Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Influence of Reverse Transcriptase Variants, Drugs, and Vpr on
Human Immunodeficiency Virus Type 1 Mutant Frequencies
Louis M. Mansky,
1* Erwann Le Rouzic,
2Serge Benichou,
2and Lisa C. Gajary
1Department of Molecular Virology, Immunology, and Medical Genetics, Center for Retrovirus Research, Comprehensive Cancer Center, Ohio State University Medical Center, Columbus, Ohio 43210,1
and Department of Infectious Diseases, Institut Cochin, INSERM U567, CNRS UMR 8104, Paris, France2
Received 18 July 2002/Accepted 23 October 2002
The evolution of drug resistance is a major complication of human immunodeficiency virus type 1 (HIV-1) chemotherapy. HIV-1 reverse transcriptase (RT) is a major target of antiretroviral therapy and ultimately the target of drug resistance mutations. Previous studies have indicated that drug-resistant HIV-1 RTs can alter HIV-1 mutant frequencies. In this study, we have tested a panel of HIV-1 RT variants for their ability to influence virus mutant frequencies. The RT variants tested included drug-resistant RT variants as well as other
variants analyzed in enzyme fidelity studies with thelacZ␣gene as a mutation target and/or implicated as being
important for enzyme fidelity by structural studies. Combinations of mutations that alone had a statistically significant influence on virus mutant frequencies resulted in different mutant frequency phenotypes.
Further-more, when virus replication occurred in the presence of drugs [e.g., 3ⴕ-azido-3ⴕ-deoxythymidine, (ⴚ)2/,3ⴕ
-dideoxy-3ⴕ-thiacytidine, hydroxyurea, thymidine, or thioguanine] with selected RT variants, virus mutant
frequencies increased. Similarly, Vpr variants deficient for binding to the uracil DNA glycosylase repair enzyme were observed to influence HIV-1 virus mutant frequencies when tested alone or in combination with RT variants. In summary, these observations indicate that HIV-1 mutant frequencies can significantly change by single amino acid substitutions in RT and that these effects can be altered by additional mutations in RT, by drugs, and/or by expression of Vpr variants. Such altered virus mutant frequencies could impact HIV-1 dynamics and evolution in small population sizes.
The continuous treatment of human immunodeficiency virus type 1 (HIV-1) infection with the combination of three or more antiretroviral drugs has significantly reduced morbidity and mortality (22, 49). Combination antiretroviral therapy typically contains at least two reverse transcriptase (RT) inhibitors as well as a protease inhibitor. The risk in the emergence of drug-resistant HIV-1 increases when there is poor patient compliance to prescribed drug regimens. This risk may also be of great concern during structured treatment interruptions (1). The viral polymerase encoded by HIV-1 and other retrovi-ruses, i.e., RT, is highly error prone due to the lack of proof-reading ability. The high rates of mutation and recombination that occur during the conversion of the single-stranded viral RNA to a double-stranded DNA is thought to play an impor-tant role in generating diversity in HIV-1 and other retrovirus populations (41). RNA polymerase II is thought to also con-tribute to HIV-1 mutagenesis but to a smaller degree (48). Analysis of the fidelity of HIV-1 RT in cell-free reactions and the HIV-1 mutation rate in cells indicate differences that may be due to protein factors not present in the cell-free reactions or altered in vitro assay conditions (45). The general mecha-nism used by RT to maintain fidelity likely includes the proper positioning of the template-primer complex, the local
geome-try of the active site, and the global influence of different conformational states of the enzyme.
HIV-1 RT is a heterodimer that is composed of two sub-units, p66 and p51 (32, 35, 64). The p66 subunit contains both polymerase and RNase H catalytic sites while the p51 subunit is a proteolytic cleavage product of p66 that lacks the RNase H domain. The overall folding of both subunits is similar, but the spatial arrangements of their subdomains are very different, which prevents p51 from having polymerase activity. The na-tive structure of the p66 subunit folds into a conformation that resembles the right hand (24, 26, 31). The subdomains of p66 are referred to as the fingers, palm, thumb, and connection. Structural studies in conjunction with phylogenetic analyses have implicated conserved amino acid residues and motifs in HIV-1 RT as being important for enzyme fidelity. The RT active site is located in the palm subdomain within conserved residues 185, 186, and 110. Highly conserved amino acid resi-dues in two alpha-helices of the thumb subdomain along with the fingers and palm subdomains of the 66-kDa subunit act as a clamp to position the template-primer complex relative to the polymerase active site (24). In general, the interactions between RT and its substrates (i.e., the template-primer com-plex and deoxynucleoside triphosphates [dNTPs]) are impor-tant determinants of enzyme fidelity.
Additional factors, including the viral protein R (Vpr), can also influence HIV-1 mutant frequencies. HIV-1 Vpr is a 96-amino-acid nonstructural protein that is associated with virus particles and can accumulate in the nuclei of infected cells (9, 38, 50). The HIV-1 Vpr protein has been found to interact with several cellular partners (53, 68), including the DNA repair
* Corresponding author. Mailing address: Department of Molecular Virology, Immunology, and Medical Genetics, The Ohio State Uni-versity, 2078 Graves Hall, 333 W. 10th Ave., Columbus, OH 43210. Phone: (614) 292-5525. Fax: (614) 292-9805. E-mail: mansky.3@osu .edu.
2071
on November 8, 2019 by guest
http://jvi.asm.org/
enzyme uracil DNA glycosylase (UNG) (4). Vpr has been found to recruit the nuclear form of UNG into HIV-1 virions (44). This recruitment is required for Vpr to modulate the in vivo mutation rate (44). Vpr was found to influence the mu-tation rate of HIV-1, and the Vpr-UNG interaction was in-volved in modulating the mutation rate (39, 44, 56).
The goal of this study was to investigate the impact of single amino acid substitutions in RT and to test the potential inter-play of RT variants, drugs, and Vpr on HIV-1 mutant frequen-cies. HIV-1 mutant frequencies were measured with an HIV-1 vector, containing the lacZ gene as a reporter gene, which undergoes one round of replication. The RT variants analyzed included those conferring drug resistance to various drugs as well as variants implicated from structural studies and cell-free fidelity studies as being important in enzyme fidelity. One drug-resistant RT analyzed was observed to significantly alter virus mutant frequencies. In addition, significant changes in virus mutant frequencies were observed with many RT variants that significantly altered the cell-free fidelity of purified RT with the lacZ␣gene as a mutation target. These mutant fre-quencies were influenced by additional mutations in RT, by various drugs, and by the expression of Vpr variants.
MATERIALS AND METHODS
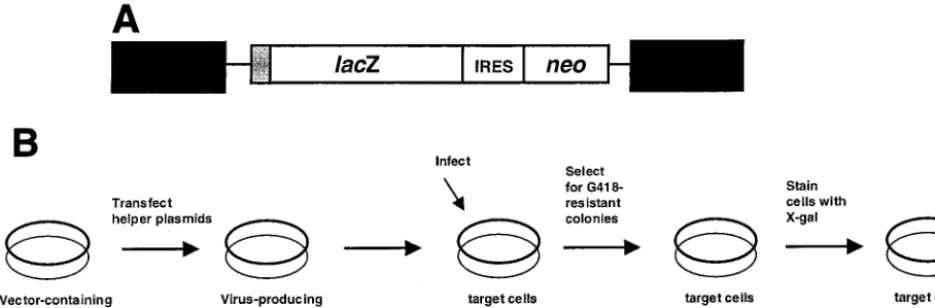
Retroviral vectors and expression plasmids.The HIV-1 vector used in these
studies is shown in Fig. 1A. The vector cassette containing thelacZgene, an
internal ribosomal entry site (IRES) element, and the neomycin
phosphotrans-ferase gene (neo) was introduced into pGEM-NL4-3 (full-length molecular clone
of NL4-3) to create pHIVLacZ-IRES-neo. In order to produce vector virus, the
HIV vector was complemented in trans with pSVgagpol-rre-MPMV (kindly
provided by David Rekosh, University of Virginia), the amphotropic murine
leukemia virusenvexpression plasmid, pSV-A-MLV-env (34), and a Vpr
expres-sion plasmid derived from pAS1B (44). The RT variants analyzed in these experiments were constructed by a primary combinatorial, two-step PCR proto-col (25, 39) or by using the Quickchange XL mutagenesis kit (Stratagene). All RT variants made were sequenced to verify the proper introduction of mutations. Vectors for expression of the hemagglutinin (HA)-tagged forms of wild-type (wt) or mutated Vpr were constructed in the pAS1B plasmid as described previously (44).
