and Type II Interferon during Rhesus Macaque
Rhadinovirus Infection
Bridget A. Robinson,a,bRyan D. Estep,bIlhem Messaoudi,a,b,cKelsey S. Rogers,band Scott W. Wonga,b,c
Department of Molecular Microbiology and Immunology, Oregon Health & Science University, Portland, Oregon, USAa; Vaccine and Gene Therapy Institute, Oregon
Health & Science University, Beaverton, Oregon, USAb; and Division of Pathobiology and Immunology, Oregon National Primate Research Center, Beaverton,
Oregon, USAc
Kaposi’s sarcoma-associated herpesvirus and rhesus macaque rhadinovirus (RRV), two closely related gammaherpesviruses, are
unique in their expression of viral homologs of cellular interferon regulatory factors (IRFs), termed viral IRFs (vIRFs). To assess
the role of vIRFs during
de novo
infection, we have utilized the bacterial artificial chromosome clone of wild-type RRV
17577(WT
BACRRV) to generate a recombinant virus with all 8 of the vIRFs deleted (vIRF-ko RRV). The infection of primary rhesus
fibroblasts and peripheral blood mononuclear cells (PBMCs) with vIRF-ko RRV resulted in earlier and increased induction of
type I interferon (IFN) (IFN-␣/) and type II IFN (IFN-␥). Additionally, plasmacytoid dendritic cells maintained higher levels of
IFN-␣
production in PBMC cultures infected with vIRF-ko RRV than in cultures infected with WT
BACRRV. Moreover, the
nu-clear accumulation of phosphorylated IRF-3, which is necessary for the induction of type I IFN, was also inhibited following
WT
BACRRV infection. These findings demonstrate that during
de novo
RRV infection, vIRFs are inhibiting the induction of IFN
at the transcriptional level, and one potential mechanism for this is the disruption of the activation and localization of IRF-3.
T
he interferon (IFN) response is integral to a host’s antiviral
defenses. IFNs are divided into three distinct types (types I to
III), characterized by sequence, receptor usage, and biological
ac-tivity (18). There are two well-studied type I IFNs (IFN-␣
and -).
IFN-
␣
, which is expressed as multiple subtypes (13 in humans), is
produced primarily by plasmacytoid dendritic cells (DCs) (pDCs)
(19). IFN-

, on the other hand, is produced by a wide variety of
cell types, including fibroblasts and epithelial cells (18). One of the
most important and earliest-studied functions of type I IFNs is the
promotion of an antiviral environment within a virus-infected cell
as well as surrounding cells (26). Additionally, type I IFN signaling
also stimulates the adaptive immune response through the
pro-motion of T cell survival, effector function, and proliferation (25,
46, 65); the activation of natural killer (NK) cells (50); and the
maturation and activation of DCs (27, 44, 48). Type II IFN
(IFN-
␥
) is produced by activated T cells and NK cells, promotes
the activation of monocytes and macrophages (68), and is crucial
for an effective Th1 adaptive response. Type III IFNs (IFN-
1,
-
2, and -
3) are the most recently identified IFNs. They exhibit
innate and antiviral properties similar to those of type I IFNs, but
they signal through a different receptor, so their biological impact
is likely distinct (15).
The expression of IFNs is governed by a family of transcription
factors known as interferon regulatory factors (IRFs) (24, 64). In
particular, the transcription of type I IFN (IFN-␣/) relies on the
activation of IRF-3 and IRF-7, highly homologous proteins that
become activated via C-terminal phosphorylation (58). IRF-3 is
constitutively expressed in most cell types and becomes
phos-phorylated following the recognition of a variety of
pathogen-associated molecular patterns (PAMPs) (33), including viral
nu-cleic acids (74). Following the C-terminal phosphorylation of
IRF-3, homodimeric complexes accumulate in the nucleus, where
an interaction with the transcriptional cofactor p300/CBP
poten-tiates the transcription of IFN-
(30, 40, 75), human IFN-␣1 (23,
38), and murine IFN-␣4 (58). The activation of IRF-7 occurs in a
similar manner, except that it has a more restricted expression
profile: IRF-7 is constitutively expressed in some lymphoid cells
and pDCs and is transcriptionally upregulated following type I
IFN signaling in a variety of cell types (45, 57). Because of specific
cell type expression and regulation, IRF-7 plays a vital role in the
induction of IFN-␣
in pDCs (12) and is crucial for the induction
of most IFN-␣
subtypes that constitute the second wave of type I
IFN production (45, 57, 58). To efficiently withstand
IRF-dependent antiviral responses and establish an infection within
the host, viruses have evolved a number of mechanisms to
inter-fere with cellular IRFs, the induction of IFN, and subsequent
IFN-induced signaling (3, 6).
Kaposi’s sarcoma-associated herpesvirus (KSHV) and rhesus
macaque (RM) rhadinovirus (RRV) are two highly related
gam-maherpesviruses and the only viruses known to carry genes with
significant homology to cellular IRFs, aptly named viral interferon
regulatory factors (vIRFs) (2, 54, 56, 60). Because of their unique
homology to cellular IRFs, it has been hypothesized that the vIRFs
employ novel mechanisms of immune evasion. Indeed, 3 of the 4
vIRFs encoded within KSHV can individually inhibit the
induc-tion of IFN and IFN-induced signaling by disrupting the funcinduc-tions
of cellular IRF-1, IRF-3, IRF-5, and IRF-7 (9, 10, 20–22, 29, 43, 71,
76). The inhibitory mechanisms employed by each of the vIRFs
are varied, suggesting that each vIRF plays a unique and significant
role. For example, KSHV vIRF-1 binds to the transcriptional
co-Received6 May 2011Accepted17 November 2011
Published ahead of print7 December 2011
Address correspondence to Scott W. Wong, [email protected].
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.05047-11
on November 7, 2019 by guest
http://jvi.asm.org/
Downloaded from
on November 7, 2019 by guest
http://jvi.asm.org/
Downloaded from
on November 7, 2019 by guest
http://jvi.asm.org/
activator p300/CBP and blocks the necessary interaction between
p300/CBP and cellular IRF-3, effectively inhibiting the
transcrip-tion of type I IFNs and other p300-dependent cytokines (9, 36, 39,
61). KSHV vIRF-3 can interact directly with cellular IRF-7 to
block IRF-7 binding at promoter regions, thus inhibiting the
sub-sequent transcription of several subtypes of IFN-␣
(29).
More-over, KSHV vIRFs also interfere with cell cycle control proteins
and apoptosis (reviewed in reference 52). The above-mentioned
studies have provided insight into the molecular strategies
em-ployed by KSHV vIRFs to inhibit IFN induction and signaling but
have not thoroughly defined their roles in the context of
de novo
KSHV infection due to inefficient lytic replication
in vitro
(re-viewed in reference 51). In contrast, RRV displays robust lytic
replication
in vitro
and encodes 8 vIRFs (open reading frames
[ORFs] R6 to R13) with similarity to KSHV vIRF-1 (60),
provid-ing a unique opportunity to study the roles of vIRFs durprovid-ing the
early phase of
de novo
infection. Sequence analysis suggests that
RRV likely acquired the first 4 vIRFs (ORFs R6 to R9) initially and
that these genes later underwent a duplication event to result in a
total of 8 vIRFs within the RRV genome (60). Therefore, it is
plausible that the duplicated vIRFs (R10 to R13) share redundant
functions with their respective antecedents. Even in the absence of
the duplication of these genes, the vIRFs potentially have
overlap-ping functions, as is the case for the KSHV vIRFs. For example,
KSHV vIRFs 1 to 3 have disparate molecular functions, but they
collectively interfere with IFN induction and signaling (52).
In this study, we tested the hypothesis that RRV vIRFs have
antagonistic effects on cellular IRFs and the IRF-dependent
in-duction of IFN. To do so, we have generated a recombinant RRV
clone lacking all 8 vIRF genes (vIRF-ko RRV) utilizing a
patho-genic molecular bacterial artificial chromosome clone of
wild-type RRV
17577(WT
BACRRV
17577) (16). The results presented
herein demonstrate that the deletion of the vIRFs resulted in an
increased induction of type I and type II IFN in RRV-infected
rhesus macaque fibroblasts (RFs) and rhesus macaque peripheral
blood mononuclear cells (PBMCs) along with an increased
pro-duction of IFN-
␣
within pDCs. The induction of type I IFN was
preceded by increased IRF-3 phosphorylation and nuclear
accu-mulation, which was notably inhibited during WT
BACRRV
infec-tion. The ectopic expression of a single vIRF, encoded by ORF R6,
was able to significantly inhibit IRF-3-mediated transcription, as
induced by poly(IC), and to specifically localize with endogenous
IRF-3. These data suggest that vIRFs play a critical role in
damp-ening antiviral responses early during RRV infection and
demon-strate that at least one vIRF could be targeting IRF-3 to modulate
the type I IFN response.
