required for DNA replication from the BKVorigin (47). For experiments done under nonrepli-catingconditions, stable cell lines that expressed the ,-globin gene under the control of the BKV promoter-enhancerwere made.
mRNA frommock-transfected cells andtRNAwereused as negative controls. These RNAs protected a group of RNA fragments of size less than 210 bp. The nonspecific protected fragments could be due to the RNA probe hybridizing with itself, nonspecifichybridization of tRNA with the RNA probe, orincompletecleavage of the RNA probe by RNase A.
The levels of cytoplasmic actin geneexpression were approx-imately the same in the mock-, pWT-Globin-, pAB-Globin-, andpGardner-Globin-transfected cellsunder replicating con-ditions (Fig. 3b). This result demonstrates that approximately equal amounts ofpoly(A)+ RNA were present in all assays. mRNA frompWT-Globin-,pAB-Globin-,and pGardner-Glo-bin-transfected cells specifically protected a pair of RNA fragments ofapproximately 220 bp and a pair of fragments of approximately 250 bp in length (Fig. 3). Both sets of globin transcripts were much lessabundant than the actin transcript (positive control) (Fig.3b).
The sizes of the protected RNA fragments correspond to start sites within the promoter atthe second inverted repeat
on November 9, 2019 by guest
http://jvi.asm.org/
6-globin PAL IR IR AT P Q S
protected
fragments
GIob;nLrobe
Tr M F NM WT B G T 51
7-50
6--3 9
6-
344-
298-AT * I
2 20
JR
201-
154*-1
34-Actinprobe
I I
hM WTr G T
4495bp
265bp >
143bp >
non-replicating replicating
a
b
FIG. 3. Accumulation ofwild-type,mutant,and Gardner strain BKVmRNAinVerocells. RNase protection analysis of mRNA extracted from astable Verocelllineundernonreplicatingconditions (a) and transiently transfectedVerocells underreplicating conditions(b)is shown. The sizes ofundigested globin (495-bp)andactin(265-bp) probesaremarked with arrowheads. The specific transcriptionalstartsitesfor the globingene
within theinvertedrepeat(IR) (two bandsat220 bp) andATblock(AT) (twobandsat250 bp)arealso shown with arrowheads. The size of the
protected actinRNAfragment is 143 bp. Sizes of the protectedRNAfragmentsweredetermined by comparison with molecular weight markers runinparallel. The negative control lanesareT(tRNA only) and M(mock-transfected cells).Lane F, RNAfragmentsprotectedfrom digestion
bymRNA isolated fromthe stablecell lineexpressingpA&F-Neo-Globin; lanes WT, B, and G, RNA fragments protected fromRNasedigestion bymRNA isolated fromcellstransiently transfected withpWT-Globin, pAB-Globin, and pGardner-Globin. Thetwobandsatapproximately 250 bpin lanes Fand Gareatslightly differentpositions because thesetwosampleswere run ontwoindependent polyacrylamide gels.
(nucleotides3381to3408)andthe AT-richregion(nucleotides 3431to3455).Thisfindingindicatesthat theboundaries of the promoterfor thePQstrain in Vero cellsarethesame asthose
previously defined in vitro and underreplicatingconditions in othercelltypeswithother BKV strains(13, 21). Furthermore, the initiation sites found forglobin genetranscription in the stable cell line expressing pAF-Globin under nonreplicating conditions (Fig. 3a) were the same as those found under replicatingconditions(Fig.3b).
There are no in- or out-of-frame translational start sites upstream of the rabbit ,-globinAUG codon in the mRNAs initiating at the inverted repeat or in the ATblock. Because
theGardner strain, PQ, and mutant mRNAs tested have the same5' termini and donothaveotherAUGs upstream ofthe authentic initiator codon for the globingene, different
trans-lation efficiencies cannot account for the differences in early
geneexpression.Therefore,thequantitationofprotein activity should be a direct measurement of transcription efficiency. These results suggest that the specific organization of the enhancer is notinvolved inposttranscriptional regulation.
Functional analysis of the BKV early enhancer. (i) Single mutations. Many sequences in the BKV early promoter-enhancer are similar to known transcription factor binding sites(Fig. 1B).TheentirePQenhancerwasscreenedby linker-scanmutations, deletions, andmutations madebyPCR. Each of these mutated controlregions inthe early orientationwas
usedtoexpress theCATgeneintransientlytransfectedVero cells. Rather than presenting raw data from a single experi-ment,we averagedtheresults from five ormoreexperiments and calculated the standarderror.Individual mutation of sites A, B, C, D,and F ledtobetweenatwo-tothreefoldchangein CAT activity (Fig. 2A). The effect of each mutation on the
495bp F
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.147.457.77.464.2]TABLE 2. Interactions betweenproteinsbindingto the five sites in the BKVenhancerl
Interaction Site
Positive with: Negativeor nonewith:
A (D, F, BF,CF), DF,BDF, CDF (B, C, BC,BD), CD, BCD, BCF,BCDF
B (D, F,
AF),
DF,ADF (C,CF),
A, AC, AD, ACD, ACF, ACDF, CD,CDFC (D, AD),DF, ADF (A,B, F, AF),AB, BF, BDF, ABF,ABDF
D (C,AC,BC), ABC, ACF,ABCF (A,B, F, AB, AF, BF,ABF), CF,BCF
F All None
a Informationwascompiledfromboththe invivo data ofFig.2and thestatisticalanalysisdescribedin Results. All letters denotethecis-actingsites andanyprotein
interactionsoccurringatthe sites.Interactions at each of thefive sites thatpositivelyornegativelyinfluenceorhavenoeffectoninteractionsatothersitesarelisted.
