0022-538X/11/$12.00 doi:10.1128/JVI.01997-10
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Herpes Simplex Virus 2 MicroRNA miR-H6 Is a Novel Latency-Associated
Transcript-Associated MicroRNA, but Reduction of Its Expression Does
Not Influence the Establishment of Viral Latency or the
Recurrence Phenotype
䌤
Shuang Tang,
1Andrea S. Bertke,
1,2Amita Patel,
1Todd P. Margolis,
2and Philip R. Krause
1*
Division of Viral Products, Office of Vaccines Research and Review, Center for Biologics Evaluation and Research, Food and
Drug Administration, Bethesda, Maryland 20892,1and F. I. Proctor Foundation, University of California, San Francisco, California2
Received 20 September 2010/Accepted 7 February 2011
The herpes simplex virus 2 (HSV-2) viral microRNA (miRNA) designated miR-H6 is located upstream of the latency-associated transcript (LAT) promoter region on the strand opposite the LAT. Deletion of the LAT promoter and part of LAT exon 1 abolished HSV-2 miR-H6 expression in acutely and latently infected guinea pig dorsal root ganglia (DRG), suggesting that this region is needed both for the expression of
LAT-encoded miRNAs and for miR-H6 expressionin vivo. Relative to cells infected with a viral rescuant,
miR-H6 expression was significantly reduced in cells infected with a mutant HSV-2 virus, NotPolyA, with an insertion of a simian virus (SV40) polyadenylation signal sequence between the LAT promoter and miR-H6 sequences. In addition, expression of miR-H6, but not LAT or viral DNA, was significantly reduced in both mouse trigeminal ganglia (TG) and guinea pig DRG latently infected with the NotPolyA mutant. Guinea pigs infected with NotPolyA experienced reduced neurological complications of acute infection relative to those infected with the rescuant, but the recurrence phenotype of the NotPolyA mutant was similar to those of its rescuant and wild-type HSV-2, indicating that reduction of miR-H6 expression is not by itself able to alter the establishment of latency for the wild-type virus or the recurrence phenotype. Furthermore, the mutation in NotPolyA did not affect the propensity of wild-type HSV-2 to establish latency in neurons positive for subtype marker KH10. In contrast to published reports regarding its HSV-1 homolog, HSV-2 miR-H6 did not affect ICP4 expression in transfected or infected cells. We hypothesize that viral miRNAs associated with LAT expression are likely to work collectively, contributing to the phenotype attributed to the LAT.
Herpes simplex virus 1 (HSV-1) and HSV-2 are closely re-lated herpesviruses. HSV-1 typically infects the facial region and establishes a lifelong latent infection in sensory neurons of the trigeminal ganglion (TG), while HSV-2 typically infects the genital region and establishes a lifelong latent infection in sensory neurons of the sacral dorsal root ganglia (DRG). Pe-riodically, either virus may reactivate to cause symptomatic or asymptomatic recurrences in the area served by these sensory neurons. Both HSV-1 and HSV-2 have similar latent transcrip-tion patterns, in which the latency-associated transcript (LAT) is transcribed from within the genomic long repeats. In con-trast to other viral promoters, the LAT promoter is highly active during latency. Prior to discovery of virus-encoded microRNAs (miRNAs) (19, 25), LAT was the only viral gene product that was readily detectable during latency (18, 25). The LATs play an important role in HSV latency and reactivation. Deletion of the LAT promoter in both HSV-1 and HSV-2 reduces the efficiency of reactivation (10, 12, 16, 17, 22, 24, 27). The HSV-1 LAT is currently believed to act at least in part by increasing the establishment or maintenance of latency (16, 22), likely via an effect on the survival of acutely infected neurons (21). Animals infected with an HSV-1 LAT deletion
mutant virus are more likely to have apoptotic neurons during the acute infection (15, 23).
Recently, both HSV-1 and HSV-2 LATs have been found to be primary miRNA genes that encode multiple functional miRNAs (19, 20, 25, 26). The relative genomic locations of the miRNAs encoded within the LAT region are illustrated in Fig. 1. miRNAs are a family of 21- to 24-nucleotide (nt) noncoding RNAs that regulate gene expression based on sequence simi-larity to their target (1, 6, 7). For HSV-2, miR-I and miR-II, encoded by HSV-2 LAT on the strand opposite ICP34.5 exon 1 and its 5⬘ untranslated region (5⬘ UTR), efficiently silence ICP34.5, a major viral neurovirulence factor (19, 20). HSV-2 miR-I was abundantly expressed and detectable in every hu-man DRG tested (19, 26). HSV-2 miR-III is able to downregu-late ICP0 expression, a key viral transactivator (20). Recently, two more viral miRNAs encoded by the HSV-2 LAT region on the strand opposite ICP0 exon 2 and intron 1 were reported (9, 26). On the basis of similar sequence location (although not seed sequence homology) to HSV-1 miRNAs, some investiga-tors have applied the subsequently developed HSV-1 nomen-clature of miR-H4, miR-H3, and miR-H2 to HSV-2 miR-I, miR-II, and miR-III, respectively. To reduce confusion, for this article, we retain the original nomenclature used in the description and functional studies of the HSV-2 miRNAs, rec-ognizing that the miRNAs from different viral species may ultimately turn out to have different cellular targets.
* Corresponding author. Mailing address: FDA/CBER, HFM-457, 29 Lincoln Drive, Bethesda, MD 20892-4555. Phone: (301) 827-1914. Fax: (301) 496-1810. E-mail: [email protected].
䌤Published ahead of print on 16 February 2011.
4501
on November 7, 2019 by guest
http://jvi.asm.org/
On the basis of the homology between HSV-2 and HSV-1 in the region of ICP34.5, we predicted and confirmed HSV-1 miRNAs miR-LAT-ICP34.5 and a miR-I homolog encoded by HSV-1 LAT sequences, also named HSV-1 H4 and miR-H3, respectively (25). HSV-1 miR-H3 and -H4 are able to silence HSV-1 ICP34.5 (S. Tang et al., unpublished data). HSV-1 miR-H2 silences HSV-1 ICP0 (25). In general, miRNAs encoded by HSV-1 and HSV-2 LAT sequences are more conserved in location than in sequence, suggesting that major targets for the miRNAs are likely to be the genes on the strand opposite the miRNAs that are perfectly complementary to the miRNAs, and in cases where miRNA seed sequences are not conserved, that any cellular target sequences are more likely to differ between HSV-1 and HSV-2.