MATERIALS AND METHODS
Cells, virus, drugs, and cytokines.Primary rhesus fibroblasts (RFs), te-lomerized RFs (tRFs) (32), and HEK293 cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech, Herndon, VA) supple-mented with 10% fetal bovine serum (FBS) (HyClone, Ogden, UT). Pe-ripheral blood mononuclear cells (PBMCs) were isolated from the whole blood of expanded-specific-pathogen-free (ESPF) rhesus macaques (RMs) by using Histopaque (Sigma-Aldrich, St. Louis, MO) according to the manufacturer’s guidelines. RM splenocytes were collected from spleens, and red blood cells (RBCs) were subsequently lysed with RBC lysis solution (Five Prime, Gaithersburg, MD) (10 min at room tempera-ture [RT]). RRV infection of RM PBMCs and splenocytes was carried out with serum-free RPMI (Mediatech). ESPF RMs are serologically negative for RRV, simian immunodeficiency virus (SIV), type D simian retrovirus
(SRV), herpesvirus simiae (B virus), simian T-lymphotropic virus 1 (STLV-1), rhesus cytomegalovirus (RCMV), and simian foamy virus (SFV).
These studies utilized plaque-purified isolates of bacterial artificial chromosome (BAC)-derived RRV17577(WTBACRRV17577) (16) and WT RRV17577-GFP. WT RRV17577-GFP was generated via homologous re-combination in RFs to express green fluorescent protein (GFP) via the EF-1␣promoter and is located in the intergenic space between ORF 57 and ORF R6. All RRV stocks were purified through a 30% sorbitol cushion and resuspended in phosphate-buffered saline (PBS), and viral titers were determined by using a serial dilution plaque assay with RFs. Herpes sim-plex virus 1 (HSV-1) strain F was a generous gift from David Johnson (Oregon Health & Science University).
Poly(IC) (Sigma) was resuspended in PBS and transfected into cells by using TransIT LT1 transfection reagent (Mirus, Madison, WI). Recombi-nant human IFN-␣2 (PBL, Piscataway, NJ) was resuspended in PBS with 0.1% bovine serum albumin (BSA) and used at a final concentration of 10 U/ml.
Construction of a vIRF-ko RRV clone using the WT RRV17577BAC.
The construction of vIRF-ko RRV was performed by using WTBAC RRV17577DNA as a template (16). First, an Flp recombination target (FRT)-flanked kanamycin resistance (Kanr) cassette was engineered with 40-bp arms of homologous RRV sequence to target the 10.9-kb region (nucleotides [nt] 78436 to 89386) encoding the 8 vIRFs (ORF R6 to ORF R13) within RRV. The Kanrcassette with RRV homology arms was then cloned into pSP73 and sequenced before the linearized cassette was elec-troporated into recombinogenicEscherichia colistrain EL250 (35), which contains the WT RRV17577BAC. Recombinant EL250 clones were selected for kanamycin resistance, and BAC DNA was subsequently isolated by using a standard alkaline lysis method to identify potential clones contain-ing the Kanrcassette in place of the vIRFs. EL250 clones with a successful recombination of the vIRF region were then treated with arabinose to induce Flp recombination within the EL250 system for the removal of the Kanrcassette within the BAC.
The construction of vIRF-ko RRV-GFP was also done by using WTBAC RRV as a template but with thegalKrecombination system inE. coli SW105 cells, and vIRF deletion clones were identified via growth on plates with galactose as the sole carbon source (69). ThegalKcassette was sub-sequently removed via recombination with a GFP cassette and excised from pQ100 and consisted of the open reading frame of GFP driven by the constitutive promoter EF-1␣. GalK-negative recombinants were selected for resistance to 2-deoxygalactose in minimal medium with glycerol as the sole carbon source. Individual vIRF-ko RRV-GFP clones were analyzed via restriction digestion and Southern analysis and were sequenced across the vIRF region.
For Southern blot analysis, BAC-derived DNA was isolated from EL250 clones, digested with BamHI overnight at 37°C, visualized on a 0.7% agarose gel, and subsequently transferred onto a Duralon-UV mem-brane (Stratagene, La Jolla, CA). Probes were labeled with digoxigenin (DIG) by using the DIG-High Prime kit (Roche, Indianapolis, IN), and hybridization, washing, and detection were done according to kit proto-cols. Probes were made for the vIRF deleted region (Kanrcassette), the intact vIRF region (ORF R9), and an additional RRV gene (ORF 4), as a control.
Generation of infectious virus using vIRF-ko RRV BAC DNA. Infec-tious virus was isolated from RRV17577BAC DNA as previously de-scribed (16). Briefly, vIRF-ko RRV17577BAC DNA from EL250 clones was transfected into RFs by using TransIT LT1 reagent (Mirus, Mad-ison, WI). After the development of an RRV-associated cytopathic effect (CPE) (73), the supernatant and cells were collected, freeze-thawed once, and sonicated to release any cell-associated virus. The virus collected after this transfection underwent two rounds of infec-tion in RFs transiently transfected with Cre recombinase to remove the loxP-flanked BAC cassette. To confirm the removal of the BAC cas-sette, the BAC cassette insertion site between ORF57 and R6 was
on November 7, 2019 by guest
http://jvi.asm.org/
plified and sequenced by using purified viral DNA as a template. The recombinant virus subsequently went through two rounds of plaque purification in RFs to obtain a purified clone of vIRF-ko RRV.
Viral DNA isolation and complete genome hybridization.As previ-ously described (16), RFs were infected with BAC-derived WT or vIRF-ko RRV (multiplicity of infection [MOI] of 0.01) and allowed to progress to complete CPE before the supernatant and cells were collected and di-gested overnight with proteinase K. Viral DNA was then isolated via ce-sium chloride centrifugation (77,400⫻gfor 72 h). Fractions containing viral DNA were pooled and dialyzed against Tris-EDTA. Viral DNA from vIRF-ko RRV-infected cells was subsequently analyzed via comparative genome hybridization (CGH) at NimbleGen Systems, Inc. (Madison, WI), as described previously by Estep et al. (16). WTBACRRV17577DNA was used as a reference genome, and data were analyzed by using Signal-Map software (NimbleGen Systems, Inc.) to identify potential nucleotide changes in vIRF-ko RRV.
In vitrogrowth curves.Single-step (MOI ⫽2.5) and multistep (MOI⫽0.1) growth curve analyses were carried out with RFs. Cells were seeded into culture tubes (Corning, Aston, MA) at a density of 2⫻105cells per tube and infected in duplicate on the following day. After a 2-h adsorption period, the tubes were washed twice with phosphate-buffered saline to remove any unbound virus, and fresh medium was added. Cells and supernatants were collected at 2, 12, 24, 48, 72, and 96 h postinfection (hpi). Samples were freeze-thawed once and sonicated to release any remaining cell-associated virus, and titers were determined via a plaque assay with RFs.
RNA isolation, RT-PCR, and real-time RT-PCR.RNA was isolated from tRFs and rhesus macaque PBMCs by using the RNeasy kit (Qiagen, Valencia, CA) and DNA endonuclease, RQ1, was used to remove DNA from RNA preperations (Promega, Madison, WI) according to kit proto-cols. Reverse transcription-PCR (RT-PCR) was performed by using Su-perscript III one-step RT-PCR with PlatinumTaq(Invitrogen, Carlsbad, CA). Transcripts were detected with the specific oligonucleotide pairs listed in Table 1.
First-strand cDNA synthesis was carried out by using the High Capac-ity cDNA RT kit (ABI, Foster CCapac-ity, CA), and cDNA was subsequently amplified by using TaqMan PreAmp master mix (ABI) and TaqMan gene expression assays specific for the following transcripts: IFN- (ABI catalog no. Rh_03648734), IFN-␣1/13 (Rh_03456606), IFN-␥ (Rh_02621721), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Rh_02621745) (ABI). All data were normalized to the levels of GAPDH in each sample, and normalized levels of IFN transcripts were made relative to a standard, positive sample included on each plate: either poly(IC)-stimulated tRFs or IFN-␣-stimulated PBMCs.
3=RACE.Primary rhesus fibroblasts were infected (MOI⫽2) for 72 h, followed by RNA extraction and first-strand cDNA synthesis. Rapid am-plification of 3=cDNA ends (3=RACE) was performed by using the Gene-Racer kit (Invitrogen) with gene-specific primers upstream of the stop codon of ORF 57 (ORF-57gsp [5=-ACG CGC AAA AAC ACG CTA GCG-3=]) or ORF 58 (ORF-58gsp [5=-GCT CCT CGG ACT TGT ACA CTA TT-3=]). 3=RACE products were analyzed on a 1% agarose gel, purified, and cloned into pCR4-TOPO (Invitrogen), and at least 3 clones of each product were subsequently sequenced.