Parenthesesindicate interactionsatthe BKV enhancerwhicharepredictedbythe statisticalanalysis.Threefoldorgreatereffectsareconsideredstatistically significant.
consensus
sequence
fortranscription factorbinding was also examined. For the sites thatare similar to the NF-1 binding motif, mutations within plasmids LS 4 (B),LS 34 (C), pAE-CAT, pAF-CAT, andpAG-CAT alter theimportant TGG or CCA half-site andareexpectedtoaffectproteinbindingtothe sites (10, 40). Alteration of the B(222%),
C(304
to337%),
andF(57%)half-sites ledto two- tothreefoldchangesin CAT activity,but alteration of theE(68%) andG(74%) half-sites only ledto aslight decrease inactivity. Inaddition, adeletion that removedsites E, F, and G(pAEFG-CAT)ledtothesame twofold decrease inCAT activity
(50%)
asdid thespecific
F mutation within pAF-CAT (57%). This result demonstrates that sitesEand G are not important for enhancer activityin Verocells.The site D mutation disrupted the perfect SP-1 binding motif, so it was expected that proteins would bind very differentlytothemutated site (2, 19).
pAD-CAT
had twofold-lower CATactivity (45%) than pBK68, which demonstrated theimportanceof theSP-1-like site. Thereare two setsoftwooverlapping AG-rich sequences in the early enhancer (over-lappingcircles inFig.1B). Althoughthesecond motif(labelled AG rich in Fig. 1B) appeared to be nonfunctional (LS 39D, 117%),mutation ofsiteA(LS 2)led toatwofold decrease in CATactivity(53%).
Itis of particular interest that mutation of two of the three NF-1-like sites, B (LS 4) and C (LS 34), led to a two- to threefold increase in activity (222 and 337%, respectively), while mutation of the other site, F (pAF-CAT), led to a comparable twofold decrease in CAT activity(57%).
(ii) Multiple mutations. Results from the analysis of multi-ple mutations are shown in Fig. 2B. Capital letters in the second column denote sites that have not been altered by mutation, and this is how themutant enhancerDNAwill be referred to hereafter. AB3454 (enhancerless promoter) (13) has 1% of the pBK68 (wild-type promoter-enhancer) gene activity.Early geneactivity ranged from this background level (DF = 3%) to over300% (ABDF = 337%) compared with
wild-type gene activity (ABCDF = 100%).
Conclusions were basedon thevisual inspection and inter-pretation of the data and confirmed by the statistical approach explained in Materials and Methods. One hundred thirty-six datum points obtained from the gene activities of all of the promoter-enhancer-CAT constructs represented in Fig. 2 were fitted to an exponential regression. Using the equation and independent variables generated by the regression, one can calculatethe effectthat interactions at one wild-type site have oninteractionsatother wild-type sites. This model reflects the gross impact of these interactions, and it is stressed that it is onlyanapproximation. However, the paradigm can be used to summarize thedata and make predictions, which are shown in Table 2. Only positive or negative effects that were greater
thanthreefold are viewed asstatistically significant. Less than threefold effects are considered without effect. Romancapital letters denote thecis-acting sites within thePQenhancer, and underlined letters denote anyprotein interactions thatoccurat theseDNA sites.
First,
A interacts unfavorably with B and C and positively with other combinations of sites. When A was coupled with both B andC,
we observed a substantial decrease in gene activity (compare ABDF[337%]withABCDF[100%])orno significant change in activity (compare AB [8%] and ABC [6%]).However, A actedpositively with D and F(comparethe approximately 17-fold difference in geneactivity between DF [3%] and ADF[50%]). A also actedpositively with BorC as long as site Fwas notmutated. Forinstance,
Ahasaneightfold effectonBDF (compareBDF[43%] andABDF[337%])and a fivefold effect on CDF (compare CDF [43%] and ACDF[222%]).
Second,
B andC interactunfavorably. Withoneexception,B wasantagonisticwithC,but B hadastimulatoryeffect inmost othercontexts.Examplesof unfavorable interactionswerethe similar gene activities ofACDF (222%)andABCDF (100%) and of CDF(43%) and BCDF(53%).Alternatively, B hada 6-fold stimulatory effect on ADF (compare AD [50%] and ABDF[337%]) and 14-fold effect onDF (compare DF [3%] and BDF [43%]).Reciprocally,ChinderedinteractionsinvolvingB, but C had astimulatory influenceonD aslongassite Bwasmutated.The antagonism betweenC and Bwasshownby thedifference in the gene activity ofABDF
(337%)
andABCDF (100%) and similar gene activities of BDF (43%) and BCDF (53%). In contrast, Chada 14-fold positive effecton DF(compare DF [3%] and CDF [43%]) and a 4-fold positive effect on ADF (compareADF [50%] andACDF [222%]).Third,
D and Cinteract synergistically. In contrast to the negative influence that B hadonC,D hadonly a stimulatory effecton C. Thispositive effect is indicated by the threefold effect of DonACF(compareAC [69%]andACDF[222%]) andthe 9.5-fold effect ofDonABC(compareABC [6%] and ABCD [57%]). Incomparison,thenegative influence ofD,in the absence ofC,waspredicted bythe statistical analysis. Forinstance,
D had negative interactions withA, B,
F,
AB,
AF,
and BF.
Fourth, the effect of F is always positive. F appears to act independently ofA_ B,
C,
and D; it stimulates activity in any context.Examples of this activationincludethe fourfold effect ofF onCD(compare CD [10%]andCDF[43%]),the 7.5-fold effect ofF onABC (compareABC [6%] andABCF[45%]), and the 14-fold effect on ACD (compare ACD [16%] and ACDF[222%]).Insummary,the results suggest
B}
and Cinteract unfavorably witheach otherandwhenthey are together with A. In contrast,on November 9, 2019 by guest
http://jvi.asm.org/
inmostcombinations,Dand FinteractfavorablywithB andC
(Table 2).