Two additional HSV-1 miRNAs, miR-H1 and miR-H6, have been reported upstream of the LAT promoter (5, 25). miR-H1 is encoded by sequences upstream of the LAT promoter in the LAT sense direction, while miR-H6 is upstream of the LAT promoter sequences in a LAT antisense direction. HSV-1 miR-H6 was reported to silence the major viral transactivator ICP4, suggesting a contribution to the establishment and main-tenance of viral latency (25). Recently, Jurak et al. reported the identification of an HSV-2 miRNA sequence in a genomic location similar to that of HSV-1 miR-H6 (9), designated HSV-2 miR-H6. Given the prominent location within the ge-nome and the potential roles of HSV-1 miR-H6 during latency and reactivation, we thought it important to study the expres-sion and biological function of HSV-2 miR-H6in vitroandin vivo. In this study, we show that the LAT promoter and part of the LAT exon 1 sequences, which are critical for HSV latency establishment and reactivation, are also crucial for miR-H6
expression in latently infected guinea pigsin vivo. A mutant virus with reduced miR-H6 expression was studied in infected-cell culture and in infected mice and guinea pigsin vivo. Po-tential effects of HSV-2 miR-H6 on the expression of ICP4 and ICP0 were also investigatedin vitro.
MATERIALS AND METHODS
Cells, viruses, and antibodies.HSV-2 strain HG52 (GenBank accession no. NC_001798) and HSV-1 strain 17syn⫹(GenBank accession no. NC_001806) genomic sequences were used as reference sequences. Vero, HEK 293, and U2OS cell lines were obtained from ATCC. HSV-2 strain 333 was obtained from Gary Hayward (Johns Hopkins University, MD). HSV-1 McKraedLAT371 and dLAT371R were obtained from Steven Wechsler (UCLA School of Medicine, Los Angeles, CA) (14). HSV2⌬R was a rescuant virus for HSV2⌬LAT. HSV2
⌬LAT mutant virus and⌬R rescuant virus have been previously described (10, 19). Briefly, HSV2⌬LAT contained a 624-bp NotI (nt 119109)-NotI (nt 119732) deletion in both the LAT promoter and adjacent 218 bp of LAT exon 1 region. The NotPolyA mutant virus was constructed previously by homologous recom-bination of pSph-Bam/NotPA (see below for details) and HSV-2 strain 333. The NotPolyA rescuant virus (designated NotPolyA-R) was made through recombi-nation of the NotPolyA mutant DNA and a pSph-Bam plasmid. All mutants and rescuant viruses were verified by sequencing and Southern blotting. Rabbit polyclonal anti-HSV-2 ICP4 antibody was raised against synthetic peptides cor-responding to both the N and C termini of HSV-2 ICP4 (AADGVVSPRQL ALLA and GGVEVVGTAAGLATP, respectively). Rabbit polyclonal anti-HSV-2 ICP0 antibody was raised against synthetic peptides corresponding to both the N and C termini of HSV-2 ICP0 (APPLRCQSFPCLHPFC and HAR NCVRPPDYPTPP, respectively). Anti--tubulin was obtained from BD Biosci-ence (San Jose, CA).
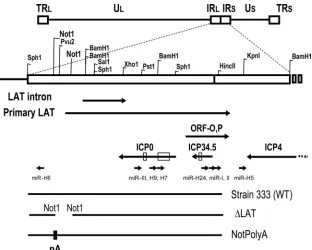
[image:2.585.136.448.67.317.2]Plasmids, oligonucleotide probes, and RNA oligonucleotides.To construct the HSV-2 miR-H6 expression clone, pmiR-H6, a PCR fragment from oST608 (5⬘-GCCTCTCTTTCCCGTTGCTTTC-3⬘) and oST609 (5⬘-CACAGACGAAC ACACGGTGGCGAT-3⬘) with HSV-2 333 DNA as the template was first cloned into a pCR4 TOPO vector (Invitrogen, CA) and then subcloned into a pFlag expression vector (Sigma, St. Louis, MO). To construct pSph-Bam/NotPA, a FIG. 1. Schematic diagram of herpes simplex virus 2 (HSV-2) latency-associated transcript (LAT) region and HSV-2 LAT-associated microRNAs (miRNAs). Enzymes used to create mutant viruses and plasmids are labeled. Potential transcripts, including LAT primary transcripts, ICP0, ICP34.5, ICP4, ORF-O,P, and HSV-2 mature miRNA sequences are also shown (9, 19, 20, 26). Abbreviations: TRL, terminal repeat long; IRL, internal repeat long; IRS, internal repeat short; US, unique short; TRS, terminal repeat short; WT, wild type; pA, polyadenylation site.
on November 7, 2019 by guest
http://jvi.asm.org/
192-bp bidirectional simian virus 40 (SV40) poly(A) (pA) signal was digested at the BamHI site from pNASSB and blunted. A subclone containing the HSV-2 LAT promoter and partial LAT exon 2 sequences from SphI to BamHI (nt 117963 to 120479) was partially digested with NotI and blunted. The blunted vector and insert were ligated to form pSph-Bam/NotPA, encompassing the HSV-2 region from nt 6771 to 8144, with the SV40 pA signal introduced at the NotI site (at nt 119109). NotPolyA was rescued with pSSB (19) to make a NotPolyA rescuant virus (NotPolyA-R). The plasmid was sequenced to confirm the direction and sequence of the SV40 poly(A) signal. RNA oligonucleotides corresponding to the sequences of HSV-2 miR-H6-5p and miR-H6-3p were synthe-sized (Dharmacon, CO) and annealed in 1⫻annealing buffer (Dharmacon, CO) to generate the miR-H6 duplex. The HSV-2 miR-H6-3p inhibitor, a 2⬘ -O-methyl-modified RNA oligonucleotide with a sequence completely complementary to HSV-2 miR-H6-3p, was also synthesized by Dharmacon. An HSV-2 miR-H6 lucif-erase reporter was constructed by insertion of two annealed oligonucleotides (5⬘-ct agaGACTCCCATCTTCTGCCCTTCCATCCTCCt-3⬘and 5⬘-ctagaAGGAGGAT GGAAGGGCAGAAGATGGGGAGTCt) with XbaI compatible overhangs (lowercase letters) into the XbaI site of the pGL3 promoter vector (Promega, WI). The miR-H6 luciferase reporter was confirmed by sequencing. HSV-2 pICP4, an HSV-2 ICP4 expression plasmid that contains the HSV-2 ICP4 open reading frame sequence and its adjacent 3⬘polyadenylation signal sequence in a pcDNA3 expres-sion vector, was obtained from Jeff Cohen and Kening Wang (NIH/NIAID) (19).