Transfections, infections, and cocultivations.The COS-1 and HeLa cell lines used were obtained from the American Type Culture Collection (Manassas, Va.) and were maintained in Dulbecco’s modified Eagle’s medium containing 10% calf serum or 10% fetal bovine serum, respectively. HIV-1 vector and expression plasmids were transfected into HeLa cells by using the Superfect reagent (Qia-gen). HeLa cells were infected in the presence of Polybrene (23). Infection of HeLa target cells was also done by cocultivation of virus-producing cells with target cells (40, 46).
The influence of the antiretroviral drugs on HIV-1 mutant frequencies was determined by postinfection treatment of cells with drug. Postinfection treatment refers to maintaining HeLa target cells in medium supplemented with drug for 2 h before cocultivation and continued until 24 h after cocultivation. Postinfec-tion treatment with drug influences the HIV-1 mutant frequency only during reverse transcription (42).
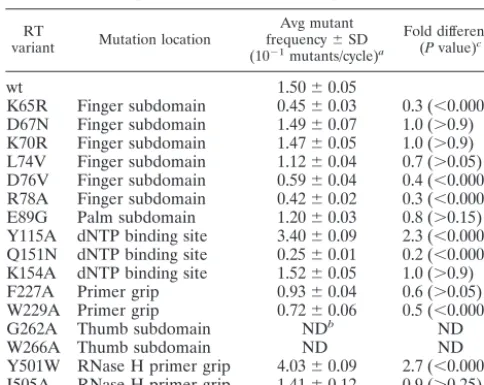
Experimental protocol for generating a single round of HIV-1 vector replica-tion.The experimental protocol developed to generate a single round of HIV-1 vector replication is shown in Fig. 1B. In this protocol, HeLa cells containing the HIV vector provirus was created by transiently transfecting the HIV vector and helper plasmids into COS-1 cells, harvesting virus 2 days posttransfection, and infecting HeLa cells. G418-resistant clones were isolated and characterized for the presence of single proviruses. The cells were also stained with
5-bromo-4-chloro-3-indolyl--D-galactopyranoside (X-Gal) to ensure that no mutations had
occurred in thelacZgene. Selected clones were then used to generate a single
round of HIV-1 vector replication. HeLa cell clones with single integrated proviruses were transiently transfected with helper plasmids, treated with mito-mycin C at 48 h posttransfection, and then mixed with fresh HeLa cells. G418-resistant cells resulting from virus infection of fresh HeLa cells were selected, and cells were then stained with X-Gal. The ratio of white- plus light blue-stained colonies to total colonies observed provided a forward mutant frequency. In each experiment, similar numbers of colonies were screened for control and experi-mental samples. Titers for control experiments (wt RT in the absence of drug)
were typically 500 to 1,000 CFU per 5⫻105target cells. The vector system and
protocols for analysis of mutant frequencies with thelacZ␣peptide gene as a
mutational target have been previously described (45).
Assay for Vpr virion incorporation into HIV-1 particles.Incorporation of the Vpr variants was analyzed by using a packaging assay in which HA-tagged Vpr
was expressed intransand incorporated into virions (57). Briefly, 293T cells were
cotransfected with 10g of the HIV-1-based packaging vector pCMV⌬R8.9
(lacking theenvand auxiliary genes), 5g of the pMD.G plasmid for expression
of the vesicular stomatitis virus G protein, and 10g of pAS1B-Vpr (wt or
mutated). Cell culture supernatants were collected 48 h after transfection and
filtered through 0.45-m-pore-size filters, and virions were collected by
[image:2.603.58.526.75.229.2]ultra-centrifugation and suspended in ice-cold lysis buffer (10 mM Tris [pH 7.6], 150 mM NaCl, 2 mM EDTA, 0.5% Triton X-100). For preparation of cell lysates,
FIG. 1. Assay system used to analyze HIV-1 mutant frequencies in vivo during one round of HIV-1 replication with thelacZgene as a reporter gene. (A) HIV-1 vector used to measure virus mutant frequencies. The proviral DNA form of the vector is shown. The large black rectangular boxes are the long terminal repeats. The small gray box is the simian virus 40 promoter. ThelacZgene, the IRES sequence, and theneogene are indicated. (B) Single-cycle replication assay for mutant frequencies. HeLa cell clones with single integrated vector proviruses were transiently transfected with helper plasmids, and the produced virus was used to infect fresh HeLa cells. G418-resistant cells resulting from virus infection of fresh HeLa cells were selected, and cells were then stained with X-Gal. The ratio of white- plus light blue-stained colonies to total colonies was used to determine the forward mutant frequency.
on November 8, 2019 by guest
http://jvi.asm.org/
cells were trypsinized, collected by centrifugation, and suspended in ice-cold lysis buffer. Cell lysates were incubated for 5 min and clarified by centrifugation. Proteins from cell and virion lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and analyzed by Western blotting with an-ti-HA 3F10 (Boehringer) or anti-CA-p24 antibodies (63).
RESULTS
Analysis of HIV-1 RT single-amino-acid variants on virus
mutant frequencies. Previous studies have shown that some
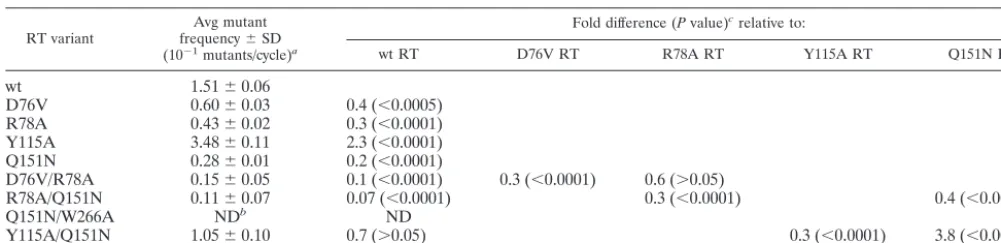
drug-resistant RT enzymes can influence virus mutant frequen-cies and the virus mutation rate (42, 43). In particular, two zidovudine (AZT)-resistant enzymes with multiple mutations, i.e., M41L/T215Y and M41L/D67N/K70R/T215Y, were found to increase the mutation rate by factors of 3.3 and 4.3, respec-tively (42). We thus analyzed a series of drug-resistant enzymes for their influence on virus mutant frequencies. The residues mutated were located in the finger and palm subdomains. The mutant enzymes K65R and L74V confer resistance to the drugs dideoxyinosine, dideoxycytosine, and lamivudine (3TC); D67N and K70R confer resistance to AZT; and E89G confers resistance to multiple drugs (18, 19, 51, 62). K65R also confers resistance to 9-R-2-phosphonomethoxypropyl adenine (tenofo-vir) and abacavir (1592U89) (21, 61). K65R has been previ-ously reported to increase the fidelity of purified RT eightfold while L74V had no affect on cell-free fidelity when thelacZ␣
gene was employed to detect ct mutations (58). The cell-free fidelity of the E89G RT variant increased the dNTP insertion fidelity but did not decrease the cell-free fidelity of the enzyme (11, 12). Analysis of these RT variants for their influence on virus mutant frequencies indicated that the D67N, K70R, L74V, and E89G RT variants did not significantly alter virus mutant frequencies, whereas the K65R RT variant decreased virus mutant frequencies (Table 1). These data indicate that
drug resistance conferred by single-amino-acid substitutions in RT can significantly alter HIV-1 mutant frequencies.