In vivo competition analysis shows the same pattern of
synergyandantagonism.Thereis anotherwaytodemonstrate
that cooperativity exists among the proteins that bindto the
PQ enhancer. Previously, the in vivo competition technique wasusedtoshowcompetitionfor factorsbindingto theSV40
enhancer(45).Theprocedure involvedconstructing
competi-torplasmids that contained different parts of the BKV early
enhancerwith thepromoterlinkedtoaCATgene,which had
a frameshift mutation in the middle of the gene so that the
active protein was not synthesized (see Materials and
Meth-ods). The testDNA usedwaspBK68 (15). We cotransfected
different ratios of competitor to test DNA into Vero cells,
extracted proteins fromthecellsapproximately48 hlater,and
assayed the proteinsforCATactivity.WeexpectCATactivity
of thetestplasmidtochangeiftheenhancerDNAcompeted
forthetranscriptionfactorsthat bindtothe testDNAandare
responsible for its gene activity. We used this method to
determine whethertheeffects ofmutatingsiteswere thesame
as the effects of using excess enhancer DNA sequences to
titratethe factorsthat bindto the sites.
pComp-prom-CAT-, the enhancerless promoter plasmid,
wasused as anegativecontrol. Itwasnotexpected tocompete
for enhancer-binding proteins, and test gene activity is not
expectedtochange (i.e.,itremainsat thearbitrarilydesignated
100%CATactivity). All otherresultswere comparedwiththe
leveloftestgeneactivityobtainedbycompetitionwith
pComp-prom-CAT-.Whenthisplasmidwasusedasthecompetitor,it
caused little or no decrease in test gene activity even at
maximummolarexcess(datanot shown),soit wasusedasthe
carrier DNA in the remaining experiments.
pComp-WT-CAT- wasused as apositivecontrol, sinceit wasexpected to
yield full competition if used in high enough molar excess.
Twentymicrogramsof DNAwasusedfor eachcotransfection
experiment (28),sothemaximummolarratio ofcompetitorto
wild-type DNA used was 3.5 to 1. Under these conditions,
pComp-WT-CAT- diminished test gene activity by almost
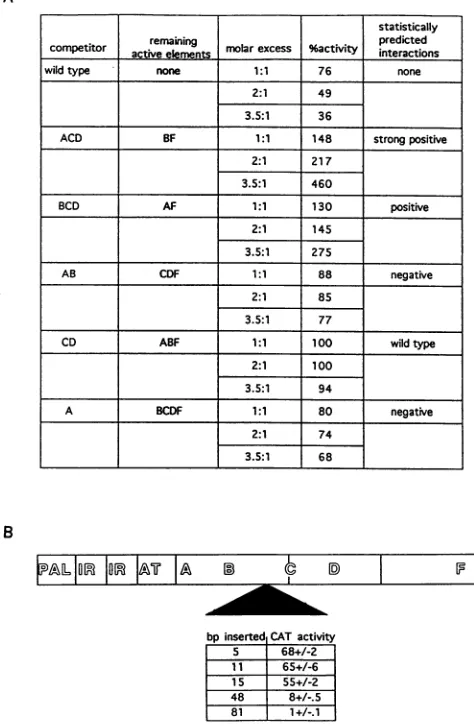
threefold (Fig.4A).
Areductionof 1.3- to 1.5-foldin testgene activityoccurred
when a3.5-to-1 molarratio ofpComp-A-CAT- and
pComp-AB-CAT- wastransfected withthe testplasmid (68 to77%).
Competition withenhancerDNAcontainingsite Aand
muta-tionofsiteAboth ledtoadecrease in testgeneactivity(LS2
has53% CAT activity). Similarly, competition with enhancer
DNAcontainingsites Aand Band mutation of sitesAandB
bothresulted in a decrease in testgene activity (pCDF-CAT
has 43% activity).
We did not observe a change in test gene activity when
pComp-CD-CAT- was used as the competitor DNA. This
resultsupportsthe statisticalprediction thatinteractionsofA,
B, and F will have wild-type gene activity (Fig. 4A). When
pComp-BCD-CAT- wasusedasthecompetitorDNA, Aand
Fwereexpectedtoremain functionalin thetestplasmid. We
observed an almost threefold increase in test gene activity,
whichsupports the statisticallypredictedpositive interactions
between A and
E
(Fig. 4A). Similarly, whenpComp-ACD-CAT- was used as the competitor DNA,B and
E
wereexpectedto remainfunctionalin the testplasmid, and we saw
up to a 4.6-fold increase in test gene activity. This result
supportsthestatistical analysis,whichpredicts strongpositive interactions betweenB and F (Fig. 4A).
These data show reductions and increases of test (CAT)
geneactivitycorrespondingtotheamountandtypeof
cotrans-fected enhancer competitor DNA. Overall, results of the in vivocompetitions supporttheresultsof themutational
analy-A
B
bpinsertedlCATactivity
5 68+/-2
1F
1 65+/-6iS 55+/-2
48 8+/-.5
[81 1+/-.1
FIG. 4. (A) Effect of in vivo competition on CAT activity. All
competitor plasmids contain the wild-type promoter with various
segments of the early enhancer linked to the CAT gene with a frameshift mutationinitscodingregion(see MaterialsandMethods).