Transfection, infection, and Western blotting.Cultures of U2OS cells were transfected with 20 nM concentration of the nonspecific small interfering RNA (siRNA) (NS-siRNA) control (Dharmacon, CO), with synthetic HSV-2 miR-H6 with or without 40 nM miR-H6 inhibitor, or with 2g of pmiR-H6 or pFlag vector, respectively. At 16 h posttransfection, cells were infected with HSV-2. In the cotransfection experiment, 0.5g of HSV-2 pICP4 were cotransfected with 20 nM nonspecific siRNA, synthetic miR-H6 with or without miR-H6 inhibitor, or with 2 g of pmiR-H6. Total protein was extracted at 24 h posttransfection. Western blots were performed with cor-responding anti-HSV-2 ICP4, HSV-2 ICP0, or anti--tubulin antibodies on the same membrane after stripping.
Growth curve of HSV-2.One-step growth of HSV-2 in Vero cells was studied as previously described (10). Briefly, Vero cells were infected in duplicate with NotPolyA, NotPolyA-R, and HSV-2 strain 333 at a multiplicity of infection (MOI) of 0.1, and total virus was collected from cells at 0, 2, 5, 15, and 20 h postinfection (hpi). HSV-2 was quantified by plaque assay. One-step growth of HSV-2 in Vero cells pretransfected with miR-H6 or NS-siRNA was also studied. Briefly, Vero cells were transfected with 20 nM miR-H6 or NS-siRNA in dupli-cate 16 h before the cells were infected with HSV-2 strain 333 at a MOI of 0.1, and total virus was collected and quantified as described above.
Vaginal HSV infection of guinea pigs.Female Hartley guinea pigs (250 to 350 g) (Charles River, Wilmington, MA) were inoculated intravaginally with 2⫻105
PFU of HSV-2 strain 333, NotPolyA, or NotPolyA-R (a rescuant virus of NotPolyA), as described previously (3). Briefly, guinea pigs were monitored and scored daily during acute infection (14 days) and latent in-fection (42 days) for the severity of lesions around the external genitalia. The severity of the lesions were scored on a scale from 0 to 4 as follows: 0 for no disease, 1 for redness/swelling, 2 for one or two lesions, 3 for three to five lesions, and 4 for six or more lesions or coalescence of lesions. For analysis during latency, infected animals were sacrificed on day 42 postinoculation. Lumbosacral dorsal root ganglia (DRG) (L1 through S2) were collected from each animal immediately after sacrifice and snap-frozen on dry ice. DNA and RNA were extracted from ganglia in parallel using the Qiagen AllPrep DNA/RNA minikit (Valencia, CA) after homogenization with an Omni rotor-stator homogenizer (Omni International, Marietta, GA). 18S rRNA was used to normalize RNA loading. Animals were housed in American Association for Accreditation of Laboratory Animal Care-approved facilities and cared for in accordance with institutional guidelines.
Infection of mice with HSV-2.Six-week-old female Swiss Webster mice (Si-monsen Laboratories, Gilroy, CA) were anesthetized by intraperitoneal injection with sodium pentobarbital, followed by topical corneal administration of 0.5% proparacaine hydrochloride. Following corneal scarification, the eyes of the mice were inoculated with 10l of viral stock (NotPolyA, 4.5 ⫻107
PFU/ml; NotPolyA-R, 2⫻107 PFU/ml; HSV-2 strain 333, 5⫻106PFU/ml; HSV-1
McKraedLAT371, 106
PFU/ml;dLAT371R, 106
PFU/ml). Mice were treated with acyclovir starting at 40 h postinfection. The trigeminal ganglia (TG) were removed at 21 days after inoculation and snap-frozen. One mouse infected with NotPolyA died at 16 days postinfection and was excluded from further analysis. Total DNA and RNA were extracted from ganglia in parallel using the Qiagen AllPrep DNA/RNA minikit (Valencia, CA) after homogenization with an Omni rotor-stator homogenizer (Omni International, Marietta, GA). 18S rRNA in
these RNA samples was quantified by real-time PCR with a TaqMan ribosomal control kit (Applied Biosystems, CA) and used to normalize RNA loading.
Dual fluorescent staining of tissue sections for monoclonal antibodies A5 and KH10 and FISH for LAT.Dual fluorescent staining of frozen mouse TG tissue sections was carried out as previously described (13). Mouse monoclonal anti-bodies A5 and KH10 were obtained from Developmental Studies Hybridoma Bank (Iowa City, IA). Fluorescencein situhybridization (FISH) for HSV-2 LAT was performed as previously described (13). Labeled riboprobe for the HSV-2 stable LAT intron was prepared using a 782-bp (StuI-XhoI) fragment of pBam/ Xho4Z as a template.
Detection of HSV-2 miR-H6 by real-time PCR.Total RNAs from infected or transfected cells were prepared with Trizol (Invitrogen, CA). Fifty or 100 ng of total RNA was used in each miRNA reverse transcription (RT) reaction mixture. HSV-2 miR-H6 was reverse transcribed with 1 nM reverse transcriptase primer (oST595, 5⬘-GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGAT ACGACGATGGA-3⬘) and then detected with 1.2M forward primer (oST594 forward primer, 5⬘-CACTGGCCCATCTTCTGCCCTT-3⬘), 0.6 M reverse primer (oST408, 5⬘-GTGCAGGGTCCGAGGT-3⬘), and 0.1M TaqMan probe (oST596, 6-FAM-TGGATACGACGATGGAA-MGB where 6-FAM stands for 6-carboxyfluorescein [FAM] and MGB stands for dihydrocyclopyrroloindole tri-peptide minor groove binder). The real-time PCRs were performed with an ABI 7900 real-time thermal cycler (Applied Biosystems, CA).
Detection of HSV-2 LAT and DNA copies by real-time PCR.HSV-2 LAT and viral DNA copy numbers were quantified by real-time PCR as previously de-scribed (18).
RESULTS
HSV-2 miR-H6 is not detectable in DRGs of guinea pigs latently or acutely infected with a LAT promoter deletion
mu-tant.Previously described LAT-encoded miRNAs are
detect-ablein vivoin the DRG or TG of infected animals or humans.