Recent reports have implicated residues of the finger sub-domain (that are not associated with drug resistance) in alter-ing RT fidelity in cell-free systems usalter-ing the lacZ␣gene. In particular, the D76V and R78A substitutions have each been found to increase the cell-free fidelity of purified HIV-1 RT by a factor of 9 (29, 30). D76 has been found in structural studies to interact with the template nucleotide that base pairs with the incoming dNTP while R78 interacts with the template nucle-otide that base pairs with the nuclenucle-otide at the 3⬘ primer terminus (24). In our experiments, D76V and R78A were ob-served to decrease HIV-1 mutant frequencies (Table 1).
The amino acid residues that interact with the incoming dNTP and form the dNTP binding site have been identified in structural studies. Two mutations in the dNTP binding site of HIV-1 RT, Q151N and K154A, have been reported to increase fidelity by factors of 13 and 2.1, respectively, usinglacZ␣(65). Interestingly, the Q151 residue in RT has been implicated in drug resistance to multiple drugs (i.e., the Q151M RT variant) (59). The Q151N RT variant significantly decreased virus mu-tant frequencies while the K154A RT variant was not found to have a significant influence on virus mutant frequencies (Table 1). Intriguingly, another substitution in the dNTP binding site, Y115A, has been previously reported to decrease fidelity by a factor of 4 using thelacZ␣gene (27). However, the Y115F and Y115V RT variants were found inlacZ␣cell-free fidelity as-says to have slightly lower error rates than that of wt RT (5). The Y115F mutation confers resistance to abacavir (21). Non-conservative changes at the Y115 residue have been found to result in a dramatic reduction in the ability of purified HIV-1 RT to discriminate against ribonucleotides in the presence of both magnesium and manganese cations (6). In our experi-ments, we observed that the Y115A RT variant significantly increased (2.3-fold) virus mutant frequencies (Table 1).
The ␣H helix motif of the HIV-1 RT thumb subdomain binds to the minor groove of the template-primer complex and is associated with alterations in RT processivity and fidelity. The 3⬘end of the primer is positioned near the RT active site by conserved residues 224 to 335 on the11b loop and the12 and13 hairpins. The region bounded by residues 227 to 235 is referred to as the primer grip (26). The F227A and W229A mutations in the␣H helix have been reported to alter misin-corporation and mispair extension frequencies, presumedly through an increase in the rate of RT dissociation from the template DNA or through an increase in strand slippage re-sulting in lower enzyme fidelity (66). These mutations in RT were therefore tested to determine what influence they would have on virus mutant frequencies by using an assay for mea-suring the HIV-1 mutant frequency in one round of replication (Fig. 1). As indicated in Table 1, the F227A mutation did not significantly alter the virus mutant frequencies, but the W229A RT variant did influence virus mutant frequencies. The G262 and W266 residues are also located in the thumb domain and have been previously found to increase HIV-1 RT cell-free fidelity in the lacZ␣ reporter assay (2). In particular, the G262A and W266A variants have reduced fidelity for tem-plate-primer slippage errors, such as frameshift and deletion mutations. However, these RT variants lowered virus
infectiv-TABLE 1. Influence of HIV-1 RT variants on virus mutant frequencies in one round of replication
RT
variant Mutation location
Avg mutant
frequency⫾SD
(10⫺1mutants/cycle)a
Fold difference
(Pvalue)c
wt 1.50⫾0.05
K65R Finger subdomain 0.45⫾0.03 0.3 (⬍0.0001) D67N Finger subdomain 1.49⫾0.07 1.0 (⬎0.9) K70R Finger subdomain 1.47⫾0.05 1.0 (⬎0.9) L74V Finger subdomain 1.12⫾0.04 0.7 (⬎0.05) D76V Finger subdomain 0.59⫾0.04 0.4 (⬍0.0005) R78A Finger subdomain 0.42⫾0.02 0.3 (⬍0.0001) E89G Palm subdomain 1.20⫾0.03 0.8 (⬎0.15) Y115A dNTP binding site 3.40⫾0.09 2.3 (⬍0.0001) Q151N dNTP binding site 0.25⫾0.01 0.2 (⬍0.0001) K154A dNTP binding site 1.52⫾0.05 1.0 (⬎0.9) F227A Primer grip 0.93⫾0.04 0.6 (⬎0.05) W229A Primer grip 0.72⫾0.06 0.5 (⬍0.0005)
G262A Thumb subdomain NDb ND
W266A Thumb subdomain ND ND
Y501W RNase H primer grip 4.03⫾0.09 2.7 (⬍0.0001) I505A RNase H primer grip 1.41⫾0.12 0.9 (⬎0.25)
aThe average mutant frequencies⫾standard deviations were determined
from three independent experiments.
bMutant frequency and standard deviation were not determined (ND) due to
low virus titers.
cPvalues were determined by chi-square analysis. Differences (n-fold) are
given relative to the wt RT.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:3.603.42.284.89.281.2]ity to a level at which their influence on virus mutant frequen-cies could not be readily determined (Table 1).
The RNase H domain is responsible for the RNase H activ-ity required for the degradation of the viral RNA present in the RNA-DNA replication intermediate, allowing the DNA to act as a template for the DNA-dependent DNA polymerase activ-ity of RT to complete synthesis of the double-stranded viral DNA. The RNase H activity of RT is crucial for the strand transfers that occur during reverse transcription. Recent struc-tural studies of HIV-1 RT complexed with a polypurine track-containing RNA-DNA hybrid has suggested that an RNase H primer grip exists that interacts with the DNA primer strand and could influence the trajectory of the RNA template rela-tive to the RNase H catalytic center (55). Amino acid residues in the RNase H domain that are involved in the RNase H primer grip include residues 473 to 476, 501, and 505. To test whether residues in the RNase H primer grip could influence virus mutant frequencies, mutations at Y501 and I505 (i.e., Y501W and I505A) were analyzed. Analysis of these RT vari-ants on virus mutant frequencies indicated that Y501W, but not I505A, significantly influenced virus mutant frequencies compared to wt RT (Table 1). Interestingly, a recent study of a murine leukemia virus RT variant, Y586F (the equivalent of Y501 in HIV-1 RT), showed a 5-fold increase in the in vivo mutation rate and a 17-fold increase in the frequency of sub-stitution mutations within 18 nucleotides of adenine-thymine tracts (which are known to induce DNA bending) (67). This observation suggests a mechanism for how the Y501W mutant, a residue in the RNase H domain, may influence HIV-1 mu-tant frequencies. In summary, several RT variants with single-amino-acid changes have been characterized in both the poly-merase and RNase H domains that influence virus mutant frequencies.
Analysis of combined mutations in HIV-1 RT on virus
mu-tant frequencies. The results from Table 1 identified several
amino acid residues in RT which, when mutated, can signifi-cantly alter virus mutant frequencies. To test whether mutated residues could act together to alter virus mutant frequencies, a series of RT variants with combined mutations were analyzed (Table 2). The variants tested included the D76V/R78A, R78A/Q151N, Q151N/W266A, and Y115A/Q151N mutations, which corresponded to mutations in the finger subdomain, the finger subdomain and the dNTP binding site, the dNTP
bind-ing site and the thumb subdomain, and the dNTP bindbind-ing site, respectively.
The HIV-1 RT variants containing either the D76V/R78A or the R78A/Q151N double mutation led to mutant frequen-cies that were significantly lower than the mutant frequenfrequen-cies of wt HIV-1 (Table 2). The mutant frequency of R78A/Q151N was significantly lower than the variants with each mutation alone while the mutant frequency of D76V/R78A was signifi-cantly lower than that of the R76V RT variant but not that of the R78A RT variant. The RT variant with the Q151N/W266A double mutation did not produce high enough levels of infec-tious virus in order to determine the effect of the two muta-tions on the virus mutant frequency. Finally, the Y115A/ Q151N double mutation in RT led to a mutant frequency that was significantly different than the mutant frequencies after one round of HIV-1 vector replication with RT variants con-taining the Y115A mutation, the Q151N mutation, or wt RT (Table 2). A recent study with cell-free RT found that the M230I/Y115W RT variant had an altered phenotype com-pared to that of the Y115W RT variant in misinsertion fidelity assays, which may be associated with our observations here (20). In summary, the data support the conclusion that resi-dues in RT can act together in altering virus mutant frequen-cies.