"Remaining activeelements"indicatesthefunctionalelementsin the
testplasmid enhancerexpected toremainboundbyVero proteins if
complete competition were to occur. "Molar excess" refers to the
molarratio ofcompetitortopBK68testplasmid.Percentactivityis the
activity of pBK68 when it is transfected with competitor plasmid
compared with the activity of pBK68 when it is transfected with
pComp-prom-CAT- (pComp-prom-CAT- should not compete for
enhancer-bindingproteins,sopBK68CATactivityremains at 100%).
Statistically predicted interactions were determined by using the
equation and coefficients generated by the exponential regression
described in Materials andMethods. (B) Spacing mutants and their
effects on early gene expression. The wild-type early
promoter-en-hancerwith insertionsbetween sites BandCwas linked tothe CAT
gene(LS 21,5-bpinsertion;pBCMX.11, 11-bpinsertion; pBCMX.15,
15-bp insertion; pBCMX.48,48-bp insertion; and pBCMX.81, 81-bp
insertion [see Materials and Methods]). The transfection and assay
procedures usedwere the same as those described for the multiple
mutations. Designations areas in Fig. 1.
sis.Adecrease inactivitywas detectedwhether siteA orsites
Aand Bwere mutated orused as competitor DNAs. Activity
higher than for pBK68, which waspredicted for AF andBF,
was observed with pComp-BCD-CAT- and
pComp-ACD-CAT- usedascompetitorDNAs.
Efects ofspacingoninteractions at the enhancer.Another
statistically
competitor reinin molarexcess %activity predicted
activeelements interactions__
wildtype none 1:1 76 none
2:1 49
3.5:1 36
ACD BF 1:1 148 strongpositive
2:1 217
3.5:1 460
BCD AF 1:1 130 positive
2:1 145
3.5:1 275
AB CDF 1:1 88 negative
2:1 85
3.5:1 77
CD ABF 1:1 100 wildtype
2:1 100
3.5:1 94
A BCDF 1:1 80 negative
2:1 74
3.5:1 68
LION
JON
I&T
I&
0Iy
IFon November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.316.553.86.449.2]4282 FERGUSON AND SUBRAMANI
A
B
GGW.LWA
ED CDL
A
,...
.: ii,a" $
s;,., .?. 4*
_ * i'
.. r 'e .e.:
i?
.:?.' :r ___ .^
_
.zei
__g w
ailijL'....8
_?:^
-
...eSt
_-
*'_,-w...;
.<.iv;=lFS ?
_ ..
^aQ.
B
=...
.:w. ....
- .g.
.
g.w_i.$..$.'
_....
- Wo :. .
A !!k 9 "' ; ;
3 _0mm
-1
_.
.7,s,.
P.
F
.00
.0
X,
L_
lk ...FIG. 5. DNase Ifootprintingof the BKVearlyenhancer.(A)The277-bp BamHI-HindIIIearlypromoter-enhancer codingstrand(nucleotides 3362to3639 inFig. 2)wasradiolabelledattheHindlIl end.Thecis-actingsites of the enhancerarebracketed.Arrowsindicate theareasof DNase Ihypersensitivitymentioned in Results. Lanes: G andG/A,sequenceladdergenerated by cleavageatthesenucleotidesof thewild-type enhancer DNAbythe method of Maxam andGilbert(36);L, ladderofDNase Isensitivityof thewild-typeenhancer DNAprobewithout the presenceof protein;WT, wild-type DNAprobe;A, DNAprobe containing theAmutation(LS 2); B,DNAprobecontainingthe Bmutation(LS4); C,DNA probe containingthe Cmutation (LS 34);F, DNAprobecontainingthe Fmutation (pAF-CAT).Doubling of lanesrepresentstwohalves ofone
sample. (B)The277-bpBamHI-HindIII early promoter-enhancer noncodingstrandwasradiolabelledattheBamHI end.LaneD, DNAprobe containingthe Dmutation(pAD-CAT). Mutationatsite Bincludesa2-bp deletion,whichcauses ashiftinthe DNaseIdigestionpatternin lane Bof panel A. This was taken into account when the autoradiographs were visually inspected.
way to demonstrate cooperativity among the five proteins binding to the PQ enhancer is to show that changing the spacingbetween thesites affects enhancer activity(Fig.4B). It is known that interactions between transcription factors are affectedby the number of helical turns between them (32, 36, 50). Sinceanimportantphysical and functional interaction was occurring between B and C, a spacer DNA sequence was inserted between these two NF-1-like motifs. In the PQ enhancer, sitesBand C areseparated by 31 bp. There was no significant change in CAT activity whether one half (LS 21), one full (pBCMX.11), or one and a half (pBCMX.15) addi-tional helical turns were inserted between sites B and C (Fig. 4B).These data suggest that interactions are not affected if the proteinsbinding to the sites are forced to interact on opposite sides of the DNAhelix.
Furthermore, insertion of an additional 48 bp between sites B andC(pBCMX.48) led to a 10-folddecrease in gene activ-ity, and insertion of an additional 81 bp between the sites (pBCMX.81) abolished gene activity. There are two possible
models for interactions at the enhancer, independent and cooperative. IfAB and CDF acted as independent enhancer elements,thenABwouldindependentlystimulate geneactivity (AB = 8% of wild-type activity; Fig. 2B) and CDF would independentlystimulate geneactivity (CDF = 43%; Fig. 2B). Since itisknownthatenhancers, inparticular the enhancer of the Gardnerstrain, canaffect transcription from up to 1.6 kb awayfrom a promoter (13), theseparation of AB and CDF should have little effect on gene activity. However, basal activity is observed when 48 and 81 bp are inserted between sitesBandC, which strongly suggests that cooperativity among the five DNA sites and the proteins that bind to them are necessary for enhancer function. Results of the mutant and competition analysis have already lended support for this proposal.