Immunoprecipitation (IP), PAGE analysis, and immunoblotting. Cell lysates were immunoprecipitated with an anti-hemagglutinin (HA) monoclonal antibody (MAb) or an anti-IRF-3 polyclonal antibody (pAb) (FL-425) in native lysis buffer (50 mM Tris-Cl [pH 8.0], 1% NP-40, and 150 mM NaCl supplemented with phosphatase inhibitors [100⫻cocktail; Sigma] and protease inhibitors [100⫻cocktail; Sigma]), followed by in-cubation with protein A/G Plus-agarose (Santa Cruz), and lysates were finally collected in radioimmunoprecipitation assay (RIPA) buffer (native lysis buffer with 0.1% SDS and 0.5% sodium deoxycholate). Whole-cell extracts were also collected in RIPA buffer, nuclear and cytoplasmic ly-sates were collected according to kit protocols (NE-PER; Thermo-Scientific), and all samples were analyzed by 10% SDS-PAGE.
Protocols for native PAGE analysis of dimeric forms of cellular IRF-3 were originally described elsewhere (28). Briefly, 7.5% Ready Gels (Bio-Rad, Hercules, CA) were prerun (30 min at 40 mA at 4°C) with 25 mM Tris (pH 8.4) and 192 mM glycine with and without 1% sodium deoxy-cholate (Sigma) in the cathode and anode chambers, respectively. Lysates were resuspended in native sample buffer and electrophoresed for 60 min at 25 mA. Proteins were then transferred onto a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) via semidry transfer (60 min at 100 mA at RT).
Membranes were probed with anti-human IRF-3 pAb (FL-425) (Santa Cruz), anti-human phospho-IRF-3 (Ser396) (4D4G) (Cell Signaling Technology), anti-human GADPH MAb (SC 51906) (Santa Cruz), and anti-human poly(ADP-ribose) polymerase 1/2 (PARP1/2) pAb (H-250) (Santa Cruz). Data were analyzed by using densitometry.
Immunofluorescence.Cells were grown on glass coverslips in 12-well plates and fixed with 4% paraformaldehyde in PBS (20 min at RT). Cells were then permeabilized and blocked in 5% normal goat serum (NGS)– 0.1% Triton X in PBS (PBST) (1 h at RT) prior to staining, and all subse-quent steps were performed with 1% NGS–PBST. Cells on coverslips were stained with anti-human IRF-3 MAb (clone SL012.1) (BD Pharmingen, San Diego, CA) overnight at 4°C and subsequently stained with anti-mouse IgG-Texas Red (Vector Labs, Burlingame, CA). Subsequently, cells were stained with anti-HA-fluorescein isothiocyanate (FITC) (HA-7) (Sigma) (2 h at RT), and nuclei and/or DNA was detected by using Hoechst 33258 dye. Cells on coverslips were mounted onto slides by using Vectashield (Vector Labs) and examined on a Zeiss Axio Imager.M1 mi-croscope (Zeiss Imaging Solutions, Thornwood, NY). Images were ac-quired by using a Zeiss Axiocam camera (MRm) with Axiovision software (version 4.6) and subsequently processed by using Adobe Photoshop (Adobe Systems, San Jose, CA).
Intracellular cytokine staining (ICCS).Freshly isolated PBMCs or splenocytes were maintained in serum-free medium. Under each condi-TABLE 1Oligonucleotide sets used for RT-PCR
ORF Oligonucleotide Oligonucleotide sequence (5=–3=)
R6 R6 for ATC GTT ACT CAC GCA AAT CTT R6 rev TGA AGA CCA CGT TTG CAA ATG
R7 R7 for TGG AAC AAA GTA ACT GCA GAT R7 rev AAC TCA CTG ATT AAA CCA AGG
R8 R8 for TGC TGC GAG ACA GGT CGT CAT R8 rev CAG TAG TGC CGG AGG CCT GAT
R9 R9 for TCC GAC GAG ATC AGC GTA C R9 rev CCC TCG TTA TAC GCG ACC A
R10 R10 for CGT TTC CCA ATT ATG ATT ATC R10 rev CCG ATA CCG TCT CTC TTG ATC
R11 R11 for AAC CGG TGC ACC GAC AGT CGC R11 rev CCG TGT CCT CTC GAA AAC ATC
R12 R12 for ATT GTT GCG ATA ATG ATA AGC R12 rev CCG GTG GCA TCC GCT TCG TTA
R13 R13 for ATG CAA CCT GTG GTT GCG TAC R13 rev CTG GCG GCC CTG GCA TAT A
R3 (vMIP) R3 for CCT ATG GGC TCC ATG AGC R3 rev ATC GTC AAT CAG GCT GCG
GAPDH GAPDH for GTG GAT ATT GTT GCC ATC AAT GAPDH rev ATA CTT CTC ATG GTT CAC ACC
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.298.545.78.372.2]tion, 4 million cells were infected (MOI⫽1) and treated with brefeldin A (0.02g/l) (Sigma) during the final 6 h of each experiment to block cytokine secretion. PBMCs were then surface stained with fluorescently labeled antibodies, CD3 (SP34), CD20 (B9E9), CD14 (M5E2), HLA-DR (L243), CD11c (3.9), and CD123 (6H6), to delineate specific populations, including plasmacytoid DCs (CD3⫺, CD20⫺, CD14⫺, HLA-DR⫹,
CD11c⫺, and CD123⫹). Cells were subsequently fixed in fixation buffer
(BioLegend) for 20 min at 4°C, permeabilized, and stained for intracellu-lar IFN-␣(MMHA-2). IFN-␣was labeled by using Zenon labeling tech-nology according to kit protocols (Molecular Probes, Eugene, OR). Sam-ples were acquired on an LSRII instrument (BD, San Jose, CA), and data were analyzed by using FlowJo software (TreeStar, Ashland, OR).
Cloning of vIRFs for transient-expression assays.RRV vIRFs were amplified from purified WTBAC RRV17577 DNA, engineered with a C-terminal HA tag, and cloned into pcDNA3.1(⫺) (Invitrogen). Oligo-nucleotide primers utilized for amplification are listed in Table 2. Each vIRF clone was sequenced and analyzed via immunofluorescence and Western blot analysis to verify expression.
Luciferase assays.IRF-3-mediated transcription was measured by us-ing a reporter plasmid encodus-ing firefly luciferase driven by 5 copies of the interferon-stimulated response element (ISRE) in the promoter
(pISRE-LUC) (Promega, Madison, WI). Firefly luciferase readings were normal-ized via the constitutive expression ofRenillaluciferase (pRL-SV40) (Pro-mega). HEK293 cells were transfected overnight with 500 ng total DNA [250 ng pISRE-LUC, 10 ng pRL-SV40, 50 ng vIRF-HA-pcDNA3.1, and 190 ng empty pcDNA3.1(⫺)]. As a negative control, HEK293 cells were transfected overnight with 250 ng pISRE-LUC, 10 ng pRL-SV40, and 240 ng empty pcDNA3.1(⫺). Cells were then transfected with poly(IC) (10
g) for 6 h and analyzed with the Dual-Glo luciferase assay according to the manufacturer’s protocol (Promega). Data were made relative to lucif-erase units recorded for an empty vector stimulated with poly(IC).
Statistical analysis. Data were analyzed by using GraphPad Instat (GraphPad Software, La Jolla, CA), and significant differences were deter-mined by a pairedttest, withPvalues ofⱕ0.05 being considered significant.
RESULTS
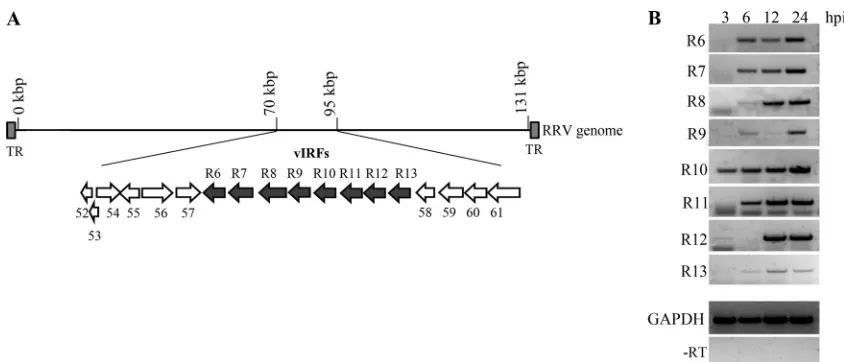
RRV vIRFs are expressed early during infection.