In vivoexpression of miR-I, -II and -III is dependent on the
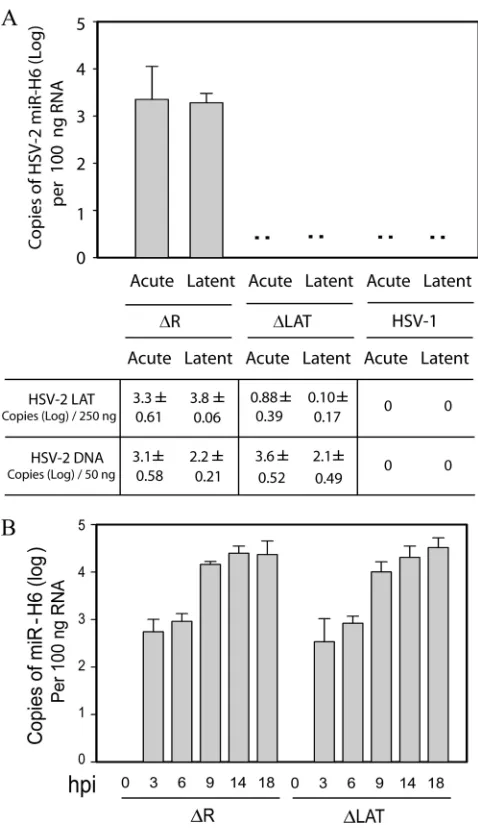
LAT promoter (19, 20). To determine whether HSV-2 miR-H6, which is located upstream of the LAT promoter on the strand opposite the LAT (and opposite miR-I, -II, and -III), is detectablein vivoand transcriptionally controlled by LAT re-gion sequencesin vivo, we infected guinea pigs with⌬LAT, a reactivation-deficient mutant virus with a 624-bp deletion in the promoter and exon 1 of the HSV-2 LAT, or its rescuant virus,⌬R, which has a wild-type recurrence and LAT expres-sion phenotype (10) (Fig. 2A). Otherwise identically treated HSV-1-infected animals were included as negative controls. Total RNA and DNA were extracted from DRG harvested at 8 days postinoculation (acute) or 28 days postinoculation (la-tent). miR-H6 was detectable in all ganglia from both acutely and latently infected guinea pigs infected with⌬R (Fig. 2A). However, miR-H6 expression was not detected in the ganglia acutely or latently infected with HSV-1 or with⌬LAT. Quan-tification of LAT and viral DNA showed a dramatic reduction of LAT in DRG acutely or latently infected with⌬LAT com-pared to the rescuant or⌬R. However, as previously reported (10), viral DNA levels were not significantly different in the groups infected with⌬LAT or⌬R. This suggests that the LAT promoter region and the first 230 bp of LAT exon 1 are im-portant for miR-H6 expressionin vivo.
In HSV-2-infected cell culture, HSV-2 miR-H6 is expressed
independently of the LAT promoter and the 5ⴕ end of LAT
exon 1.HSV-2 miR-H6 was detected as early as 3 h hpi in
infected Vero cells (Fig. 2B). However, miR-H6 expression did not reach its peak until at least 14 h into the infection. Al-though miR-H6 expression was not detected in⌬LAT-infected guinea pigs, no difference in miR-H6 expression was observed in cell cultures infected with⌬LAT or⌬R (Fig. 2B). This result
on November 7, 2019 by guest
http://jvi.asm.org/
is consistent with miR-H6 expression in cell culture via read-through transcription from an upstream promoter, suggesting that the promoter for miR-H6 expression in infected-cell cul-turesin vitromay be different from the promoter responsible for miR-H6 expression in guinea pig DRGin vivo.
The HSV-2 viral mutant NotPolyA expresses much less miR-H6 in infected-cell cultures but retains growth properties
similar to those of rescuant and wild-type viruses.To study the
biological function of miR-H6, we used a mutant HSV-2 virus, NotPolyA, in which a SV40 polyadenylation signal was inserted into the NotI site (nt 119109) upstream of the LAT promoter sequences. Transcription is expected to be largely terminated after RNA polymerase II passes the polyadenylation signal. Thus, miR-H6 expression is expected to be sharply reduced in the NotPolyA mutant virus. As shown in Fig. 3A, in infected Vero cell culture, HSV-2 miR-H6 was reduced approximately 100-fold in NotPolyA-infected Vero cells relative to NotPolyA-R, the corresponding rescuant virus.
To determine whether downregulation of miR-H6 af-fected the in vitrogrowth properties of HSV-2, a one-step growth curve experiment was conducted in Vero cells. HSV-1 miR-H6 has been reported to reduce the expression of HSV-1 ICP4 (25). If HSV-2 miR-H6 targets HSV-2 ICP4, the NotPolyA mutant might be expected to display differ-ences in growth relative to wild-type or rescuant virus, be-cause ICP4 is critical for viral replication. However, we found that the growth of NotPolyA in Vero cells was similar to that of NotPolyA-R and wild-type HSV-2 (Fig. 3B), sug-gesting that downregulation of miR-H6 does not affect viral replication in infected-cell cultures.
miR-H6 expression is dramatically reduced in mouse TG
latently infected with the HSV-2 mutant NotPolyA.To
deter-mine whether miR-H6 expression is reduced in NotPolyA-infected animalsin vivo, we infected mice via the ocular route with NotPolyA, NotPolyA-R, or HSV-2 strain 333. HSV-1 McKraedLAT371 and HSV-1dLAT371R (14) were also used to infect mice as negative controls (Fig. 4A). TG were ex-tracted at 21 days postinfection and pooled with other TG from mice infected with the same virus. miR-H6 was detected in RNA extracted from the TG latently infected with both wild-type HSV-2 and NotPolyA-R. miR-H6 expression in NotPolyA-infected TG was at the assay background level (as assessed using uninfected and HSV-1 McKraedLAT371 and HSV-1dLAT371R-infected TG). The quantities of LAT RNA and HSV-2 DNA as assessed by TaqMan PCR were similar among TG infected with NotPolyA, NotPolyA-R, and HSV-2 strain 333.
The HSV-2 mutant NotPolyA has the wild-type HSV-2 phe-notype of preferential establishment of latency in
KH10-ex-pressing neurons.Latent infection with HSV-1 and HSV-2, as
[image:4.585.42.281.67.481.2]detected by LAT expression, occurs preferentially in different sensory neuron subtypes, as assayed by neuronal staining with monoclonal antibodies to neuron subtype-specific marker A5 or KH10 (13). Latent HSV-1 is associated with neurons ex-pressing A5, while latent HSV-2 is more frequently observed in neurons expressing KH10. This phenotype is dependent upon the virus species-specific LAT sequences present in HSV-1 and HSV-2 (3, 13). On the basis of mutant virus studies, LAT exon 1 appears to play an important role in this virus species-specific preference. To investigate any potential effect of miR-H6 on this phenotype, neuronal subpopulations that harbored latent virus were identified by dual fluorescencein situhybridization for the LAT and immunohistochemistry for the neuronal sub-type markers A5 and KH10 in trigeminal ganglia from mice latently infected with HSV-2 strain 333, NotPolyA, and
FIG. 2. The LAT promoter and the first 220 bp of LAT exon I region are required for efficient expression of miR-H6 in both acutely and latently infected guinea pig dorsal root ganglia (DRG), but not in infected culture. (A) HSV-2 miR-H6 is highly expressed in guinea pig ganglia latently or acutely infected with rescuant virus (equivalent to wild-type HSV-2), but not with LAT promoter deletion mutant. Total RNA was prepared from guinea pig DRG acutely or latently infected with⌬LAT, HSV-2⌬R, or HSV-1 17syn⫹(n⫽3 per group). HSV-2 miR-H6-specific real-time PCR was used to detect HSV-2 miR-H6 and HSV-1 viral DNA, and LAT copies were measured with HSV-1-spe-cific primers and TaqMan probes. HSV-2 miR-H6 is not detectable in the DRG from animals latently or acutely infected with⌬LAT or HSV-1 strain 17. The copy numbers of LAT RNA and virus DNA (on a log scale) are shown below the graph. (B)Time course of HSV-2 miR-H6 expression in LAT mutant virus- and wild-type virus-infected cells. Vero cells were infected in triplicate with⌬LAT or ⌬R (the rescuant virus of⌬LAT) at a MOI of 2. Total RNA was prepared at 0, 3, 6, 9, 14, and 18 hpi. Fifty nanograms of total RNA was used for miR-H6 specific real-time PCR at each time point for each virus.