Vero cell proteins that bind to the early enhancer of BKV strainPQ. The wild-typePQenhancer was analyzed by DNase Ifootprinting todetermine whether the DNA sequences that were functionally important for early gene expression were
J. VIROL.
on November 9, 2019 by guest
http://jvi.asm.org/
alsobinding sites forVero nuclear proteins. Figure5 demon-strates that Vero cell nuclear proteins clearly protected the NF-1-like sites B, C, and F from DNase I digestion. Mobility shiftassayswereperformedtoverifyVero cell protein binding tosites A and D. Wedetectedspecific DNA-proteincomplexes between oligonucleotides containing the Aand D motifs and Vero cell nuclear proteins (data not shown). These results show that Vero cell nuclear proteins bind to each ofthefive cis-actingsequencesof theenhancer, althoughprotein binding
tosites B, C,and Fis moreprominent and easilydetectable.
DNase I footprint analysis was done with both purified
HeLacell NF-1 and heparin-agarose-purified HeLa cell
pro-tein extract by using the wild-type DNA as the probe. Both
protein samples protected the NF-1-like sites, B and C (data not shown). The protection of sites B and C resembled that whichwas observedpreviously (21, 29).
Alterations of DNase I sensitivity upon mutation of
tran-scriptionfactorbindingsites. Itwasimportanttodemonstrate thatthe mutation ofaDNA motif thatalteredthe level ofgene
activityalso alteredthe ability ofproteinstobindtothat site. To showthis, DNase I footprint analysis wasperformed with
five different DNAprobes, each containing oneof the
single-site mutations. These analyses were repeated at least twice, and resultswere similar each time. The results ofone set of DNase I footprint analyses are shown in Fig. 5. Careful examination of the autoradiographs in Fig. 5 allowed us to identifyhow eachmutation affected the DNase Isensitivityat
the DNAsite(s). It isimportant tonote thatthe mutation at
site B included a 2-bp deletion, which causes a shift in the
DNase Idigestionpatternin lane B ofFig.5A.Thiswastaken
into account when the autoradiographs were visually
in-spected.
Nucleotides that are underlined below indicate those that arehypersensitivetoDNase Idigestion.Resultsof the DNase Ifootprint analysisshown inFig. 5A, usingtheLS 2 enhancer (mutationatsiteA)as aDNAprobe,showedhypersensitivities at site A(AGGGAGGAG), which areindicated by arrows 1 and 2.Results with theLS 4 enhancer(mutationatsiteB)used
as a DNA probe indicated a decrease in protection from
DNase IdigestionatsitesA, B, C,and Fshown inFig. SA,as
wellashypersensitivitiesatsites A(AGGGAGGAG,indicated
by arrows 1 and2), B (ATGGAATGCAGCCA, indicated by
arrow3),C(TGGGCAGCCAGCCAG,indicatedbyarrow5),
D (CCCCGCC, indicated byarrow6), andF (AAACTGGA CAAAGGCCATG, indicated by arrow 8) shown in Fig. 5. Interestingly, a novel hypersensitivity was generated at the perfect estrogen-responsive element (ERE) consensus site (CCAGTTAAACTGG), which overlaps site F (Fig. 1B). To test whether this hypersensitivity was due to opportunistic binding by a new protein, the estrogen-estrogen receptor complex,or achangein bindingofprotein(s) tothenearbyF site, the enhancer DNA probe from LS 4 was used with purified NF-1 in a DNase I footprint assay. Purified NF-1 causedhypersensitivity atthe ERE thatwasverysimilar to the hypersensitivity generated using Vero cell proteins (data not
shown). This result indicates that the hypersensitivity at the ERE is due to binding of an NF-1 family member to the neighboringFsite.
Decreasedprotection at sites B and Cwas observed when the LS 34 enhancer (mutation at site C) wasused as a DNA
probe (Fig. 5A). In addition, hypersensitivities were seen at two sites, B (ATGGAATGCAGCCA, indicated by arrow 3) and C (TGGGGCAGCCAGCCCA, indicated by arrow 4). When the pAD-CAT enhancer was used as a DNA probe, results demonstrated a hypersensitivity at site D, which is
shown in Fig. SB (CCCCGCC,indicatedbyarrow7). Results
MUT B WT B MUTC
0 0 co oo0o
rx CO et O0N03 r0 NCO' 0
_ N _NN -N
WTC MUT F
00 0o 000 0
NOC '3s0 N l:Oe 0
co co
WT F
0 o0
NOD v O
[image:10.612.317.557.80.241.2]_ CI
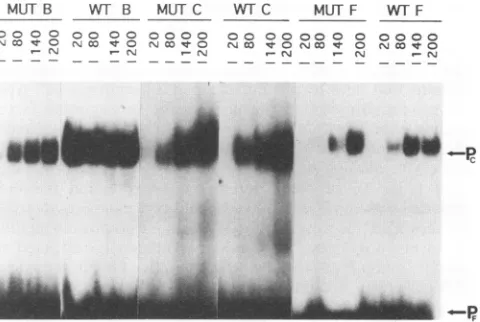
FIG. 6. Mobilityshiftanalysisformutantand
wild-type
B,C,and Foligonucleotides.MUT,mutant
oligonucleotide;
WT7,
wild-typeoligo-nucleotide for the respective sites. Each
oligonucleotide
was titrated with20, 80, 140,and 200 ng of Vero cell nuclearprotein. Pf,freeprobe; Pc,protein-bound
probe.Thespecific activityof the mutantCprobewassevenfoldhigherthan that ofthewild-typeCprobe;thiswastaken into accountafter densitometricscanningwas performed.
of assays
using
thepAF-CAT
enhancerindicatedadecrease inprotection
from DNase Idigestion
atsiteF,
asshown inFig.
5B.