RRV encodes 8
[image:4.585.39.537.77.231.2]vIRFs (ORFs R6 to R13), encoded in a cluster between ORF 57 and
ORF 58 (Fig. 1A). To analyze the temporal expression of the 8
RRV vIRFs, reverse transcription-PCR (RT-PCR) was performed
to analyze the expression of the vIRF transcripts during the first 24
TABLE 2Oligonucleotide pairs used to generate vIRF constructsvIRF Oligonucleotide Oligonucleotide sequence (5=–3=)a
R6 R6 for ctt ggc agtgcggccgcATG GCT ACC TGG CGC CCA CCT
R6-HA rev cgtgaattctca agc gta gtc tgg gac gtc gta tgg gta TTC AAA GTG CCG ATA TAT TTC
R7 R7 for ctt ggc agtgcggccgcATG GCG GGC CGT GGA GTC GAT
R7-HA rev cgtgaattctga agc gta gtc tgg gac gtc gta tgg gta CGA GCA GGC CAC CCC ATC ATC
R10 R10 for ctt ggc agtgcggccgcATG GCC GCT GGG GAA TCG AGA
R10-HA rev agcggatcctta agc gta gtc tgg gac gtc gta tgg gta TTC AAA GTG CCT ATA GAT TTC
R11 R11 for ctt ggc agtgcggccgcATG GCG GAA CGC GAT ATG GAT
R11-HA rev cgtgaattctta agc gta gtc tgg gac gtc gta tgg gta CTT CCT CCC ATA CGG TGC GAG
K9 K9 for ctt ggc agtgcggccgcATG GAC CCA GCC AAA GA
K9-HA rev cgtgaattccta agc gta gtc tgg gac gtc gta tgg gta TTG CAT GGC ATC CCA TAA
aRestriction enzyme sites are in boldface type; gene-specific sequences are in uppercase type.
FIG 1Expression of 8 vIRFs during rhesus rhadinovirus infection. (A) Schematic of the RRV genome showing the positions and orientations of the eight vIRFs (ORFs R6 to R13) encoded within a 10.95-kbp region (nucleotides 78436 to 89386) between ORF 57 and ORF 58. The arrows point toward the 3=end of each ORF. TR, terminal repeat. (B) tRFs were infected (MOI⫽2), and RNA was extracted at 3, 6, 12, and 24 hpi and analyzed by RT-PCR using 8 vIRF-specific oligonucleotide pairs (Table 1). As controls for RNA input and purity, levels of GAPDH in each sample were analyzed, and a reaction was run without reverse transcriptase (⫺RT). ORF, open reading frame; hpi, hours postinfection; GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.79.501.492.673.2]h of WT
BACRRV infection. RT-PCR revealed an early and
sus-tained expression of all eight vIRFs during the first 24 h of
infec-tion (Fig. 1B). Specifically, R10 was expressed as early as 3 hpi, and
R6, R7, and R11 transcripts were all present by 6 hpi (Fig. 1B).
Moreover, the expression pattern of the first four vIRFs (R6 to R9)
closely paralleled the expression pattern of the duplicated vIRFs
(R10 to R13) (60), demonstrating that the sequence-related vIRFs
are similar in their temporal expressions. Overall, these data
dem-onstrate the early expression of the vIRFs during RRV infection of
RFs, which would position their respective proteins to be present
during the virus-stimulated induction of IFN.
Generation of vIRF-ko RRV using WT
BACRRV
17577.
To
de-termine the collective role of the vIRFs during RRV infection, an
infectious BAC-derived clone of RRV
17577(16) was utilized to
generate a recombinant virus lacking all 8 vIRFs, designated
vIRF-ko RRV. Because the vIRFs are encoded within a cluster in
the RRV genome, we were able to delete all 8 vIRFs concurrently.
To achieve this, we replaced the vIRFs with a single kanamycin
resistance (Kan
r) cassette via homologous recombination using
the Red recombination system within
E. coli
strain EL250 (35). To
verify the targeted recombination of the vIRFs, BAC-derived DNA
was digested with BamHI (Fig. 2A and B) and examined via
Southern analysis (Fig. 2C) after the initial recombination of the
vIRF region and again after the Flp removal of the Kan
rcassette.
The deletion of the vIRFs and the Flp removal of the Kan
rcassette
resulted in a 199-bp lesion, including a single FRT site (48 bp), in
the genomic region where the vIRFs were deleted. To verify this,
BAC DNA from the vIRF-ko RRV clone was isolated, and the
genomic region containing the vIRF deletion was amplified and
sequenced to confirm targeted deletion and that there were no
changes in the surrounding genomic sequence (data not shown).
BAC DNA from the vIRF-ko clone was subsequently
trans-fected into RFs to produce infectious virus. This virus was then
used to infect RFs transiently expressing Cre recombinase to
re-move the LoxP-flanked BAC cassette (16) and then subjected to
two rounds of plaque purification in RFs to obtain a purified
iso-late of vIRF-ko RRV. Viral DNA was purified and analyzed via
comparative genomic hybridization (CGH) to compare the
com-plete genomic sequence of vIRF-ko RRV viral DNA to that of
WT
BACRRV
17577(Fig. 2D). CGH is a sensitive, array-based
anal-ysis used to identify single-nucleotide changes,
insertions/dele-tions, and rearrangements between two highly similar genomes.
FIG 2Construction and molecular characterization of vIRF-ko RRV using WTBACRRV17577. (A) Schematic representation of the RRVBACDNA highlighting theBamHI restriction sites (B1 to B4) used to monitor the recombination of the vIRF region and removal of the Kanrcassette. (B) WT
BACand vIRF-koBACDNAs
were digested with BamHI before and after Flp recombination. The 15.7-kbp band (⬍) in WTBACDNA shifts upwards to 16.3 kbp (ⴱ) after the recombination
of the vIRF region and migrates with a similar-sized fragment. After the Kanrcassette is removed via Flp recombinase, a new band appears at 14.7 kbp ( °). (C) Southern blot analysis performed on BamHI-digested DNA using DIG-labeled probes against the vIRF region, the Kanrgene, and another RRV gene (ORF4), as
a control. (D) Comparative genome hybridization used to directly compare viral DNA from the vIRF-ko RRV recombinant clone to that from WTBACRRV. The
schematic of the RRV genome shows the vIRFs (ORFs R6 to R13) shaded in gray. Any alterations within the vIRF-ko RRV genome resulted in incomplete hybridization to the array, depicted by the ratio of vIRF-ko to WT RRV, and signaled a potential nucleotide mismatch between the two viruses. This comparison identified mismatches only within the region of the vIRF deletion (nt 78436 to 89386). TR, terminal repeats; Flp, flippase.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.49.542.65.382.2]The effectiveness and validity of CGH have been demonstrated by
its use in comparisons of different strains of WT RRV and WT
BACRRV
17577isolates (16), in comparisons of coronavirus isolates
(72), and in studies of bacterial evolution (1). The CGH analyses
did not detect any changes in the vIRF-ko RRV isolate compared
to WT
BACRRV viral DNA (Fig. 2D), indicating that WT
BACRRV
and vIRF-ko RRV have identical sequences outside the deleted
vIRF region.
Deletion of vIRFs did not affect growth kinetics or inhibit
transcription of neighboring ORFs during RRV infection of
RFs.
To evaluate viral replication in the absence of the vIRFs,
single-step (MOI
⫽
2.5) and multistep (MOI
⫽
0.1) growth
curves were performed to assess viral growth and spread,
respec-tively. In both the single-step (Fig. 3A) and multistep (Fig. 3B)
growth analyses, vIRF-ko RRV displayed growth kinetics and
magnitude similar to those of WT
BACRRV. These data show that
the vIRFs are not essential for RRV infection, replication, or
spread during RRV infection of RFs
in vitro
. Additionally, the
GFP-expressing clones of WT and vIRF-ko RRV were further
an-alyzed to validate GFP expression as a suitable marker for RRV
infection. WT RRV-GFP and vIRF-ko RRV-GFP infections
re-sulted in 16.8% and 21.6% GFP-positive (GFP
⫹) cells at 36 hpi,
FIG 3In vitrocharacterization of vIRF-ko RRV. (A and B) RFs were infected with recombinant RRV isolates in single-step (MOI⫽2.5) (A) and multistep (MOI⫽0.1) (B) growth curves. Infected RFs were harvested at the specified time points and subjected to a serial-dilution plaque assay on RFs to ascertain viral titers, displayed as PFU/ml. The data from 2 separate experiments were averaged (⫾standard errors of the means [SEM]). (C) RFs infected with GFP-expressing clones of RRV were fixed at 48 hpi, and GFP expression was analyzed via immunofluorescence (green, RRV-GFP; blue, Hoechst 33342). This experiment was repeated three times, and a representative image is shown (magnification,⫻200), with the percentage of GFP-expressing cells (⫾SEM) included within each image. (D to G) RFs were infected with WTBACRRV or vIRF-ko RRV (MOI⫽1), and RNA was harvested at 72 hpi and analyzed via 3=RACE to verify thetranscription of ORF 57 and ORF 58. (D) Two ORF 57 products were detected (arrows labeled a and b), and a third transcript was also detected in vIRF-ko RRV infection but resulted in a truncated transcript (#). (E) ORF 57 3=RACE products were cloned and sequenced, and the full-length transcripts (a and b) utilized 2 distinct poly(A) sites (shaded in gray). The stop codon is underlined, and the addition site of the poly(A) tail is noted (⬎). (F and G) 3=RACE analysis of the ORF58 transcript. The most prevalent transcript is marked (arrow) (F) and was subsequently cloned and sequenced to identify a single poly(A) site (shaded in gray) (G). The stop codon (underlined) and the addition of the poly(A) tail (⬎) are noted.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.74.511.65.477.2]respectively (Fig. 3C). The similar expression intensities and
per-centages of GFP positivity suggest that GFP is an adequate marker
for comparing the efficiency of RRV infection.