Influence of drugs and HIV-1 RT variants on virus mutant
frequencies.Previous studies have indicated that both RT
[image:4.603.42.543.80.201.2]vari-ants and drugs could increase virus mutant frequencies (42, 43). Several of the RT variants identified in Table 1 were found to significantly decrease virus mutant frequencies. To deter-mine if virus mutant frequencies during virus replication with RT variants is influenced by the presence of drugs, selected RT variants (i.e., R78A and Q151N) were used for virus replica-tion in the presence of several different drugs. First, AZT and 3TC were used because of previous work showing that these drugs could increase virus mutant frequencies (42). Virus rep-lication with the R78A or Q151N RT variant in the presence of 0.4M AZT increased virus mutant frequencies to those ob-served during virus replication with each of the HIV-1 RT variants in the absence of drug (Table 3). HIV-1 replication with the R78A or Q151N RT variant in the presence of 0.3M 3TC also led to higher mutant frequencies than that observed during virus replication with the RT variants in the absence of drug (Table 3).
TABLE 2. Analysis of RT variants with combined mutations on HIV-1 mutant frequencies
RT variant frequencyAvg mutant⫾SD
(10⫺1mutants/cycle)a
Fold difference (Pvalue)crelative to:
wt RT D76V RT R78A RT Y115A RT Q151N RT
wt 1.51⫾0.06
D76V 0.60⫾0.03 0.4 (⬍0.0005)
R78A 0.43⫾0.02 0.3 (⬍0.0001)
Y115A 3.48⫾0.11 2.3 (⬍0.0001)
Q151N 0.28⫾0.01 0.2 (⬍0.0001)
D76V/R78A 0.15⫾0.05 0.1 (⬍0.0001) 0.3 (⬍0.0001) 0.6 (⬎0.05)
R78A/Q151N 0.11⫾0.07 0.07 (⬍0.0001) 0.3 (⬍0.0001) 0.4 (⬍0.0005)
Q151N/W266A NDb ND
Y115A/Q151N 1.05⫾0.10 0.7 (⬎0.05) 0.3 (⬍0.0001) 3.8 (⬍0.0001)
aThe average mutant frequencies⫾standard deviations were determined from three independent experiments.
bMutant frequency and standard deviation were not determined (ND) due to low virus titers.
cPvalues were determined by chi-square analysis.
on November 8, 2019 by guest
http://jvi.asm.org/
Hydroxyurea (HU) and thymidine (Thy) have been previ-ously shown to alter nucleotide pools and to increase the virus mutant frequencies of different retroviruses, including HIV-1 (3, 8, 15–17, 28, 37, 43, 47, 54, 60). We hypothesized that virus replication with the R78A and Q151N RT variants in the presence of either HU or Thy would increase virus mutant frequencies compared to those in the absence of drug because of the ability of HU and Thy to alter nucleotide pools. HU (2 mM) treatment during HIV-1 replication with the R78A RT resulted in an increased mutant frequency compared to that of the R78A RT in the absence of drug. Similarly, an increased mutant frequency was also observed with HIV-1 replication with the Q151N RT along with HU treatment of cells com-pared to virus replication in the absence of drug. Treatment of cells with 50M Thy during virus replication with the R78A or Q151N RT variant led to significant increases compared to those of virus replication with these RT variants in the absence of drug. The increased mutant frequencies observed during virus replication in the presence of HU or Thy were pre-sumedly due to the alteration of nucleotide pools. These data
support the conclusion that RT variants with single-amino-acid substitutions and drugs can act together in modulating virus mutant frequencies.
Effect of TG on HIV-1 mutant frequencies. Thioguanine
(TG) has been widely used as an antileukemic agent for many years (13). Recently, it has been reported that the active me-tabolite of TG, 2⬘-deoxy-6-thioguanosine 5⬘-triphosphate, can inhibit HIV-1 replication by preventing RNase H activity (33). We were interested in testing whether TG could influence virus mutant frequencies. First, we tested various concentra-tions of TG, ranging from 0.5 to 10 M, on virus mutant frequencies by using the same postinfection treatment strategy described earlier for other drugs (see Materials and Methods). We observed that there was a corresponding increase in virus mutant frequencies (Fig. 2). This indicates that TG influences the virus mutant frequency in a dose-dependent manner. The maximum increase in virus mutant frequency was three times that of the mutant frequency in the absence of the drug. We next tested whether TG treatment could influence virus mutant frequencies during replication with selected HIV-1 RT vari-ants. Interestingly, 5M TG treatment was found to signifi-cantly increase the mutant frequencies when virus replication occurred with either the R78A or the Q151N variant compared to that of wt RT (Table 4). This indicates that TG and RT variants can act together to modulate virus mutant frequencies. Combined influence of Vpr and HIV-1 RT variants on virus
TABLE 3. Influence of various drugs and selected RT variants on HIV-1 mutant frequencies
RT
variant Drug dose
Avg mutant
frequency⫾SD
(10⫺1mutants/cycle)a
Fold difference
(Pvalue)b
wt None 1.60⫾0.07
R78A None 0.55⫾0.03 0.3 (⬍0.0001)
Q151N None 0.35⫾0.02 0.2 (⬍0.0001)
wt 0.4M AZT 10.1⫾0.11
R78A 0.4M AZT 4.12⫾0.04 0.4 (⬍0.0005) Q151N 0.4M AZT 3.03⫾0.03 0.3 (⬍0.0001) wt 0.3M 3TC 4.97⫾0.04
R78A 0.3M 3TC 0.98⫾0.03 0.2 (⬍0.0001) Q151N 0.3M 3TC 0.75⫾0.02 0.2 (⬍0.0001)
wt 2 mM HU 4.88⫾0.03
R78A 2 mM HU 0.96⫾0.03 0.2 (⬍0.0001) Q151N 2 mM HU 0.81⫾0.02 0.2 (⬍0.0001)
wt 50M Thy 5.03⫾0.04
R78A 50M Thy 1.04⫾0.02 0.2 (⬍0.0001) Q151N 50M Thy 0.77⫾0.02 0.2 (⬍0.0001)
aThe average mutant frequencies⫾standard deviations were determined
from three independent experiments.
bPvalues were determined by chi-square analysis. Differences are relative to
[image:5.603.43.284.89.263.2]the wt RT plus drug.
FIG. 2. Dose-dependent effect of TG on HIV-1 mutant frequencies. Various concentrations of TG were analyzed for their influence on HIV-1 replication in one round of replication. Mutant frequencies were determined as described in the legend to Fig. 1. The data were collected from three replicate experiments and are presented as average mutant frequencies⫾standard deviations.