Mutations,
inand ofthemselves,
maymodify
the patternof DNase Icleavage
of free DNA. To ensure that thechanges
observed were due to altered
protein binding,
the ladderproduced by
DNase Idigestion
of free LS 4 DNA wascompared
with thedigestion
pattern of the same DNA incu-bated with Vero cellprotein.
The samepatterns ofhypersen-sitivities and
protections
were observedonly
when Vero cellproteins
were included in the assay(data
notshown).
These data indicate that
proteins
binddifferently
to the mutated sitecompared
with thewild-type
site. Mutation of sitesA, D, and F affectsprotein binding
toeach mutatedsite. Incontrast, mutation of sites B and C affectsprotein binding
to the mutated siteaswellastoother DNAsites. Thispleiotropic
effect indicatessome
physical cooperativity
amongtheproteins
that bind tothe DNAsites in the BKV enhancer.
Does mutationcausealteredor new
protein
binding?
Wild-type and mutantoligonucleotides representing
the NF-1-like sites B,C,
and F(see
Materials andMethods)
were used inmobility
shiftexperiments
to observe whether the mutationalteredthe
affinity
ofproteins
for the DNA siteorwhetheranew
protein-DNA complex
formed with the mutated DNA site(Fig. 6).
Thespecific
activity
of the mutant Cprobe
wasconsistently
sevenfoldhigher
than that of the wild-type Cprobe,
so this was taken into account after densitometricscanning
wasperformed.
Afternormalizing
the specific activ-itiesof the sixprobes,
wefound thatVero cell nuclearproteins had a loweraffinity
for each of the mutantoligonucleotides
than for the wild-type
oligonucleotides.
At the concentration of 80 ng,proteins
had an 8-fold loweraffinity
for the mutant site B, a 13-fold loweraffinity
for the mutant site C, and a7-fold lower
affinity
for the mutant site F. We didnot observe mutantoligonucleotide-protein
complexes
that hadadifferentmobility
in thepolyacrylamide gel
matrix than thewild-type
oligonucleotide-protein
complexes,
which indicates that newproteins arenot
binding
tothe mutated DNAsites.on November 9, 2019 by guest
http://jvi.asm.org/
DISCUSSION
The PQ strain early enhancer was studied inthe context of Verocells to understand the interactionsoccurring among the proteins that bind to this enhancer in a permissive cell type. We generated mutations in many of the transcription factor binding motifs within the enhancer. The DNA sequences alteredincluded the NF-1-like sites E and G, the AP-1, AP-3, andc-Myc binding sites, and the cyclic AMP-responsive ele-ment (23). All of these mutant gene expression plasmids exhibited between 70 and 100% of wild-type gene activity (data notshown). Mutations in the c-Myb and NFKB protein binding sites were not made. Since c-Myb synthesis is restricted to hematopoieticcells (18) and NFKB is active only in the nuclei of Bcells (39), it is unlikely these two proteins play a role in early BKV transcription in Vero cells. In addition, we do not detectbinding of Vero nuclear proteins to these sites in DNase I footprint analysis.
Mutational analysis demonstrates that proteins binding to sites A, B, C, D, and F cooperatively contribute to early activity. In particular, the results reveal that B and C interact unfavorably in some contexts and favorably in others. In nature, a balance of synergistic and antagonistic interactions may make iteasier, through amplification and deletion events (42), to create new viruses that are more suitable for growth in new celltypes and altered environments. This may explain why both negative and positive interactions (Table 2) occur at the BKV early enhancer.
Thecooperativeinteractions of theNF-1-like sites, B and C. TheNF-1 family of proteins is conserved in higher vertebrates and found in many tissue types (1, 9, 17, 20, 35, 44). NF-1 proteins areimportantfor BKV early transcription in a variety of cell types (6, 15, 21, 29, 33). Results of DNase I footprint analysis using proteins from HeLa, 293, MK2, and retinoic acid-differentiated (glial and neural cells) embryonal carci-noma(EC) cells demonstrate protection of site B from DNase Idigestion (6, 15, 21, 29, 33). The only cell lines found that lack proteins that bind to the B motif are undifferentiated and dimethyl sulfoxide-differentiated (muscle) EC cells (33). The low ornonexistent transcriptional activity of the enhancer in these two celltypes is attributed to the lack of a B-site-specific transcriptional activator (33). The C motif is protected from DNase I digestion by using proteins from HeLa cells (15, 21, 29) and retinoicacid-differentiated EC cells (38). It is postu-lated that low enhancer activity in these cells is due to a transcriptional repressor that binds to site C (21). Conversely, nuclear proteins from two cell lines in which the BKV en-hancer is very active, 293 and MK2, do not protect site C from DNase Idigestion. Thus, it is proposed that proteins that bind tositeBactivatetranscription, while proteins that bind to site C represstranscription.
Incontrast, Vero cell nuclear proteins bind to sites B and C of the PQ enhancer, and yet the early enhancer is active and provides a selective advantage for growth in Vero cells. Mutation of either siteBorsite Cleads to an increase in CAT activity, which suggests that proteins binding to the two sites act as repressors. This is in contrast to results obtained for CV-1 cells, which show that the NF-1-like sites play a role in transcriptionalactivation (15). DNase I footprint analysis using Veroproteins shows that mutation of site C leads to altered DNase I sensitivity at both sites B and C.
Furthermore,
mutation of siteB leads to changes in the pattern of protein binding to all five DNA sites. Taken together, the functional andprotein binding results suggest that there exists cooperat-ivityamong theproteins that bind to the PQ enhancer in Vero cells.Model for the cooperative interactions that occur at the BKV enhancer. There may be many models that can be used to explain the functional and proteinbinding interactions occur-ring at the enhancer. One possible model is that mutation of site B alters the ability of proteins to bind to the other four sites but leads to new interactions among the proteins in the presence of a wild-type F site, so that enhancer function is maintained.