To further characterize the vIRF-ko RRV recombinant clone,
gene expression of the ORFs directly upstream (ORF 57) and
downstream (ORF 58) of the deleted vIRFs during vIRF-ko RRV
infection was verified. RRV ORF 57 encodes a protein with
immediate-early expression and shares similarity with ORF 57
genes of other gammaherpesviruses, which function in the nuclear
export of unspliced viral mRNA (7, 51, 63). RRV ORF 58 is also
conserved among gammaherpesviruses (51) and encodes a
pro-tein with a putative epithelial binding function, as was shown
previously for Epstein-Barr virus (67). The coding regions for
ORF 57 and ORF 58 are oriented in opposing directions, both
being transcribed toward the vIRF region (Fig. 1A), so we utilized
3
=
RACE with gene-specific primers within ORF 57 and ORF 58 to
detect both transcripts during vIRF-ko RRV infection. We
iden-tified two poly(A) sites that yielded full-length ORF 57 transcripts
in WT
BACRRV- and vIRF-ko RRV-infected tRFs (Fig. 3D, arrows
a and b, and E). The poly(A) site further downstream of the stop
codon is preferentially used during vIRF-ko RRV infection, while
the poly(A) site that overlaps with the stop codon is preferentially
used during WT
BACRRV infection (Fig. 3E). There was also a
third, less abundant product identified for ORF 57 in vIRF-ko
RRV-infected cells (Fig. 3D), which resulted in an abbreviated
transcript. The transcription of ORF 58 also utilizes multiple
poly(A) sites during WT
BACand vIRF-ko RRV infections, but the
dominant transcript used the same poly(A) site (Fig. 3F) and
re-sulted in a full-length transcript (Fig. 3G). Collectively, these data
suggest that the removal of the vIRFs did not alter the growth of
RRV in RFs or considerably impact the transcription of adjacent
ORFs.
vIRFs are necessary for efficient infection of rhesus macaque
PBMCs.
Although RRV infection of fibroblasts is easily studied,
peripheral blood mononuclear cells (PBMCs), and B cells
specif-ically, represent an important cellular target for RRV in the rhesus
macaque (4). Because such a small percentage of PBMCs becomes
infected with RRV
in vitro
, it is difficult to compare growth
prop-erties within PBMCs by using a standard plaque assay. Thus,
RT-PCR was initially utilized to verify the presence of RRV transcripts
as a marker of RRV infection of rhesus macaque PBMCs. RRV
ORF R3, which encodes viral macrophage inflammatory protein
(vMIP), was examined due to the ease of detection (DNA and
RNA), even at low levels of infection (data not shown). In fact, the
vMIP transcript was present in both WT
BACRRV- and vIRF-ko
RRV-infected PBMCs throughout the 48-h time course (Fig. 4A).
Additionally, we looked for the presence of one of the vIRF
tran-scripts to verify that the vIRFs are also expressed during RRV
infection of PBMCs. We chose the vIRF encoded by ORF R10,
because it was transcribed early and persisted during the first 24 h
in RRV-infected RFs (Fig. 1B). As expected, the vIRF R10
tran-script was present only in WT
BACRRV-infected PBMCs, and the
transcript was present at 3 hpi and persisted through 48 hpi
(Fig. 4A).
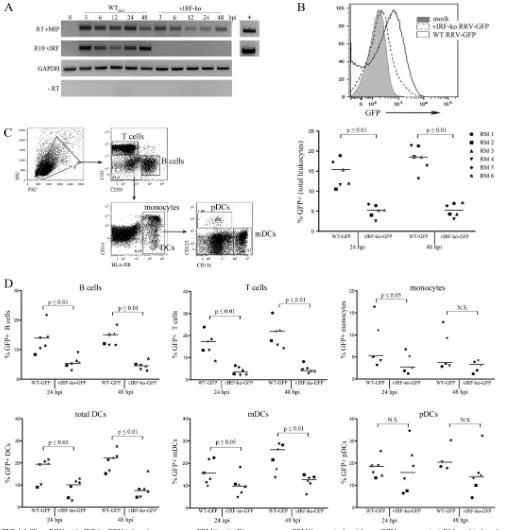
To evaluate the infection efficiency within total PBMCs as well
as within specific immune cell populations in the blood, PBMCs
or splenocyte populations from 6 different RMs were infected
with GFP-expressing clones of WT and vIRF-ko RRV. The
fre-quency of GFP
⫹cells was significantly lower in vIRF-ko
RRV-infected cultures. Approximately 5% of PBMCs were RRV-infected
with vIRF-ko RRV, compared to 15% and 19% of PBMCs in WT
RRV-infected cultures at 24 and 48 hpi, respectively (Fig. 4B). To
determine if the deletion of the vIRFs equally reduced infection in
specific immune cell subsets, infected PBMCs were also surface
stained to delineate T cells, B cells, monocytes, myeloid DCs
(mDCs), and plasmacytoid DCs (pDCs) (8) (Fig. 4C). Similar to
total PBMC cultures, these analyses revealed that vIRF-ko RRV
infects T cells, B cells, monocytes, and mDCs at a reduced
effi-ciency (Fig. 4D). Interestingly, we did not detect any difference in
the frequencies of GFP
⫹pDCs between WT- and vIRF-ko
RRV-infected cultures (Fig. 3D).
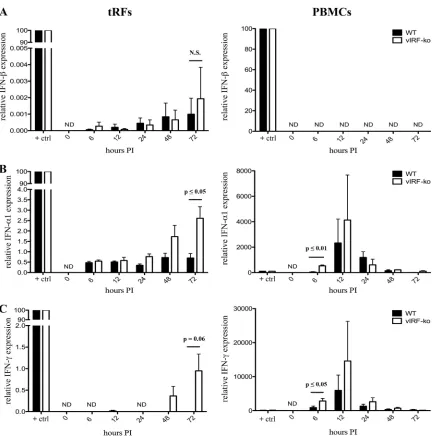
Deletion of vIRFs results in increased induction of type I and
type II IFNs during RRV infection.
To examine whether the
vIRFs collectively function to inhibit the induction of IFNs, we
measured the induction of type I (IFN-

and IFN-
␣
) and type II
(IFN-␥) IFN in WT
BACRRV- and vIRF-ko RRV-infected tRFs and
rhesus macaque PBMCs using real-time RT-PCR. Gene
expres-sion was normalized to GAPDH expresexpres-sion and determined
rela-tive to the posirela-tive control. The induction of IFN-

occurs early
during viral infection in a variety of cell types, including
fibro-blasts, but WT
BACRRV infection did not result in a robust
induc-tion of IFN-, especially within PBMCs (Fig. 5A). This is
partic-ularly evident compared to poly(IC), which induced 10
5times
more IFN-
transcript than did RRV infection (Fig. 5A).
How-ever, the deletion of the vIRFs resulted in an average 2-fold
in-crease in IFN-
transcript levels at 72 hpi compared to those of
WT
BACRRV, although these differences were not statistically
sig-nificant (Fig. 5A).
To further assess the type I IFN response during RRV
infec-tion, we also examined the induction of IFN-␣1
,as it is the only
IFN-
␣
subtype that is immediately induced upon viral
infec-tion (23, 38). As measured by real-time RT-PCR, the inducinfec-tion
of IFN-
␣
1 in WT
BACRRV-infected tRFs was unremarkable
throughout the 72-h time course, with levels between 0.5 and
1% of the magnitude of IFN-
␣
1 induction after poly(IC)
stim-ulation, the positive control (Fig. 5B). In contrast, vIRF-ko
RRV infection of tRFs resulted in a progressive increase in
IFN-␣1 transcript levels, which culminated in a statistically
significant 3-fold-higher induction at 72 hpi than that with
WT
BACinfection (Fig. 5B). For rhesus macaque PBMCs, the
levels of the IFN-
␣
1 transcript peaked much earlier, and the
induction of IFN-␣1 at 6 hpi in vIRF-ko RRV-infected PBMCs
was 12-fold higher than that during WT
BACRRV infection (Fig.