on November 7, 2019 by guest
http://jvi.asm.org/
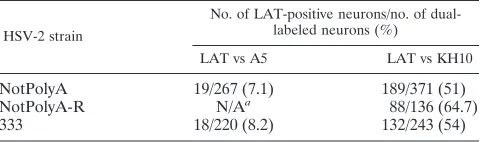
NotPolyA-R (Table 1). The NotPolyA virus was more fre-quently observed in KH10-positive cells, as was the case with wild-type HSV-2 and its rescuant. There was no observed in-crease in detection of NotPolyA in A5-positive cells relative to wild-type HSV-2, unlike the previously described results for HSV-2 chimeric viruses that include HSV-1 LAT sequences. Because the mutation in the NotPolyA virus did not influence this phenotype, these results indicate that miR-H6 is not re-sponsible for the virus species-specific neuronal subtype phe-notype.
miR-H6 expression is dramatically reduced in guinea pig
DRG latently infected with NotPolyA.To determine whether
HSV-2 miR-H6 was also downregulated in DRG infected with NotPolyA, the viruses were evaluated in the guinea pig genital model. Female guinea pigs were inoculated intravaginally with NotPolyA (n⫽ 7) or NotPolyA-R (n⫽6). miR-H6 was
ex-pressed at considerably lower levels in guinea pig DRG latently infected with NotPolyA compared to NotPolyA-R (Fig. 4B). Only one guinea pig latently infected with NotPolyA was pos-itive for miR-H6 at a low level, while the other six guinea pigs were miR-H6 negative. HSV-2 LAT and viral DNA copy num-bers were not significantly different in the two groups, suggest-ing that NotPolyA was able to normally establish latency in the infected DRG.
The HSV-2 mutant NotPolyA gives rise to less severe acute neurological symptoms, but its recurrence phenotype is
simi-lar to those of NotPolyA-R and wild-type HSV-2.To determine
whether HSV-2 miR-H6 contributes elements essential for viral species-specific reactivation of HSV-2, the viruses were evaluated in the guinea pig genital model. The severity of lesions was compared during the acute phase of infection through day 14 postinoculation, and recurrences were
enumer-FIG. 3. An HSV-2 mutant virus, NotPolyA, expresses a significant lower level of miR-H6 in infected Vero cells but maintains wild-typein vitro
[image:5.585.133.450.70.193.2]viral replication characteristics. (A) Expression of miR-H6 is dramatically reduced in the NotPolyA mutant in infected Vero cells. Vero cells were infected with NotPolyA or NotPolyA-R at a MOI of 1 in triplicate. At 6 hpi, total RNA was extracted, and miR-H6 expression was quantified by TaqMan PCR. 18S rRNA was used to normalize RNA loading. (B) The one-step growth curve of NotPolyA mutant was similar to the curves of the rescuant virus NotPolyA-R and the wild-type virus HSV-2 strain 333. Vero cells were infected with NotPolyA, NotPolyA-R, and HSV-2 strain 333 at a MOI of 0.1. Cells were collected, and the titer of the virus was determined by plaque assay at 0, 2, 5, 15, and 20 hpi.
FIG. 4. HSV-2 miR-H6 expression was significantly reduced in mouse trigeminal ganglia (TG) or guinea pig DRG latently infected with the NotPolyA mutant. (A) miR-H6 in mouse TG latently infected with the NotPolyA mutant was at the assay background level. Mouse TG infected with NotPolyA (n⫽10), its rescuant NotPolyA-R (n⫽8), HSV-2 strain 333 (n⫽10), HSV-1 McKrae mutant straindLAT371 (n⫽10), and its rescuantdLAT371R (n⫽10) were extracted and pooled after 21 days postinoculation. Total RNA and DNA were prepared from these pooled TG. Mice infected with HSV-1 McKraedLAT371 anddLAT371R were used as negative controls. HSV-2 LAT and HSV-2 viral DNA were not detected in the TG infected with HSV-1 McKraedLAT371 anddLAT371R. HSV-2 miR-H6 expression in the NotPolyA mutant is comparable to HSV-1 McKraedLAT371 anddLAT371R (background level). (B) HSV-2 miR-H6 expression was significantly lower in the guinea pig DRG latently infected with NotPolyA mutant (n⫽7) relative to its rescuant, NotPolyA-R (n⫽6). Total RNA and DNA were prepared from the DRG at 28 days postinoculation. miR-H6, LAT, and HSV-2 DNAs were quantified by TaqMan PCR. In panels A and B, 100 ng of total RNA, 250 ng of total RNA, and 50 ng of total DNA were applied to TaqMan PCR for miR-H6, LAT, and viral DNA, respectively.