TABLE 4. Influence of TG and selected RT variants on HIV-1 mutant frequencies
RT
variant Drug dose
Avg mutant
frequency⫾SD
(10⫺1mutants/cycle)a
Fold difference
(Pvalue)b
wt None 1.58⫾0.05
R78A None 0.59⫾0.03 0.4 (⬍0.0005)
Q151N None 0.30⫾0.03 0.2 (⬍0.0001)
wt 5M TG 3.02⫾0.11
R78A 5M TG 1.16⫾0.04 0.4 (⬍0.0005) Q151N 5M TG 0.87⫾0.03 0.3 (⬍0.0001)
aThe average mutant frequencies⫾standard deviations were determined
from three independent experiments.
bPvalues were determined by Chi-square analysis. Differences are relative to
the wt RT plus drug.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:5.603.301.542.92.182.2] [image:5.603.133.454.570.693.2]mutant frequencies. Previous studies have indicated that HIV-1 Vpr can influence the in vivo virus mutation rate (39, 44). This influence is related to the property of Vpr to recruit the nuclear form of UNG (UNG2) into virus particles. One Vpr variant, Vpr*W54R, which failed to recruit UNG2 into
HIV-1 virions, was not able to complement avpr-null mutant HIV-1 in the mutation rate assay (44, 56). To further charac-terize how Vpr may influence virus mutant frequencies, other Vpr variants that did not efficiently interact with UNG were analyzed. Specifically, two Vpr variants with single substitu-tions of H71 and H78 (i.e., Vpr*H71R or Vpr*H78R), which
bound poorly to UNG2 (56), were analyzed. Another mutant (Vpr*S79A), which interacted more efficiently with UNG2
than wt Vpr, was also included in the analysis (56). We first checked that these three Vpr variants were efficiently incorpo-rated into HIV-1 particles. As shown in Fig. 3, each Vpr vari-ant was readily detected in both cells and virus particles, indi-cating that each variant was efficiently expressed and incorporated into HIV-1 virions as efficiently as the wt Vpr protein.
We next tested how these Vpr variants influenced virus mutant frequencies during virus replication with either wt RT or an RT variant, R78A. As expected, there was a significant increase in mutant frequency during virus replication with wt RT and expression of either Vpr*H71R or Vpr*H78R than
when wt Vpr was expressed (Table 5). In addition, a significant increase in virus mutant frequency was observed with the R78A RT in combination with either Vpr*H71R or Vpr*H78R
compared to that found when wt Vpr was expressed (Table 5). These data indicate that the influence of these Vpr variants on virus mutant frequencies followed similar trends when virus replication was with wt RT or the R78A RT variant. The average mutant frequency observed during virus replication with wt RT and expression of the previously reported UNG2 binding-deficient Vpr*W54R variant was not significantly
dif-ferent from that of either Vpr*H71R or Vpr*H78R. Similarly,
when the R78A RT variant was used in virus replication, the mutant frequency with Vpr*W54R was not significantly
differ-ent from that of Vpr*H71R or Vpr*H78R. This indicates that
Vpr*H71R and Vpr*H78R influence virus mutant frequencies
in the same manner as Vpr*W54R. Since Vpr*H71R and
Vpr*H78R do not efficiently interact with UNG2, this confirms
that the poor interaction with UNG2 correlates with the al-tered mutant frequencies. The average mutant frequency ob-served during virus replication with wt RT and the expression of Vpr*S79A was not significantly different from that with wt
Vpr, and the mutant frequencies with the R78A variant and Vpr*S79A were not significantly different from those with the
R78A RT and wt Vpr. Since Vpr*S79A interacts efficiently
with UNG2, these data further support the observation that the Vpr-UNG interaction can modulate virus mutant frequen-cies. Furthermore, this indicates that the Vpr-UNG interaction has a similar impact on virus mutant frequencies with either an RT variant (R78A) or the wt RT. In particular, the ability of the R78A RT variant to increase virus mutant frequencies was counteracted by the Vpr effect, indicating that the mechanism responsible for the Vpr effects on the virus mutation rate is independent of the altered mutant frequencies associated with the RT variant.
Finally, we tested whether the Vpr*W54R variant could act
along with an RT variant (Y115A RT), which increased the mutant frequency, to alter virus mutant frequencies in the presence of drug (AZT) compared to wt RT in the absence of drug. Since the potential impact on the virus mutant frequency was anticipated to be significant, thelacZ␣peptide gene was used as a mutational target (44). ThelacZ␣peptide gene is a smaller target than thelacZgene and can more easily detect larger changes in mutant frequencies. The virus mutant fre-quency of the HIV-1 vector containing thelacZ␣peptide gene with Y115A RT in the presence of AZT was 18 times higher than that observed when wt RT was used in virus replication in the absence of drug. The increase in virus mutant frequency follows a multiplicative model (43). When Y115A RT was used with W54R*Vpr in the presence of 0.4 M AZT, the virus
mutant frequency was 25 times higher than during replication with wt RT, wt Vpr, and no drug (Table 6). The increase in mutant frequency due to Vpr*W54R was additive. These
ob-servations suggest that Vpr influences virus mutant frequencies in a manner that is mechanistically different from how the Y115A RT variant or AZT influences virus mutant frequen-cies. These observations extend previous studies on the role of Vpr on the influence of virus mutant frequencies (39, 44). In summary, these data indicate an interplay between RT vari-ants, drugs, and Vpr that can significantly alter HIV-1 mutant frequencies.
DISCUSSION
In this study, we have analyzed HIV-1 mutant frequencies in a single round of replication with an HIV-1 vector containing the lacZgene. A series of amino acid substitutions were cre-ated in RT to determine their influence on virus mutant fre-quencies. The amino acid residues that were targets for site-directed mutagenesis were chosen based upon their association with drug resistance or based upon structural and/or biochem-ical studies that implicated their interaction with the primer-template complex or with the incoming dNTP and implicated their ability to influence enzyme fidelity. One drug resistance mutation in RT (i.e., K65R) was observed to increase virus mutant frequencies. This indicates that at least some drug resistance mutations conferred by single-amino-acid substitu-tions can significantly alter HIV-1 mutant frequencies.
In many instances, RT variants analyzed in cell-free fidelity studies with thelacZ␣gene had a milder effect on virus mutant frequencies than that predicted from the cell-free studies. These differences could be due, in part, to the different muta-tion targets used. In this study, the entirelacZgene was used as a mutation target, whereas in many of the cell-free fidelity studies, thelacZ␣gene was used as a target (2, 5, 11, 27, 29, 30, 58, 65). In cell-free systems, synthesis of one DNA strand is used to determine these rates. In contrast, the assay described here includes both minus-strand (with an RNA template) and plus-strand (with a DNA template) DNA synthesis. In addi-tion, other differences between the cell-free and mutant fre-quency assays (including the presence or absence of protein factors, different RTs, and different experimental conditions) may play a role in discrepancies between the two systems.
Combining mutations in RT which alone were found to reduce virus mutant frequencies led to further reductions in the virus mutant frequency, indicating that the mutations can
on November 8, 2019 by guest
http://jvi.asm.org/
FIG.
3.
Virion
incorporation
of
Vpr
variants.
293T
cells
were
cotransfected
with
pCMV
⌬
R8.9
(lacking
the
vpr
gene)
and
pMD.G
and
either
with
or
without
a
mutated
HA-tagged
Vpr
expression
vector.
Proteins
from
cell
and
virion
lysates
were
separated
by
sodium
dodecyl
sulfate-polyacrylamide
gel
electrophoresis
and
analyze
d
by
Western
blotting
with
anti-HA
or
anti-CA-p24.
on November 8, 2019 by guest
http://jvi.asm.org/
act together to modulate virus mutant frequencies. The impact of selected RT variants that reduced virus mutant frequencies were significantly increased with AZT, while more subtle yet significant increases in virus mutant frequencies were observed with 3TC, HU, and Thy. The mechanism(s) whereby RT vari-ants and drugs alter virus mutant frequencies is under current investigation.
The active metabolite of the antileukemic agent TG was recently shown to inhibit HIV-1 replication by preventing RNase H activity (33). We tested whether TG postinfection treatment could alter HIV-1 mutant frequencies and found that TG increased (threefold maximum) virus mutant frequen-cies in a dose-dependent manner. Though the mechanism(s) for how TG influences HIV-1 mutant frequencies is unknown, previous studies suggest that inhibition of RNase H activity may increase HIV-1 mutant frequencies. Treatment of cells with TG was also found to increase the mutant frequencies of viruses with the R78A and Q151N RT variants. Mutation of the RNase H domain can also alter HIV-1 mutant frequencies, as indicated by the Y501W RT variant. These data indicate that the RNase H domain can alter virus mutant frequencies. The two Vpr variants that were previously shown to have altered binding to UNG, Vpr*H71R and Vpr*H78R, were
found to be efficiently incorporated into virus particles and increased the virus mutant frequency when wt RT and the R78A RT variant were used for virus replication. Therefore, these two new Vpr variants had a mutant frequency phenotype similar to that of the previously reported UNG2 binding-defi-cient Vpr*W54R variant (44), providing further evidence for a
role for Vpr-UNG2 association in modulating HIV-1 mutant frequencies. These data also suggest that the mechanism(s) used by Vpr to alter HIV-1 mutant frequencies is distinct from the mechanism by which the R78A mutation in RT modulates fidelity.