5B). These data demonstrate that the vIRFs inhibit the
induc-tion of type I IFNs during RRV infecinduc-tion in tRFs and PBMCs.
Finally, we assessed the impact of vIRFs on the induction of
type II IFN (IFN-
␥
) during RRV infection. In tRFs, vIRF-ko RRV
infection induced IFN-␥
expression at 48 to 72 hpi (Fig. 5C),
whereas WT
BACRRV infection failed to induce detectable levels of
IFN-␥
(Fig. 5C). RRV infection of PBMCs induced IFN-␥
with
kinetics similar to those of IFN-
␣
1 induction, peaking at 12 hpi
(Fig. 5C), and vIRF-ko RRV infection induced 3-fold more IFN-␥
at 6 hpi (Fig. 5C). Collectively, these data demonstrate the vIRFs
can significantly reduce and delay the induction of both type I and
type II IFN during RRV infection.
vIRFs limit IFN-␣
production by pDCs in RRV-infected
PBMCs.
To determine if the vIRF-dependent inhibition of IFN
was also detectable at the protein level, IFN-␣
production in
RRV-infected PBMCs was examined via intracellular cytokine staining
(ICCS) (Fig. 6). Freshly isolated PBMCs from six
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 4WTBACRRV and vIRF-ko RRV infect rhesus macaque PBMCs. (A) Rhesus macaque PBMCs were isolated from (RRV-seronegative) RMs and infected
with WTBACRRV or vIRF-ko RRV (MOI⫽1). RNA was harvested at the indicated time points and analyzed by RT-PCR for RRV vMIP (ORF R3), RRV vIRF
(ORF R10), and GAPDH, as a loading control. Oligonucleotide pairs used for each transcript are listed in Table 1; viral transcripts were detected by using 300 ng RNA, and GAPDH was detected by using 50 ng RNA per sample. Samples were run simultaneously withTaqpolymerase only (⫺RT) to control for input and purity, and purified RRV DNA was used as a positive control (⫹). (B to D) Leukocytes were isolated from the spleens of six RMs, and GFP-expressing clones of WT RRV and vIRF-ko RRV were used to infect total leukocyte cultures. (B) GFP expression levels in WT- and vIRF-ko RRV-infected cultures were compared by flow cytometry at 24 and 48 hpi, and median responses are represented by a horizontal line. Data were compared via pairedttests. (C) Surface markers were used to delineate cellular populations within the infected cultures: T cells (CD3⫹), B cells (CD20⫹), monocytes (Lin⫺MHC-II⫹CD14⫹), total dendritic cells (DCs) (Lin⫺MHC-II⫹CD14⫺), myeloid DCs (CD11c⫹CD123dim), and plasmacytoid DCs (CD11c⫺CD123⫹). (D) GFP expression in each cellular subset was analyzed at 24 and 48 hpi, as was done for the total leukocyte population in panel B. RM, rhesus macaque; GFP, green fluorescent protein; hpi, hours postinfection; FSC, forward scatter; SSC, side scatter.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.38.544.65.595.2]specific-pathogen-free (ESPF) rhesus macaques were infected
with WT
BACor vIRF-ko RRV (MOI
⫽
1) for 12, 24, and 48 h. For
all samples, brefeldin A was added during the last 6 h of incubation
to inhibit cytokine secretion. Subsequently, PBMCs were surface
stained to delineate pDCs (shown in Fig. 4C) (8), as they are the
major producers of type I IFN in PBMCs (11, 62), and IFN-␣
production within pDCs was measured by ICCS (Fig. 6). HSV-1
stimulation of rhesus macaque PBMCs was used as a positive
con-trol and resulted in a marked production of IFN-
␣
within the pDC
population (Fig. 6A), as previously described (11, 62). Likewise,
approximately 15% of the pDC population was producing IFN-
␣
at 12 hpi in both WT
BACRRV- and vIRF-ko RRV-infected PBMC
cultures (Fig. 6A and B). Interestingly, IFN-
␣
production was
sus-tained in vIRF-ko RRV-infected cultures, with approximately
17% of the pDCs still producing IFN-
␣
at 24 hpi (Fig. 6A and B).
In contrast, only 6% of the pDCs were still producing IFN-␣
at 24
hpi in WT
BACRRV-infected cultures (Fig. 6A and B). Thus, the
deletion of the vIRFs resulted in a sustained production of IFN-␣
following RRV infection.
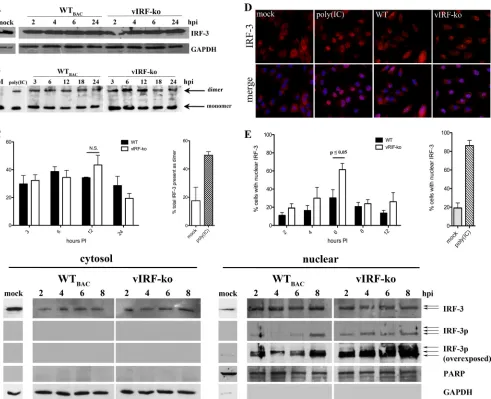
vIRFs inhibit nuclear accumulation of IRF-3 during RRV
in-fection.
IRF-3 is a vital component of the transcription machinery
that drives the transcription of type I IFN following viral infection
(23, 38, 58). Therefore, the activation of IRF-3 was examined
dur-ing the initial 24 h of infection with RRV. The total levels of IRF-3
FIG 5Induction of type I and type II IFN during RRV infection. Telomerized RFs (tRFs) and ESPF rhesus macaque PBMCs were infected with WTBACRRV orvIRF-ko RRV (MOI⫽1), and RNA was extracted at the indicated time points and used to prepare and amplify cDNA for semiquantitative real-time RT-PCR specific for IFN-(A), IFN-␣1 (B), and IFN-␥(C), using TaqMan PreAmp master mix and gene expression assays (ABI) specific for rhesus macaque transcripts. All transcripts were normalized to levels of GAPDH in each sample and made relative to a positive-control sample included on each plate. The positive control (⫹ctrl) for IFN-␣and IFN-transcripts was cDNA from tRFs transfected with poly(IC) (10g/ml for 6 h), and the positive control for IFN-␥was cDNA from PBMCs treated with recombinant human IFN-␣2 (10 units/ml for 6 h). Data are average data (⫾SEM) from at least three independent experiments each with tRFs and PBMCs. N.S., not significant; ND, not detected; PI, postinfection.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.77.514.61.497.2]present in whole-cell extracts remained unchanged after WT
BACand vIRF-ko RRV infections (Fig. 7A). Moreover, both WT
BACand vIRF-ko RRV induced similar levels of IRF-3 dimerization,
which peaked at 6 to 12 hpi, as measured by native PAGE and
Western analysis (Fig. 7B and C). However, vIRF-ko RRV
infec-tion did result in an increase in the nuclear accumulainfec-tion of IRF-3
at 6 hpi (Fig. 7D and E). Indirect immunofluorescence of total
IRF-3 showed that 61% of cells contained IRF-3 in the nucleus at
6 hpi in vIRF-ko RRV-infected cells, compared to only 30% in
cultures infected with WT
BACRRV (Fig. 7D and E). Further
char-acterization of nuclear IRF-3 revealed that vIRF-ko RRV infection
specifically results in an earlier and sustained accumulation of
hyperphosphorylated forms of IRF-3 within the nucleus from 2 to
8 hpi, as shown by Western blot analysis (Fig. 7F). In contrast,
WT
BACRRV infection did not result in a similar nuclear
accumu-lation of phosphorylated IRF-3 (Fig. 7F), nor was there direct
evidence that phosphorylated forms of IRF-3 were simply retained
in the cytoplasm (Fig. 7F). These data suggest that interference
with the activation and/or nuclear accumulation of
phosphory-lated IRF-3 is a potential mechanism by which vIRFs inhibit the
induction of type I IFN during RRV infection.
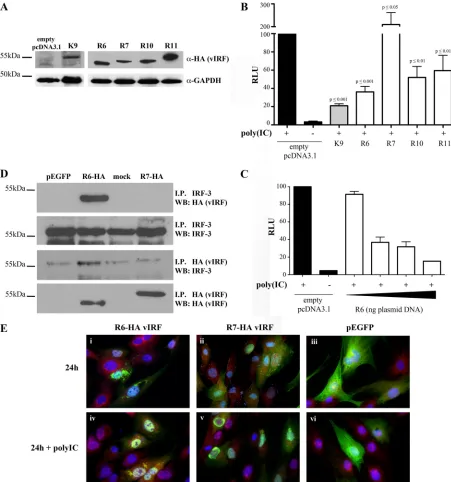
R6 vIRF independently inhibits IRF-3-mediated
transcrip-tion.