on November 7, 2019 by guest
http://jvi.asm.org/
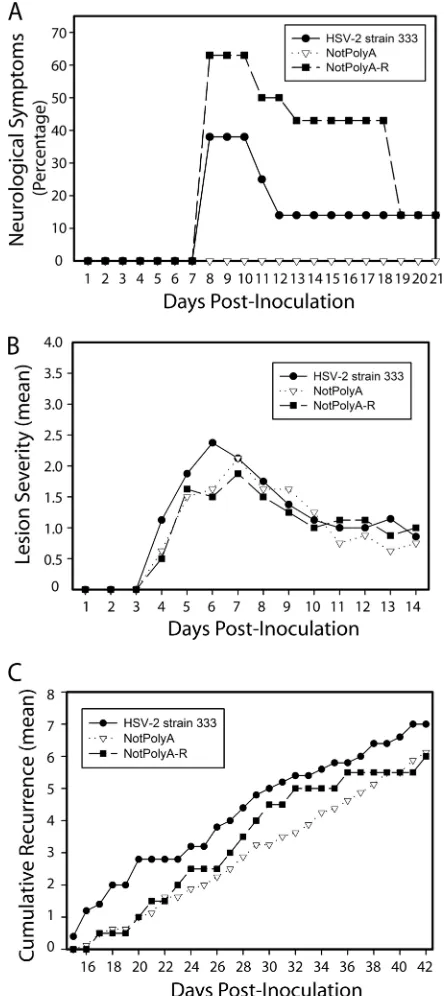
[image:5.585.114.472.478.635.2]ated during the latent infection through day 42 postinfection (Fig. 5). NotPolyA-infected guinea pigs experienced no observ-able neurological symptoms, with no paralysis or urinary tract infection (a marker for bladder paralysis), in contrast to the NotPolyA-R- and HSV-2 333-infected groups, in which 3 of 8 and 5 of 8 guinea pigs had observable neurological symptoms, respectively, suggesting that the mutation in NotPolyA caused viral neuroattenuation (Fig. 5A). However, the mean lesion severity in HSV-2 strain 333-, NotPolyA-, and NotPolyA-R-infected guinea pigs during acute infection was similar (Fig. 5B). Furthermore, the frequency of recurrences in the NotPolyA group was comparable to those in the NotPolyA-R and the wild-type HSV-2 groups during latent infection (Fig. 5C), suggesting that the reduced expression of miR-H6 in NotPolyA-infected guinea pigs does not significantly affect the recurrence phenotype of NotPolyA.
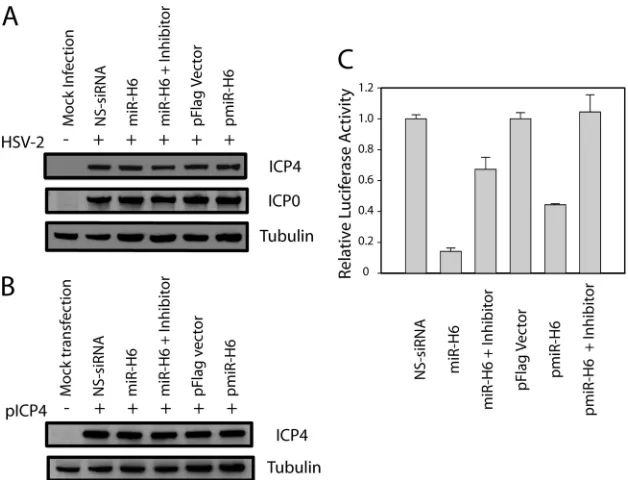
HSV-2 miR-H6 has no detectable effect on the expression of
ICP4. To determine whether HSV-2 miR-H6 downregulates
ICP4 expression, synthetic HSV-2 miR-H6 or a miR-H6 ex-pression plasmid was transfected into U2OS cells 16 h before infection with HSV-2 (Fig. 6A). Neither synthetic miR-H6 nor miR-H6 expressed from the plasmid pmiR-H6 was able to reduce ICP4 or ICP0 expression, as measured 6 h after inoc-ulation of virus. The ability of pmiR-H6 to express miR-H6 was confirmed by Northern hybridization (data not shown). Simi-larly, neither synthetic HSV-2 miR-H6 nor miR-H6 expressed from pmiR-H6 significantly reduced ICP4 expression when either HSV-2 miR-H6 duplex or miR-H6 expression plasmid was cotransfected with an HSV-2 ICP4 expression plasmid (pICP4) (Fig. 6B). The miR-H6 duplex and pmiR-H6 used in these experiments were capable of specifically silencing the miR-H6 firefly luciferase reporter (Fig. 6C). Similarly, pre-transfection of Vero cells with miR-H6 or NS-siRNA did not alter the ability of HSV-2 (inoculated at a MOI of 0.1) to replicate in a one-step growth experiment (data not shown). These findings are consistent with the absence of effect of the NotPolyA mutation on viral growth in cell culture at a low multiplicity of infection (Fig. 3B).
DISCUSSION
The mechanism by which LAT sequences influence estab-lishment of and reactivation from latency is not well under-stood. Previous studies indicated that HSV LAT-encoded miRNAs could target ICP34.5, ICP0, and ICP4 in in vitro
assays (19, 20, 25). Here we studied a recently identified miRNA, HSV-2 miR-H6, encoded by the sequences upstream of the HSV-2 LAT promoter in an antisense direction relative
to the LAT. miR-H6 was detected in guinea pig DRG latently and acutely infected with HSV-2 and in mouse TG latently infected with HSV-2. Deletion of the HSV-2 LAT promoter not only abolished the LAT-encoded miRNAs, as previously found (19), but also abolished miR-H6 expressionin vivo(Fig. 2), initially suggesting that miR-H6 might play a role in phe-notypes previously attributed to the LAT, based on a study of LAT promoter deletion mutants. A mutant virus with SV40 polyadenylation signal sequences inserted upstream of miR-H6 had significantly reduced expression of miR-H6 in infected-cell cultures and infected guinea pigs and mice. How-ever, the recurrence phenotype of the NotPolyA mutant was similar to those of its rescuant and the wild-type virus, indicat-ing that downregulation of miR-H6 is not able to change the wild-type recurrence phenotype.
Although HSV-1 and HSV-2 are different viral species, the viruses share a similar genomic organization and similar se-quences in many regions. Viral miRNAs from HSV-1 and HSV-2 are generally conserved in location, although there are significant sequence differences, including in the critical seed sequences. Both viruses express miRNAs targeting ICP34.5 (HSV-2 miR-I and miR-II and HSV-1 miR-H4 and miR-H3, which have some sequence homology) and a miRNA targeting ICP0 (HSV-2 miR-III and HSV-1 miR-H2, which are nonho-mologous but similarly located). HSV-1 miR-H6 and HSV-2 miR-H6 do not possess sufficient sequence homology to have permitted prediction of the HSV-2 miR-H6 sequence based solely on the HSV-1 miR-H6 sequence. One interesting addi-tional difference between HSV-1 and HSV-2 miRNA expres-sion is that the relative abundance of each miRNA is different for each virus. In HSV-2, miR-I is the most abundant latently expressed miRNA, while in HSV-1, miR-H2 is the most abun-dant. The levels of viral miRNAs likely affect the extent of their function, suggesting that HSV-2 may use its miRNAs primarily to target ICP34.5, while for HSV-1, it may be relatively more important to target ICP0.