We have shown that single-amino-acid substitutions in RT can change HIV-1 mutant frequencies and that these effects are influenced by additional mutations in RT, by drugs, and by Vpr variants deficient for binding to UNG2. The magnitude of these changes were all within 30-fold of the mutant frequency with wt virus in the absence of drug. The observations made in this study provide the basis for two lines of continued investi-gation. First, these data will allow for mechanistic studies of the determinants of HIV-1 mutant frequencies and the HIV-1 mutation rate. Second, these observations will allow for the analysis of altered rates of mutation on HIV-1 replication as well as on HIV-1 pathogenesis and drug therapy. Little is known about changes in fidelity during the natural course of HIV-1 infection. However, studies with simian immunodefi-ciency virus (SIV) have indicated that, during the course of disease progression, changes in fidelity occur (10). In particu-lar, an SIV clone called SIVMNE 170 was isolated from a macaque during the late symptomatic phase of infection with the parental strain SIVMNE CL8. A misincorporation assay indicated that the SIVMNE 170 RT showed much higher fi-delity than SIVMNE CL8, suggesting that the fifi-delity of len-tiviral RTs may increase during the course of viral infection. Previous studies have indicated that drugs and drug-resistant RT can significantly increase virus mutant frequencies and the HIV-1 mutation rate (42, 43). These increased mutant fre-quencies could have important implications for HIV-1 evolu-tion, population dynamics, and drug therapy regimens. How-ever, the impact of an altered HIV-1 mutation rate is dependent on the dynamics of the virus population. Determin-istic computer modeling has been used to predict the effects of mutation and selection on virus populations, particularly with HIV-1 (7, 52), while stochastic models have also been pro-posed (36). A metapopulation model for HIV-1 replication has been recently reported (14). This model found that the com-bination of founder effects and subpopulation turnover can result in an effective population size much lower than the
TABLE 5. Influence of Vpr variants and a selected RT variant on HIV-1 mutant frequencies
RT
variant Vpr variant
Avg mutant
frequency⫾SD
(10⫺1mutants/cycle)a
Fold difference (Pvalue)bof:
R78A RT relative to wt RT
wt RT Vpr variants relative
to wt Vpr
R78A RT Vpr variants relative
to wt Vpr
wt wt 1.61⫾0.06
R78A wt 0.58⫾0.03 0.4 (⬍0.0005)
wt Vpr*W54R 5.33⫾0.06 3.3 (⬍0.0001)
R78A Vpr*W54R 1.54⫾0.03 0.3 (⬍0.0001) 2.7 (⬍0.0001)
wt Vpr*H71R 4.71⫾0.11 2.9 (⬍0.0001)
R78A Vpr*H71R 1.16⫾0.04 0.2 (⬍0.0001) 2.0 (⬍0.001)
wt Vpr*H78R 4.67⫾0.09 2.9 (⬍0.0001)
R78A Vpr*H78R 1.46⫾0.04 0.3 (⬍0.0001) 2.5 (⬍0.0001)
wt Vpr*S79A 1.58⫾0.05 1.0 (⬎0.9)
R78A Vpr*S79A 0.53⫾0.04 0.3 (⬍0.0001) 1.0 (⬎0.9)
aThe average mutant frequencies⫾standard deviations were determined from three independent experiments.
bPvalues were determined by chi-square analysis.
TABLE 6. Influence of RT and Vpr variants on HIV-1 mutant frequencies during replication in the presence of AZT with the
lacZ␣peptide gene as a mutation target
RT
variant variantVpr AZT concn(M) Mutant frequency(mutants/cycle)a Fold difference(Pvalue)b
wt wt 0 0.005⫾0.002
Y115A wt 0.4 0.090⫾0.005 18 (⬍0.0001) Y115A W54R 0.4 0.124⫾0.007 25 (⬍0.0001)
aMutant frequencies are averages from three independent experiments⫾
standard deviations.
bPvalues were determined by chi-square analysis.
on November 8, 2019 by guest
http://jvi.asm.org/
[image:8.603.43.542.81.223.2] [image:8.603.42.283.644.700.2]actual population size and may contribute to the importance of genetic drift in HIV-1 evolution despite a large number of infected cells. The impact of changes in virus mutant frequen-cies due to RT, drugs, or Vpr variants on HIV-1 population dynamics and evolution could be particularly important in small population sizes.
ACKNOWLEDGMENTS
We thank D. Pearl for helpful discussion; R. Cherian, M. Mauck, M. Stachler, S. Uchida, S. Webb, S. Marie, and A. Waggoner for technical assistance; and R. Benarous for continuous support.
This research was supported by Public Health Service grant GM56615 (to L.M.M.) and by the French Agency against AIDS (ANRS) and SIDACTION (grants to S.B. and E.L.R.).
REFERENCES
1. Abbas, U. L., and J. W. Mellors.2002. Interruption of antiretroviral therapy to augment immune control of chronic HIV-1 infection: risk without reward.
Proc. Natl. Acad. Sci. USA99:13377–13378.
2. Bebenek, K., W. A. Beard, J. R. Casas-Finet, H.-R. Kim, T. A. Darden, S. H. Wilson, and T. A. Kunkel.1995. Reduced frameshift fidelity and processivity of HIV-1 reverse transcriptase mutants containing alanine substitutions in
helix H of the thumb subdomain. J. Biol. Chem.270:19516–19523.
3. Biron, F., F. Lucht, D. Peyramond, A. Fresard, T. Vallet, F. Nugier, J. Grange, S. Malley, F. Hamedi-Sangsari, and J. Vila.1996. Pilot clinical trial of the combination of hydroxyurea and didanosine in HIV-1 infected
indi-viduals. Antivir. Res.29:111–113.
4. Bouhamdan, M., S. Benichou, F. Rey, J.-M. Navarro, I. Agostini, B. Spire, J. Camonis, G. Slupphaug, R. Vigne, R. Benarous, and J. Sire.1996. Human immunodeficiency virus type 1 Vpr protein binds to the uracil DNA
glyco-sylase DNA repair enzyme. J. Virol.70:697–704.
5. Boyer, P. L., and S. H. Hughes.2000. Effects of amino acid substitutions at position 115 on the fidelity of human immunodeficiency virus type 1 reverse
transcriptase. J. Virol.74:6494–6500.
6. Cases-Gonzalez, C. E., M. Gutierrez-Rivas, and L. Menendez-Arias.2000. Coupling ribose selection to fidelity of DNA synthesis. The role of Tyr-115 of human immunodeficiency virus type 1 reverse transcriptase. J. Biol. Chem.
275:19759–19767.
7. Coffin, J.1995. HIV population dynamicsin vivo: implications for genetic
variation, pathogenesis, and therapy. Science267:483–489.
8. Cohen, A., J. Barankiewicz, H. M. Lederman, and E. W. Gelfand.1983. Purine and pyrimidine metabolism in human T lymphocytes. Regulation of
deoxyribonucleotide metabolism. J. Biol. Chem.258:12334–12340.
9. Cohen, E. A., G. Dehni, J. G. Sodroski, and W. A. Haseltine.1990. Human
immunodeficiency virusvprproduct is a virion-associated regulatory protein.
J. Virol.64:3097–3099.
10. Diamond, T. L., J. Kimata, and B. Kim.2001. Identification of a simian immunodeficiency virus reverse transcriptase variant with enhanced
replica-tional fidelity in the late stage of viral infection. J. Biol. Chem.276:23624–
23631.