To assess the role of individual RRV vIRFs in inhibiting the
induction of type I IFN, vIRF expression clones were generated
with a C-terminal HA tag for ectopic expression in cell cultures.
RRV vIRFs R6 and R10 were of specific interest, since R10 shares
the highest level of amino acid identity with KSHV vIRF-1 (ORF
K9), which is known to inhibit IRF-3-induced gene expression (9,
39), and R6 is most similar to R10. The vIRFs encoded by ORFs R7
and R11 were also included for comparison. The expression of
these vIRFs in tRFs and HEK293 cells was initially verified by
utilizing the HA tag within each construct (Fig. 8A and data not
shown). Individual RRV vIRFs were then expressed alongside an
IRF-3-responsive reporter to determine if the expression of any of
these RRV vIRFs can inhibit IRF-3-mediated transcription.
HEK293 cells were transiently transfected with a firefly luciferase
plasmid under the control of an IFN-stimulated response element
(ISRE)-containing promoter, along with a single vIRF clone. Cells
were stimulated 24 h later with poly(IC) to induce the activation
of IRF-3. Relative luciferase units (RLU) were determined by
de-fining 100% RLU as the output in poly(IC)-stimulated cells that
had been transfected with the empty vector (Fig. 8B). Similar to
previous findings, KSHV v1 was able to inhibit 80% of
IRF-3-mediated transcription, in comparison to the empty vector (Fig.
8B) (9, 39). Likewise, three of the four RRV vIRFs tested (R6, R10,
and R11) resulted in a 40 to 60% inhibition of IRF-3-mediated
transcription in these assays (Fig. 8B). The expression of R6 vIRF
resulted in the most significant effect, inhibiting IRF-3-mediated
transcription by at least 60% (Fig. 8B) in a dose-dependent
man-ner (Fig. 8C). Interestingly, R7 vIRF further enhanced
IRF-3-mediated transcription by more than 2-fold in these assays (Fig.
8B), suggesting a potentially unique role for R7 vIRF in
stimulat-ing transcription.
To further evaluate the R6 vIRF inhibition of IRF-3-mediated
transcription, the HA-tagged expression clone of R6 was
ex-pressed in tRFs and immunoprecipitated with IRF-3 pAb or HA
MAb, followed by Western blot analysis. The IP-Western data
showed that R6 vIRF interacts with endogenous, cellular IRF-3
(Fig. 8D), and the specificity of this interaction is highlighted by
the lack of an interaction between R7 vIRF and IRF-3 in these
same analyses (Fig. 8D).
The spatial relationship between R6 vIRF and IRF-3 within
the cell was also examined by immunofluorescence analysis. In
unstimulated cells, IRF-3 was detected mainly within the
cyto-plasm, as shown in Fig. 7D and 8E, and expression with a
con-trol plasmid, pEGFP, or either of the vIRF expression plasmids,
did not increase the nuclear accumulation of IRF-3 (Fig. 8Ei to
iii). R6 vIRF was expressed in both the cytoplasm and nucleus,
FIG 6Production of IFN-␣by pDCs in RRV-infected PBMC cultures. Freshly collected PBMCs from ESPF RMs were mock infected or infected with WTBACorvIRF-ko RRV (MOI⫽1) in serum-free medium. During the last 6 h of infection, brefeldin A (0.02g/l) was added to inhibit cytokine secretion, and at 12, 24, and 48 hpi, cells were surface stained for CD3, CD20, CD14, HLA-DR (MHC-II), CD11c, and CD123. PBMCs were then fixed, IFN-␣production was measured by intracellular cytokine staining, and cells were analyzed by flow cytometry. PBMCs were stimulated with HSV-1 for 12 h as a positive control for IFN-␣ production, and surface marker expression was utilized to delineate pDCs, as shown in Fig. 4C. (A) A representative experiment is shown, displaying the percentage of pDCs producing IFN-␣in each histogram. (B) This experiment was repeated with PBMCs from 6 different RMs, and the percentages of pDCs producing IFN-␣were compared via a pairedttest.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.47.543.69.253.2]with some colocalization with cellular IRF-3; however, the
co-localization of R6 vIRF and IRF-3 appeared to be enhanced
when cells were stimulated with poly(IC) (Fig. 8Eiv). The
co-localization of R6 vIRF and IRF-3 does not seem to be restricted
to either the cytoplasm or the nucleus, and there was no
appar-ent colocalization between R7 vIRF and cellular IRF-3, even
following poly(IC) stimulation (Fig. 8E). Likewise, vIRF
ex-pression did not inhibit the nuclear localization of IRF-3
fol-lowing poly(IC) stimulation, nor did poly(IC) stimulation
al-ter the expression of either vIRF (Fig. 8E). These results show
that R6 vIRF interacts and colocalizes with cellular IRF-3, and
this interaction may be enhanced when the cells are stimulated
with poly(IC) to induce IRF-3 activation.
DISCUSSION
KSHV allocates a significant portion of its genome to encoding
immunomodulatory proteins (3). Of particular interest are the
viral interferon regulatory factors (vIRFs), unique to KSHV and
RRV, a closely related simian gamma-2-herpesvirus (2, 54, 56, 60).
KSHV vIRF-1 was initially identified and named because of its
sequence similarities with cellular IRFs; specifically, it shares 13%
amino acid (aa) identity with human IRF-8 and IRF-9 (49).
More-FIG 7Nuclear accumulation of IRF-3 is inhibited during WTBACRRV infection of RFs. RFs were infected with WTBACRRV or vIRF-ko RRV (MOI⫽1). (A)Whole-cell extracts were analyzed for total IRF-3 (FL-425) and GAPDH by SDS-PAGE. (B and C) Native cellular lysates (40g per sample) were run on a 7.5% PAGE nondenaturing gel with 1% sodium deoxycholate in the cathode chamber. Native lysates were probed for IRF-3, and data were analyzed by using densitometry to quantify the amount of IRF-3 present in the dimeric form (slower-migrating band) in relation to total IRF-3 (the sum of IRF-3 present in the dimeric and monomeric forms). Data are expressed as a percentage based on this ratio (percentage of IRF3 present in the dimeric form) and are presented as averages (⫾SEM) of data from 3 independent experiments. (D) RFs were fixed and analyzed via indirect immunofluorescence for IRF-3 (SL012.1) (shown in red), and nuclei were detected by using Hoechst dye (blue). RFs were transfected with poly(IC) (10g for 6 h) as a positive control for IRF-3 activation/nuclear localization. A representative image at 6 hpi is shown (magnification,⫻400). (E) The percentage of cells expressing IRF-3 in the nucleus was calculated by counting⬃200 cells within 5 different fields under each condition. Data are represented as means (⫾SEM) of data from 5 separate experiments. (F) Cytosolic and nuclear lysates were analyzed by SDS-PAGE for total IRF-3 (FL-425) and phospho-Ser396 IRF-3 (IRF-3p). Full-length PARP and GAPDH were used as loading controls. Gel images are representative of 3 independent experiments. N.S., not significant; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PARP, poly(ADP-ribose) polymerase.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.50.541.65.464.2]over,
in vitro
studies have demonstrated that KSHV vIRF-1, -2,
and -3 possess a variety of mechanisms to independently inhibit
cellular IRFs (52). Similarly, RRV encodes eight vIRFs that also
share homology with cellular IRFs, and R6, R7, R8, R10, and R11
vIRFs share between 19 and 26% aa identity with KSHV vIRF-1
(60). However, the RRV vIRFs do not share any significant
homology with the other 3 KSHV vIRFs (60). Due to these
similarities, the RRV vIRFs are also proposed to interfere with
FIG 8R6 vIRF inhibits IRF-3-mediated transcription. RRV vIRFs (R6, R7, R10, and R11) were C-terminally tagged with HA, cloned into pcDNA3.1(⫺) using oligonucleotides listed in Table 2, and transiently transfected. (A) Whole-cell lysates were collected from RFs transfected with a single vIRF construct at 36 h posttrans-fection. Equal amounts of lysate were subjected to SDS-PAGE and probed for HA expression. (B) HEK293 cells were transfected overnight with 500 ng total DNA (vIRF-HA construct [50 ng or 0 ng], pISRE-LUC [250 ng], pRL_SV40 [10 ng], and empty pcDNA3.1 [190 ng or 240 ng]). Cells were then transfected with poly(IC) and assayed 6 h later. Firefly luciferase levels were normalized to constitutively expressedRenillaluciferase levels in each well, and all conditions were made relative to the positive control [empty vector plus poly(IC)]. KSHV vIRF-1 (K9) was included as a control. Data are average data (⫾SEM) from 4 independent experiments and are compared to the positive control via Student’sttest. (C) HEK293 cells were transfected as described above for panel B but with increasing amounts of R6-HA-pcDNA3.1 (5 ng, 25 ng, 50 ng, and 100 ng). Data are average data (⫾SEM) from 3 independent experiments. (D) RFs were nucleofected with R6-HA, R7-HA, or pEGFP or mock transfected (no DNA) for 48 h and immunoprecipitated with anti-HA or anti-IRF-3 antibody. Immunoprecipitated lysates were then analyzed by SDS-PAGE; half was used to probe for HA, and the other half was used to probe for IRF-3. The image is representative of 3 separate experiments. (E) At 40 h postnucleofection, RFs were transfected with poly(IC) for 6 h or mock transfected, fixed, and analyzed by immunofluorescence for the detection of vIRF (anti-HA) (green) and cellular IRF-3 (red) and stained with Hoechst (blue) for the detection of nuclei. I.P., immunoprecipitated; WB, Western blot.on November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.64.515.66.548.2]the transcriptional functions of cellular IRFs. The deletion of
the vIRFs using WT
BACRRV, presented herein, has finally
al-lowed an evaluation of the impact of vIRFs during
de novo
RRV
infection, something that has proven difficult with KSHV due
to its poor lytic replication
in vitro
. These data are the first to
demonstrate that the deletion of the vIRFs results in increased
gene expression and production of IFN and an increased
nu-clear accumulation of phosphorylated IRF-3 during
de novo
RRV infection.