The HSV-1 LAT exon 1 region is critical for efficient reac-tivation of HSV-1in vivo(4, 8). The 5⬘end of LAT exon 1 is significantly enriched in acetyl-histone H3 (K9, K14) (11), which is often associated with active promoters. Recently, Bertke et al. reported that the 5⬘LAT exon region is important for HSV viral species-specific phenotypes (2, 3). In the present study, a deletion encompassing the first 238 bp of LAT exon 1 and the LAT promoter abolished HSV-2 miR-H6 expression
in vivo, suggesting that the LAT promoter and part of the LAT
exon 1 region are critical for miR-H6 transcription and likely contain the promoter and transcription initiation site for the miR-H6 primary transcript. Deletion of the LAT promoter and partial LAT exon 1 region did not significantly affect miR-H6 expression in infected nonneuronal cell cultures. We previ-ously reported that LAT-encoded miRNAs, including miR-I, -II, and -III could be detected in⌬LAT-infected cell cultures but not in⌬LAT-infected guinea pigs (19, 20). These results suggest that both the LAT sense-strand miRNAs and miR-H6 promoter are expressed differently in infected DRG in vivo
than in infected epithelial cell culturesin vitro. These results further imply that the function of the LAT-associated miRNAs
in vivomight be quite difficult to evaluatein vitroin
infected-cell cultures.
[image:6.585.43.284.91.162.2]The functions of many LAT-encoded miRNAs are likely
TABLE 1. HSV-2 NotPolyA strain preferentially establishes latency in KH10-expressing sensory neurons
HSV-2 strain
No. of LAT-positive neurons/no. of dual-labeled neurons (%)
LAT vs A5 LAT vs KH10
NotPolyA 19/267 (7.1) 189/371 (51)
NotPolyA-R N/Aa 88/136 (64.7)
333 18/220 (8.2) 132/243 (54)
aN/A, not available. Experiments for detection of LAT expression in
A5-expressing cells in NotpolyA-R-infected mice were not performed.
on November 7, 2019 by guest
http://jvi.asm.org/
conserved. For example, miR-I and -II and their homologs in HSV-1 silence ICP34.5 efficiently (17; also unpublished data), and miR-III and its HSV-1 homolog, miR-H2, are suggested to target ICP0 (20, 25). However, HSV-2 miR-H6 and HSV-1 miR-H6 may have different functions. Umbach et al. reported that HSV-1 miR-H6 could target HSV-1 ICP4 in anin vitro
experiment (25). The seed sequence (2 to 8 nt) between HSV-1 miR-H6 and HSV-2 miR-H6 is quite different. In addition, HSV-1 and HSV-2 ICP4 sequences share less than 90% ho-mology. These sequence differences imply that if miR-H6 tar-gets ICP4 in both HSV-1 and HSV-2, both the miRNA seed sequence and the ICP4 target sequence would likely be differ-ent for each virus. Although Jurak et al. predicted two target sites on ICP4 for HSV-2 miR-H6 (9), we were not able to observe a significant downregulation of ICP4 by HSV-2 miR-H6 through cotransfection of ICP4 expression plasmid and miR-H6 or pretransfection of miR-H6 following infection with HSV-2.
Because the NotPolyA mutant virus had a normal recur-rence phenotype and displayed the wild-type HSV-2 pheno-type of preferential establishment of latency in KH10-positive neurons, it seems unlikely that HSV-2 miR-H6 plays a critical role in viral latency and reactivation. A potential role for miR-H6 in the observed neuroattenuation of the NotPolyA virus (Fig. 5) may still be considered, although the mechanism underlying this observation is unclear. HSV-2 miR-H6 might influence viral targets other than ICP4 and ICP0, cellular tar-gets, or could affect ICP4 expression in vivo(although noin
vitroeffect could be identified). Although the mutation in the
NotPolyA virus nearly eliminated latent miR-H6 expression (Fig. 4), we observed low-level miR-H6 expression in infected-cell cultures, likely due to incomplete polyadenylation by HSV during lytic infection (Fig. 3). Thus, we cannot rule out the possibility that trace amounts of miR-H6 could be expressed during viral replication during recurrence from infected ani-mals. We speculate that the very low level of miR-H6 in one of seven guinea pig DRG latently infected with NotPolyA may have been due to recurrence on the day when the ganglia were prepared (Fig. 4B). However, the residual miR-H6 expressed by NotPolyA was at levels approximately 2 log units below that expressed by the rescuant virus (Fig. 3A), and thus significantly below the physiological level.
[image:7.585.50.274.78.576.2]Although LAT-associated miRNAs likely contribute to the phenotype attributed to the LAT, little is known about the extent of their contribution or the role of each individual miRNA. The present study is the first to study the recurrence phenotype of a virus with a mutation reducing expression of a miRNA without reducing LAT expression (i.e., the NotPolyA virus).⌬LAT, a mutant in which the LAT promoter and part of the LAT exon 1 sequence has been deleted, has a phenotype of greatly reduced recurrence frequency in the guinea pig model. However, in infected guinea pigs and mice, the mutation in
FIG. 5. In guinea pigs, NotPolyA displayed similar acute lesion severity and recurrence frequency but was neuroattenuated relative to NotPolyA-R and HSV-2 strain 333. (A) Percentage of guinea pigs displaying signs of hind-limb paralysis or weakness on days 1 to 21 postinoculation. One guinea pig infected with HSV-2 strain 333 and one infected with NotPolyA-R were euthanized during this time pe-riod. One guinea pig infected with HSV-2 strain 333 and one infected with NotPolyA-R displayed weakness until day 33. All other animals cleared signs of paralysis and weakness by day 18. (B) Acute lesion severity in acutely infected female guinea pigs. Lesion severity is graphed as the mean for each group of guinea pigs on each day of observation from days 1 to 14 postinoculation, with 0 being no symp-toms and 4 being the most severe. The number of guinea pigs in the different groups was as follows: eight guinea pigs in the NotPolyA group, seven in the NotPoly-R group, and eight in the HSV-2 strain 333 group. (C) Cumulative recurrences per guinea pig for each group,
adjusted for the number of days of observation. The number of guinea pigs in the different groups was as follows: seven guinea pigs in the NotPolyA group, seven in the NotPolyA-R group, and eight in the HSV-2 strain 333 group. Guinea pigs that did not survive acute disease were excluded.
on November 7, 2019 by guest
http://jvi.asm.org/
⌬LAT substantially reduced expression of all of the LAT-encoded miRNAs and the upstream miR-H6. Thus, the phe-notype of⌬LAT cannot be directly attributed to an effect on miR-H6 expression but could be a collective effect of all of the HSV-2 LAT-associated miRNAs. Further in vivo studies of HSV gene-encoded miRNAs are needed to elucidate the mo-lecular functions of LAT-associated miRNAs and LAT.
ACKNOWLEDGMENTS
We thank Christine Uhlenhaut and Shasta McClenahan for critical reading. We also thank Jeff Cohen and Kening Wang of the National Institutes of Allergy and Infectious Diseases for kindly providing HSV-2 pICP4.