11. Drosopoulos, W. C., and V. R. Prasad.1998. Increased misincorporation fidelity observed for nucleoside analog resistance mutations M184V and E89G in human immunodeficiency virus type 1 reverse transcriptase does
not correlate with the overall error rate measured in vitro. J. Virol.72:4224–
4230.
12. Drosopoulos, W. C., and V. R. Prasad.1996. Increased polymerase fidelity of E89G, a nucleoside analog-resistant variant of human immunodeficiency
virus type 1 reverse transcriptase. J. Virol.70:4834–4838.
13. Elion, G. B.1989. The purine path to chemotherapy. Science244:41–47. 14. Frost, S. D. W., M.-J. Dumaurier, S. Wain-Hobson, and A. J. Leigh Brown
2001. Genetic drift and withhost metapopulation dynamics of HIV-1
in-fection. Proc. Natl. Acad. Sci. USA98:6975–6980.
15. Gao, W. Y., A. Cara, R. C. Gallo, and F. Lori.1993. Low levels of deoxynucle-otides in peripheral blood lymphocytes: a strategy to inhibit human
immu-nodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. USA90:8925–
8928.
16. Gao, W. Y., D. G. Johns, and H. Mitsuya.1994. Anti-human
immunodefi-ciency virus type 1 activity of hydroxyurea in combination with 2⬘,3⬘
-dideoxynucleosides. Mol. Pharmacol.46:767–772.
17. Giacca, M., S. Zanussi, M. Comar, C. Simonelli, E. Vaccher, P. de Paoli, and U. Tirelli.1996. Treatment of human immunodeficiency virus infection with
hydroxyurea: virologic and clinical evaluation. J. Infect. Dis.174:204–209.
18. Gu, Z., R. S. Fletcher, E. J. Arts, M. A. Wainberg, and M. A. Parniak.1994.
The K65R mutant reverse transcriptase of HIV-1 cross-resistant to 2⬘, 3⬘
-dideoxycytidine, 2⬘,3⬘-dideoxy-3⬘-thiacytidine, and 2⬘,3⬘-dideoxyinosine
shows reduced sensitivity to specific dideoxynucleoside triphosphate
inhibi-tors in vitro. J. Biol. Chem.269:28118–28122.
19. Gu, Z., Q. Gao, H. Fang, H. Salomon, M. A. Parniak, E. Goldberg, J. Cameron, and M. A. Wainberg.1994. Identification of a mutation at codon 65 in the IKKK motif of reverse transcriptase that encodes human
immu-nodeficiency virus resistance to 2⬘,3⬘-dideoxycytidine and 2⬘,3⬘-dideoxy-3⬘
-thiacytidine. Antimicrob. Agents Chemother.38:275–281.
20. Gutierrez-Rivas, M., and L. Menendez-Arias.2001. A mutation in the primer grip region of HIV-1 reverse transcriptase that confers reduced
fidelity of DNA synthesis. Nucleic Acids Res.29:4963–4972.
21. Harrigan, P. R., C. Stone, P. Griffin, I. Najera, S. Bloor, S. D. Kemp, M. Tisdale, and B. A. Larder.2000. Resistance profile of the human immuno-deficiency virus type 1 reverse transcriptase inhibitor abacavir (1592U89)
after monotherapy and combination therapy. J. Infect. Dis.181:912–920.
22. Hogg, R. S., K. V. Heath, B. Yip, K. J. Craib, M. V. O’Shaughnessy, M. T. Schechter, and J. S. Montaner.1998. Improved survival among
HIV-in-fected individuals following initiation of antiretroviral therapy. JAMA279:
450–454.
23. Hu, W.-S., and H. M. Temin.1990. Genetic consequences of packaging two RNA genomes in one retroviral particle: pseudodiploidy and high rate of
genetic recombination. Proc. Natl. Acad. Sci. USA87:1556–1560.
24. Huang, H., R. Chopra, G. L. Verdine, and S. C. Harrison.1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase:
im-plications for drug resistance. Science282:1669–1675.
25. Ito, W., H. Ishiguro, and Y. Kurosawa.1991. A general method for intro-ducing a series of mutations into cloned DNA using the polymerase chain
reaction. Gene102:67–70.
26. Jacobo-Molina, A., J. Ding, R. G. Nanni, A. D. Clark, Jr., X. Lu, C. Tantillo, R. L. Williams, R. L. Kamer, A. L. Ferris, P. Clark, A. Hizi, S. H. Hughes, and E. Arnold.1993. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0A
resolution shows bend DNA. Proc. Natl. Acad. Sci. USA90:6320–6324.
27. Jonckheere, H., E. De Clercq, and J. Anne.2000. Fidelity analysis of HIV-1 reverse transcriptase mutants with an altered amino-acid sequence at
resi-dues Leu74, Glu89, Tyr115, Tyr183 and Met184. Eur. J. Biochem.267:2658–
2665.
28. Julias, J. G., and V. K. Pathak.1998. Deoxyribonucleoside triphosphate pool imbalances in vivo are associated with an increased retroviral mutation rate.
J. Virol.72:7941–7949.
29. Kim, B., J. C. Ayran, S. G. Sager, E. T. Adman, S. M. Fuller, N. H. Tran, and J. Horrigan.1999. New human immunodeficiency virus type 1 reverse tran-scriptase (HIV-1 RT) mutants with increased fidelity of DNA synthesis.
J. Biol. Chem.274:27666–27673.
30. Kim, B., T. R. Hathaway, and L. A. Loeb.1998. Fidelity of mutant HIV-1 reverse transcriptases: interaction with the single-stranded template
influ-ences the accuracy of DNA synthesis. Biochemistry37:5831–5839.
31. Kohlstaedt, L. A., J. Wang, J. M. Friedman, P. A. Rice, and T. A. Steitz.1992.
Crystal structure at 3.5 A˚ resolution of HIV-1 reverse transcriptase
com-plexed with an inhibitor. Science256:1783–1790.
32. Kohlstaedt, L. A., J. Wang, P. A. Rice, J. M. Friedman, and T. A. Steitz.1993.
The structure of HIV-1 reverse transcriptase, p. 223–249.InA. M. Skalka
and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Plainview, N.Y.
33. Krynetskaia, N. F., J. Y. Feng, E. Y. Krynetski, J. V. Garcia, J. C. Panetta, K. S. Anderson, and W. E. Evans.2001. Deoxythioguanosine triphosphate impairs HIV replication: a new mechanism for an old drug. FASEB J.
15:1902–1908.
34. Landau, N. R., K. A. Page, and D. R. Littman.1991. Pseudotyping with human T-cell leukemia virus type I broadens the human immunodeficiency
virus host range. J. Virol.65:162–169.
35. Le Grice, S. F. J.1993. Human immunodeficiency virus reverse transcriptase,
p. 163–191.InA. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold
Spring Harbor Laboratory Press, Plainview, N.Y.
36. Leigh Brown, A. J.1997. Analysis of HIV-1 envgene sequences reveals evidence for a low effective number in the viral population. Proc. Natl. Acad.
Sci. USA94:1862–1865.
37. Lori, F., A. G. Malykh, A. Foli, R. Maserati, A. De Antoni, L. Minoli, D. Padrini, A. Degli Antoni, E. Barchi, H. Jessen, M. A. Wainberg, R. C. Gallo, and J. Lisziewicz.1997. Combination of a drug targeting the cell with a drug targeting the virus controls human immunodeficiency virus type 1 resistance.
AIDS Res. Hum. Retrovir.13:1403–1409.
38. Lu, Y.-L., P. Spearman, and L. Ratner.1993. Human immunodeficiency virus type 1 viral protein R localization in infected cells and virions. J. Virol.
67:6542–6550.
39. Mansky, L. M.1996. The mutation rate of human immunodeficiency virus
type 1 is influenced by thevprgene. Virology222:391–400.