This model is supported by the mutation and competition analyses. Enhancer constructions that contain mutations at both sites B and F have background gene activity. These mutants include CD (10%) and ACD (16%). In comparison, the gene activities of enhancers, which contain a mutation at site B and not site F, are significantly higher. Examples of these mutants are CF (38%), ACF (69%), CDF (43%), ACDF (222%), and ADF (50%). These combinations of wild-type sites retain early gene activity (at least 30% of wild-type activity). With respect to the competition analysis, competitor enhancers that contain site B are
pComp-AB-CAT-
andpComp-BCD-CAT-.
The DNA sitesexpected toremain func-tional in the test plasmid after the competitions are CDF and AF, respectively. These two combinations of cis-acting sites, which include site F, both retain test gene activity (test gene activity is significantly greater than 36%, which was obtained by competition with the wild-type promoter-enhancer).The model proposes that new interactions compensate for the loss of protein binding and function for site B. Possible cooperative compensatory protein interactions include the ability of certain transcription factors to bend DNA to make contact between proteins bound to nearby sites. This has been shown for NF-KB (46) and Fos and Jun (24). The BKV enhancer has five functional sites, and one or more of the transcription factors that bind to this region may be inducing bends that promote certain protein-protein interactions over others. For instance, it is possible that F promotes specific protein interactions among the other proteins that the B-C pair can change when the binding of F to DNA is altered by mutation. DNA bending by proteins can be influenced by spacing (50). The insertion of 48 and 81 bp between sites B and C may cause altered DNA bending, which results in new interactions among the five proteins. This may lead to the dramatic decrease to basal level of activity (Fig.
4B).
Interactions at the BKV enhancer in relation to virus growth and evolution. BKV can reproduce itself efficiently in a permissive host. This environment allows for virus evolution. In other words, DNA rearrangements that spontaneously occur will be passed on to progeny virus if the new enhancer confers a selective advantage. Over time, a new virus strain can emerge.
The biological activities of the virus, which occur during the three stages of its life cycle, depend on the function of the BKV enhancer. The enhancer DNA sequences do not directly participate in virus replication by acting as binding sites for T antigen and/or host replication proteins (14). However, the enhancer regulates the level of T-antigen gene expression, and T antigen directly stimulates replication by binding to DNA sequences in the viral origin of replication (47). The late promoter is contained within the early enhancer, so the enhancer must be controlling the balance between early and late gene expression (5). It is likely that there is competition for transcription factors required for early and late gene expres-sion. Lastly, in many cases, there is a correlation between the level of early gene expression, which is controlled by the enhancer, and the level of transformation or cell lysis resulting from viral infection (55-58).
In fact, there are examples of two- to threefold differences in
on November 9, 2019 by guest
http://jvi.asm.org/
early gene activity that exhibit a significant difference in both BKV transformation and lytic potential. These changes are similar to the ones observed with the single mutations in the PQ enhancer seen in Fig. 2. Watanabe and Yoshiike (58) demonstrated that in nonpermissive rat cells, thePQstrain has approximately 2.5-fold-higher early gene activity than the Gardner strain of BKV.PQ-infected rat cells form foci and soft agarcolonies, while Gardner-infected cells do not. In addition, thesesametwo authors(57) showed that in permissive HEK cells, the pm527 strain of BKV has approximately 2.5-fold-lower early gene activity than the Gardner strain. In these same cells,thepm527strain forms minuteplaques, while the Gard-nerstrainforms large, clear plaques.
ThereareBKVstrains whose enhancer structures resemble the enhancer constructions that contain a single mutation. Clones 108 (48) and Dik(53) lack site B, DBd182 (51) lacks site D, and AS (52) lacks site F. It would be interesting to measure the levels of early gene activity and lytic ability of these strains in Vero cells and comparethem with PQ to test whether there are any predictable correlations between en-hancer structure and virus growth. Moreover, now that the multipleinteractions occurring at thePQenhancer are known, itispossible toisolate BKVdirectly from avariety of human tumorsand normal tissueto testwhether there isarelationship between enhancer structure and tissue specificity of virus growth.
Wecompared theDNA sequences oftheknown strains of BKV. The enhancer of the progenitor strain, WW (7, 31), containsone copyofeach ofthefive DNA sites. Rearrange-mentofthe enhancer(41, 42)generatesmorecopiesof sites A, B,andCand fewercopies of siteD.SiteFispresentas asingle copyin allof the viral enhancers (information is from visual observation of thegenomicsequencesofatleast 35 strainsof BKV).Inapproximatelyhalf of thestrains,thereisanunequal number of sites B and C. Two deductions can be proposed from these patterns. First, sites A and F are important for maintenance of general enhancer activity, since they are retained in virus strains that were selected for growth in a
varietyof hosts and tissue types (54).The resultspresentedin thisreportprovideevidence that interactionsatsites B and C are themostdynamic andhave effectson the function ofthe other three DNA sites, A, D, and F. Thus, it is
likely
that different ratios of sites B and Careimportanttogeneratethe range of enhancer activitiesrequired
forgrowth
in different celltypes.ACKNOWLEDGMENTS
WethankJenniferYucel,PeterGeiduschek,andDanielDonoghue for carefulreadingof themanuscript.We aregratefultoJoanne M. Yeakley for excellenthelpwith theRNaseprotectionanalysis.
This work was supported by a grant to S.S. from the Cancer ResearchCoordinatingCommittee of theUniversityof California.