This study was supported by the intramural research program of the Center for Biologics Evaluation and Research and the Littlefield Trust.
REFERENCES
1.Bartel, D. P., and C. Z. Chen.2004. Micromanagers of gene expression: the potentially widespread influence of metazoan microRNAs. Nat. Rev. Genet. 5:396–400.
2.Bertke, A. S., et al.2009. Latency-associated transcript (LAT) exon 1 con-trols herpes simplex virus species-specific phenotypes: reactivation in the guinea pig genital model and neuron subtype-specific latent expression of LAT. J. Virol.83:10007–10015.
3.Bertke, A. S., A. Patel, and P. R. Krause.2007. Herpes simplex virus latency-associated transcript sequence downstream of the promoter influences type-specific reactivation and viral neurotropism. J. Virol.81:6605–6613. 4.Bloom, D. C., et al.1996. A 348-base-pair region in the latency-associated
transcript facilitates herpes simplex virus type 1 reactivation. J. Virol.70: 2449–2459.
5.Cui, C., et al.2006. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J. Virol.80:5499–5508.
6.Cullen, B. R.2006. Viruses and microRNAs. Nat. Genet.38(Suppl.):S25– S30.
7.He, L., and G. J. Hannon.2004. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet.5:522–531.
8.Hill, J. M., et al.1996. Quantitation of herpes simplex virus type 1 DNA and latency-associated transcripts in rabbit trigeminal ganglia demonstrates a stable reservoir of viral nucleic acids during latency. J. Virol.70:3137–3141. 9.Jurak, I., et al.2010. Numerous conserved and divergent microRNAs
ex-pressed by herpes simplex viruses 1 and 2. J. Virol.84:4659–4672. 10.Krause, P. R., et al.1995. Expression of the herpes simplex virus type 2
latency-associated transcript enhances spontaneous reactivation of genital herpes in latently infected guinea pigs. J. Exp. Med.181:297–306. 11.Kubat, N. J., A. L. Amelio, N. V. Giordani, and D. C. Bloom.2004. The
herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J. Vi-rol.78:12508–12518.
12.Leib, D. A., et al.1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol.63:2893–2900.
13.Margolis, T. P., Y. Imai, L. Yang, V. Vallas, and P. R. Krause.2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different pop-ulation of ganglionic neurons than HSV-1: role of latency-associated tran-scripts. J. Virol.81:1872–1878.
14.Perng, G. C., H. Ghiasi, S. M. Slanina, A. B. Nesburn, and S. L. Wechsler. 1996. The spontaneous reactivation function of the herpes simplex virus type 1 LAT gene resides completely within the first 1.5 kilobases of the 8.3-kilobase primary transcript. J. Virol.70:976–984.
[image:8.585.134.448.68.308.2]15.Perng, G. C., et al.2000. Virus-induced neuronal apoptosis blocked by the herpes simplex virus latency-associated transcript. Science287:1500–1503. 16.Sawtell, N. M., and R. L. Thompson.1992. Herpes simplex virus type 1 FIG. 6. HSV-2 miR-H6 had minimal effect on HSV-2 ICP4 expression. (A) HSV-2 miR-H6 did not inhibit HSV-2 ICP4 expression in cells infected with HSV-2. U2OS cells were transfected with 20 nM synthetic HSV-2 miR-H6 with or without 40 nM miR-H6-3p-specific inhibitor or 2g of pmiR-H6 (a miR-H6 expression plasmid) before infection with HSV-2 (⫹) at a MOI of 2. Twenty nanomolar nonspecific (NS) siRNA and 2g of pFlag vector were also transfected and used as negative controls, respectively. Total protein was extracted at 6 hpi and separated on an SDS-polyacrylamide gel before transfer to a membrane and incubation with an HSV-2 ICP4-specific antibody. The same membrane was stripped and incubated with an HSV-2 ICP0-specific antibody and an anti--tubulin antibody as internal controls. (B) HSV-2 miR-H6 did not inhibit HSV-2 ICP4 expression in cells cotransfected with pICP4, an HSV-2 ICP4 expression plasmid. U2OS cells were transfected with pICP4, synthetic HSV-2 miR-H6 duplex with or without miR-H6-3p-specific inhibitor, or pmiR-H6 (a miR-H6 expression plasmid), before infection with HSV-2 at a MOI of 2. Twenty nanomolar nonspecific (NS) siRNA and 2g of pFlag vector were also transfected and used as negative controls. The same membrane was stripped and incubated with an anti--tubulin antibody as a loading control. (C) Both miR-H6 duplex and pmiR-H6 were effective in specifically silencing a luciferase reporter that had miR-H6 target sequences. HEK 293 cells were cotranfected with 50 ng of HSV-2 miR-H6 firefly luciferase reporter and aRenillaluciferase reporter (RL-TS) (20) together with 20 nM HSV-2 miR-H6 duplex or 2g of pmiR-H6 with or without 40 nM miR-H6 inhibitor.
on November 7, 2019 by guest
http://jvi.asm.org/
latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J. Virol.66:2157–2169. 17.Steiner, I., et al.1989. Herpes simplex virus type 1 latency-associated
tran-scripts are evidently not essential for latent infection. EMBO J.8:505–511. 18.Stevens, J. G., E. K. Wagner, G. B. Devi-Rao, M. L. Cook, and L. T. Feldman. 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science235:1056–1059.
19.Tang, S., et al.2008. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc. Natl. Acad. Sci. U. S. A.105:10931–10936.
20.Tang, S., A. Patel, and P. R. Krause. 2009. Novel less-abundant viral microRNAs encoded by herpes simplex virus 2 latency-associated transcript and their roles in regulating ICP34.5 and ICP0 mRNAs. J. Virol.83:1433– 1442.
21.Thompson, R. L., and N. M. Sawtell.2001. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J. Virol.75: 6660–6675.
22.Thompson, R. L., and N. M. Sawtell.1997. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J. Virol.71:5432–5440.
23.Thompson, R. L., and N. M. Sawtell.2000. HSV latency-associated transcript and neuronal apoptosis. Science289:1651.
24.Trousdale, M. D., et al.1991. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J. Virol.65:6989–6993.
25.Umbach, J. L., et al.2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature454:780–783. 26.Umbach, J. L., et al.2010. Identification of viral microRNAs expressed in
human sacral ganglia latently infected with herpes simplex virus 2. J. Virol. 84:1189–1192.
27.Yoshikawa, T., et al.1996. The characteristic site-specific reactivation phe-notypes of HSV-1 and HSV-2 depend upon the latency-associated transcript region. J. Exp. Med.184:659–664.