40. Mansky, L. M.1994. Retroviral-vector-mediated gene transfer, p. 27B:5.1–
27B:5.10.InJ. B. Griffiths, A. Doyle, and D. G. Newell (ed.), Cell and tissue
culture: laboratory procedures, 6th ed. John Wiley & Sons, New York, N.Y. 41. Mansky, L. M.1998. Retrovirus mutation rates and their role in genetic
variation. J. Gen. Virol.79:1337–1345.
42. Mansky, L. M., and L. C. Bernard.2000. 3⬘-Azido-3⬘-deoxythymidine (AZT) and AZT-resistant reverse transcriptase can increase the in vivo mutation
rate of human immunodeficiency type 1. J. Virol.74:9532–9539.
on November 8, 2019 by guest
http://jvi.asm.org/
43. Mansky, L. M., D. K. Pearl, and L. C. Gajary.2002. Combination of drugs and drug-resistant reverse transcriptase results in a multiplicative increase of
human immunodeficiency virus type 1 mutant frequencies. J. Virol.76:9253–
9259.
44. Mansky, L. M., S. Preveral, L. Selig, R. Benarous, and S. Benichou.2000. The interaction of Vpr with uracil DNA glycosylase modulates the human
immunodeficiency virus type 1 in vivo mutation rate. J. Virol.74:7039–7047.
45. Mansky, L. M., and H. M. Temin.1995. Lower in vivo mutation rate of human immunodeficiency virus type 1 than predicted from the fidelity of
purified reverse transcriptase. J. Virol.69:5087–5094.
46. Mansky, L. M., and H. M. Temin.1994. Lower mutation rate of bovine
leukemia virus relative to that of spleen necrosis virus. J. Virol.68:494–499.
47. Meyerhans, A., J.-P. Vartanian, C. Hultgren, U. Plikat, A. Karlsson, L. Wang, S. Eriksson, and S. Wain-Hobson.1994. Restriction and enhance-ment of human immunodeficiency virus type 1 replication by modulation of
intracellular deoxynucleoside triphosphate pools. J. Virol.68:535–540.
48. O’Neil, P. K., G. Sun, H. Yu, Y. Ron, J. P. Dougherty, and B. D. Preston.
2002. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral
mutagenesis. J. Biol. Chem.277:38053–38061.
49. Palella, F. J., K. M. Delaney, A. C. Moorman, M. O. Loveless, J. Fuhrer, G. A. Satten, D. J. Aschman, and S. D. Holmberg.1998. Declining morbidity and mortality among patients with advanced human immunodeficiency virus
infection. N. Engl. J. Med.338:853–860.
50. Paxton, W., R. I. Connor, and N. R. Landau.1993. Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region
ofgagand mutational analysis. J. Virol.67:7229–7237.
51. Prasad, V. R., I. Lowy, T. de los Santos, L. Chiang, and S. P. Goff.1991. Isolation and characterization of a dideoxyguanosine triphosphate-resistant mutant of human immunodeficiency virus reverse transcriptase. Proc. Natl.
Acad. Sci. USA88:11363–11367.
52. Preston, B. D.1997. Reverse transcriptase fidelity and HIV-1 variation.
Science275:228–231.
53. Refaeli, Y., D. N. Levy, and D. B. Weiner.1995. The glucocorticoid receptor type II complex is a target of the HIV-1 vpr gene product. Proc. Natl. Acad.
Sci. USA92:3621–3625.
54. Reichard, P.1988. Interactions between deoxyribonucleotide and DNA
syn-thesis. Ann. Rev. Biochem.57:349–374.
55. Sarafianos, S. G., K. Das, C. Tantillo, A. D. Clark, J. Ding, J. M. Whitcomb, P. L. Boyer, S. H. Hughes, and E. Arnold.2001. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO
J.20:1449–1461.
56. Selig, L., S. Benichou, M. E. Rogel, L. I. Wu, M. A. Vodicka, J. Sire, R. Benarous, and M. Emerman.1997. Uracil DNA glycosylase specifically in-teracts with Vpr of both human immunodeficiency virus type 1 and simian immunodeficiency virus of sooty mangabeys, but binding does not correlate
with cell cycle arrest. J. Virol.71:4842–4846.
57. Selig, L., J.-C. Pages, V. Tanchou, S. Preveral, C. Berlioz-Torrent, L. X. Liu, L. Erdtmann, J.-L. Darlix, R. Benarous, and S. Benichou.1999. Interaction with the p6 domain of the Gag precursor mediates incorporation into virions
of Vpr and Vpx proteins from primate lentiviruses. J. Virol.73:592–600.
58. Shah, F. S., K. A. Curr, M. E. Hamburgh, M. Parniak, H. Mitsuya, J. G. Arnez, and V. R. Prasad.2000. Differential influence of nucleoside analog-resistance mutations K65R and L74V on the overall mutation rate and error specificity of human immunodeficiency virus type 1 reverse transcriptase.
J. Biol. Chem.275:27037–27044.
59. Shirasaka, T., M. F. Kavlick, T. Ueno, W. Y. Gao, E. Kojima, M. L. Alcaide, S. Chokekijchai, B. M. Roy, E. Arnold, R. Yarchoan, et al.1995. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides.
Proc. Natl. Acad. Sci. USA92:2398–2402.
60. Snyder, R. D.1988. Consequences of the depletion of cellular deoxynucleo-side triphosphate pools on the excision-repair process in cultured human
fibroblasts. Mutat. Res.200:193–199.
61. Srinivas, R. V., and A. Fridland.1998. Antiviral activities of 9-R -2-phospho-nomethoxypropyl adenine (PMPA) and bis(isopropyloxymethylcarbon-yl)PMPA against various drug-resistant human immunodeficiency virus
strains. Antimicrob. Agents Chemother.42:1484–1487.
62. St. Clair, M. H., J. L. Martin, G. Tudor-Williams, M. C. Bach, C. L. Vavro, D. M. King, P. Kellam, S. D. Kemp, and B. A. Larder.1991. Resistance to ddI and sensitivity to AZT induced by a mutation in HIV-1 reverse
tran-scriptase. Science253:1557–1559.
63. Steimer, K. S., J. P. Puma, M. D. Power, M. A. Powers, C. George-Nasci-mento, J. C. Stephans, J. A. Levy, R. Sanchez-Pescador, P. A. Luciw, P. J. Barr, et al.1986. Differential antibody responses of individuals infected with
AIDS-associated retroviruses surveyed using the viral core antigen p25gag
expressed in bacteria. Virology150:283–290.
64. Telesnitsky, A., and S. P. Goff.1997. Reverse transcriptase and the
genera-tion of retroviral DNA, p. 121–160.InJ. M. Coffin, S. H. Hughes, and H. E.
Varmus (ed.), Retroviruses. Cold Spring Harbor Press, Plainview, N.Y. 65. Weiss, K. K., S. J. Isaacs, N. H. Tran, E. T. Adman, and B. Kim.2000.
Molecular architecture of the mutagenic active site of human
immunodefi-ciency virus type 1 reverse transcriptase: roles of the8-␣E loop in fidelity,
processivity, and substrate interactions. Biochemistry39:10684–10694.
66. Wisniewski, M., C. Palaniappan, Z. Fu, S. F. Le Grice, P. Fay, and R. A. Bambara.1999. Mutation in the primer grip region of HIV reverse
tran-scriptase can increase replication fidelity. J. Biol. Chem.274:28175–28184.
67. Zhang, W. H., E. S. Svarovskaia, R. Barr, and V. K. Pathak.2002. Y586F mutation in murine leukemia virus reverse transcriptase decreases fidelity of DNA synthesis in regions associated with adenine-thymine tracts. Proc. Natl.
Acad. Sci. USA99:10090–10095.
68. Zhao, L.-J., S. Mukherjee, and O. Narayan.1994. Biochemical mechanism of
HIV-1 Vpr function. J. Biol. Chem.269:15577–15582.