REFERENCES
1. Amemiya, K., R. Traub, L. Durham, and E. 0. Major. 1989. Interaction ofanuclearfactor-i-like proteinwith the regulatory
region of the human polyomavirus JC virus. J. Biol. Chem. 264:7025-7032.
2. Berg, J. M. 1992. SP-1 and thesubfamilyofzincfinger proteins with guanine-rich binding sites. Proc. Natl. Acad. Sci. USA 89:11109-11110.
3. Brasier, A. R,J.E.Tate,andJ.F. Habener.1989.Optimizeduse
of thefirefly luciferaseassayas areporter gene in mammalian cell lines.BioTechniques7:1116-1122.
4. Briggs, M. R., J. T. Kadonaga, S. P. Bell, and R Tjian. 1986. Purification and biochemical characterization of the promoter-specifictranscriptionfactor, Spl.Science234:47-52.
5. Cassill, J. A., and S. Subramani. 1989. A naturally occurring deletion in the enhancerrepeatsofthe human papovavirus BK optimizes earlyenhancer functionattheexpenseof latepromoter activity. Virology170:296-298.
6. Chakraborty, I.,andG. C. Das. 1989. Identification of HeLa cell nuclearfactors that bindtoandactivatetheearlypromoterofBK invitro. Mol. Cell. Biol.9:3821-3828.
7. Chauhan, S., G. Lecatsas, and E. H. Harley. 1984. Genome analysis ofBK(WW)cloneddirectly from human urine. Intervi-rology22:170-176.
8. Chen,E.Y.,and P. H.Seeberg. 1985.Supercoil sequencing:afast and simple method for sequencing plasmidDNA. DNA 4:165-170.
9. Chodosh,L.A.,A.S.Baldwin,K. W.Carthew,and P. A.Sharp. 1988. Human CCAAT-binding proteins have heterologous sub-units. Cell 53:11-24.
10. deVries,E., W.vanDriel, S. J. L.vanderHeuvel, and P. C.van
derVliet. 1987. Contactpoint analysis ofHeLa NF-1 recognition site reveals symmetrical binding at oneside of the DNA helix. EMBO J.6:161-168.
11. de Wet, J. R, K. V.Wood, M.DeLuca, D. R. Helinski,and S. Subramani. 1987.Fireflyluciferasegene:structureandexpression inmammalian cells. Mol. Cell. Biol.7:725-737.
13. Deyerle,K.L., J.A.Cassill,andS. Subramani. 1987.Analysisof theearly regulatory regionof the humanpapovavirusBK. Virol-ogy 158:181-193.
14. Deyerle,K.L.,F.G.Sajjadi,andS. Subramani.1989.Analysisof origin ofDNA replication of humanpapovavirus BK. J. Virol. 63:356-365.
15. Deyerle,K.L.,and S. Subramani. 1988.Alinkerscananalysisof theearly regulatoryregionof the humanpapovavirusBK. J.Virol. 62:3378-3387.
16. Dignam, J. D.,RM.Lebovitz,and R G. Roeder.1983.Accurate transcriptioninitiationbyRNApolymeraseIIinasolubleextract
from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1480.
17. Dorn, A., J. Bollekens,A.Staub,C.Benoist,and D. Mathis. 1987. Amultiplicityof CCAATbox-binding proteins.Cell50:863-872. 18. Gabrielsen, 0. S., A.Sentenac,and P.Fromageot. 1991.Specific
DNAbinding by c-myb: evidence for a double helix-turn-helix-related motif. Science253:1140-1143.
19. Gidoni, D.,W. S. Dynan, and R. Tjian. 1984. Multiple specific
contactsbetween a mammaliantranscription factorandits
cog-natepromoters.Nature(London)312:409-413.
20. Goyal, N., J. Knox, and R. M. Gronostajski. 1990. Analysisof multipleforms ofnuclearfactor1in human andmurine cell lines. Mol. Cell. Biol. 10:1041-1048.
21. Grinnell, B. W., D. T. Berg, and J. D. Walls. 1988. Negative regulation of the human polyomavirus BK enhancer involves cell-specificinteractionwith anuclearrepressor. Mol. Cell. Biol. 8:3448-3457.
22. Johnson,P.F.,andS. L.McKnight. 1989. Eukaryotic transcrip-tionalregulatoryproteins. Annu. Rev.Biochem.58:799-839. 23. Jones, N. C.,P. W.J. Rigby,and E. B. Ziff. 1988. Trans-acting
protein factors and the regulation of eukaryotic transcription:
lessons fromstudies onDNAtumorviruses. Genes Dev. 2:267-281.
24. Kerppola,T.K.,andT.Curran. 1991. Fos-junheterodimersand junhomodimers bendDNAinoppositeorientations:implications fortranscriptionfactorcooperativity.Cell66:317-326.
25. Kingston,RE., C.A.Chen,and H.Okayama.1991. Transfection ofDNAintoeukaryotic cells,p.9.1.lf-9.1.9.InF. M.Ausubel,R. Brent,R. E.Kingston,D.D.Moore,J.G.Seidman,J. A.Smith, and K. Struhl (ed.),Currentprotocolsin molecularbiology,vol. 1. Greene Publishing Associates and Wiley-Interscience, New York.
26. Klemsz, M., S.McKercher, A. Celada, C. van Beveren, and R. Maki. 1990. The macrophage and B
cell-specific
transcription
factor PU.1 is relatedtotheetsoncogene.Cell61:113-124. 27. Kunkel,T.A.,J.D.Roberts,and R. A. Zakour.1987.
Rapid
andefficient site-specific mutagenesis without
phenotypic
selection. MethodsEnzymol. 154:367-382.28. Loyter,A.,G. A.Scangos,andF.H. Ruddle.1982.Mechanismsof