The IFN response is an important component of a cell’s
anti-viral defenses. To effectively circumvent the induction of IFN, an
invading pathogen must also act quickly. Accordingly,
transcrip-tional analysis has demonstrated that all 8 RRV vIRFs are detected
by 12 hpi by RT-PCR, and vIRF R10 was expressed as early as 3 hpi
in RRV-infected tRFs and PBMCs (Fig. 1B and 4A). Previous
tran-scriptional analyses of RRV
26-95also demonstrated that at least 2
vIRFs (corresponding to R6 and R10) were expressed as early as 6
to 12 hpi (14). Likewise, KSHV vIRF-1 is also transcriptionally
upregulated upon the induction of lytic replication in latently
in-fected B cells (13). Therefore, the early expression of the vIRFs
during RRV infection suggests that they also play a role in
inhib-iting the innate antiviral response, including the induction of IFN.
A comparison of WT
BACand vIRF-ko RRV
de novo
infections
revealed no difference in growth kinetics in RFs, and there were
minimal effects on the expression of ORFs adjacent to the deleted
vIRF region. The detection of an additional, truncated ORF 57
transcript in vIRF-ko RRV infection did not affect viral growth in
RFs, and because the full-length ORF 57 transcripts were still
de-tected, it is unlikely that the less abundant, truncated transcript
interferes with the efficient expression of ORF 57. Further analysis
showed that vIRF-ko RRV can also infect PBMCs albeit at a lower
infection efficiency. Despite a decrease in the infection efficiency,
vIRF-ko RRV infection resulted in an earlier and more robust
induction of type I and type II IFN during RRV infection in both
tRFs and PBMCs. Not surprisingly, the IFN-
␣
produced within
RRV-infected PBMCs was restricted to pDCs (Fig. 6 and data not
shown), which are known to produce large amounts of IFN-
␣
in
response to a number of different viruses (11, 17, 19). The
de-crease in IFN-
␣
production following WT RRV infection cannot
be attributed to reduced levels of infection, since WT RRV
in-fected PBMCs at a higher frequency (Fig. 4B), and the infection
efficiencies in pDCs were comparable for both WT RRV- and
vIRF-ko RRV-infected PBMC cultures (Fig. 4D), as was the
sur-vival of pDCs (data not shown). The data presented here do not
directly demonstrate that pDCs producing IFN-
␣
are infected,
and the utilization of GFP-expressing clones of WT and vIRF-ko
RRV suggested that only a small percentage of IFN-
␣
-producing
pDCs also express GFP (data not shown). However, it is likely that
a maximal production of IFN-
␣
precedes the detection of GFP
expression in an RRV-infected cell, as the detection of a GFP signal
is inconsistent before 24 h post-RRV infection (data not shown),
and a more sensitive detection method is not currently available. It
is important to consider, however, that pDCs are unique in their
sole expression of Toll-like receptor 7 (TLR7) and TLR9, which
specifically equips them to recognize bacterial and viral nucleic
acids, including genomic DNAs from several herpesviruses (31,
53). Moreover, it was recently demonstrated that KSHV infection
of pDCs induces the production of IFN-␣
in a TLR9-dependent
manner (70). Therefore, the detection of RRV transcripts by 3 hpi
in RRV-infected PBMC cultures (Fig. 3A) and the dramatic
in-crease in IFN transcript levels at 6 hpi imply that pDCs are likely
responding to direct infection with RRV, potentially through the
recognition of its genomic DNA.
The induction of type I IFNs is orchestrated by IRF-3,
consti-tutively expressed in most cell types, and IRF-7, consticonsti-tutively
expressed in pDCs. These IRFs are quickly activated following
viral infection and subsequently accumulate in the nucleus, where
they drive the transcription of type I IFNs. Data presented herein
suggest that vIRFs inhibit the nuclear accumulation of
phosphor-ylated IRF-3 within the first 8 h of RRV infection, preceding the
inhibition of type I IFN induction during RRV infection, which
peaked at around 12 hpi. Additionally, reporter assays were used
to examine the role of individual RRV vIRFs in inhibiting the
induction of IFN. These analyses showed that R6 vIRF and, to a
lesser extent, R10 and R11 vIRFs significantly inhibit
IRF-3-mediated transcription. Further examination of the relationship
between R6 vIRF and IRF-3 revealed that R6 vIRF can directly or
indirectly interact with endogenous IRF-3, and R6 vIRF and IRF-3
also colocalize within the cell. Therefore, R6 vIRF could be
seques-tering IRF-3, disrupting IRF-3 associations with other
transcrip-tional factors, or inhibiting IRF-3 from binding promoter regions
in target genes, such as type I IFN. Indeed, KSHV vIRFs are known
to bind cellular transcription factors and inhibit the induction of
type I IFN; KSHV vIRF-1 can bind p300/CBP and inhibit its
es-sential interaction with IRF-3 (9, 39), and KSHV vIRF-3 can bind
to and retain NF-B in the cytoplasm (61).
Interestingly, R7 vIRF enhanced IRF-3-mediated transcription
in our reporter assays. This phenotype is potentially independent
of a direct targeting of IRF-3, as a further characterization of the
relationship between R7 vIRF and IRF-3 showed that R7 vIRF
does not bind or colocalize with IRF-3 in the cell (Fig. 8).
How-ever, the R7 vIRF-mediated enhancement of transcription was not
entirely surprising, since KSHV vIRF-1 and vIRF-3 have also been
shown to enhance the transcription of cellular genes (41, 42, 55).
Moreover, a role for vIRFs in the stimulation of IRF-mediated
transcription (41) could promote the cytokine-enhanced
reacti-vation of the virus (47), providing the necessary drive to maintain
an adequate pool of infected cells within the host.
In addition to establishing an antiviral state early during viral
infection, type I IFNs (IFN-␣/) also have effects on NK cells (5,
50), T cells (25, 46, 65), and DCs (27, 44, 48), directly impacting
the adaptive immune response. Therefore, the inhibition of type I
IFNs by vIRFs could disrupt both the innate and adaptive immune
responses during
in vivo
RRV infection. Additionally, our data
indicate that RRV vIRFs also inhibit the induction of type II IFN
(IFN-␥), which is essential for promoting an efficient Th1
im-mune response. In fact, previous
in vitro
analyses demonstrated
that KSHV vIRF-1 and vIRF-3 can also interfere with type II IFN
signaling. Specifically, KSHV vIRF-1 inhibits transcription
in-duced by IFN-␥
(37, 76) and results in the decreased surface
ex-pression of major histocompatibility complex class I (MHC-I)
(34). Likewise, KSHV vIRF-3 similarly inhibits IFN-␥-responsive
promoters, resulting in a decreased expression of MHC-II on
la-tently infected B cells (59). A preliminary examination of IFN-␥
production in RRV-infected PBMC cultures, and T cells and DCs
specifically, did not show any measurable production over levels
in mock-infected cultures in the first 24 hpi (data not shown),
despite the significant induction at the transcript level in vIRF-ko
RRV infection (Fig. 5). However, the vIRF-mediated inhibition of
IFN-␥
may be important for other cell types that were not
on November 7, 2019 by guest
http://jvi.asm.org/
ined here, such as natural killer cells (5, 66). Additionally, vIRFs
may be essential for inhibiting IFN-␥
at later times during RRV
infection, potentially in response to other cytokines, such as
interleukin-12 (IL-12) (50), which would be important during the
development of the adaptive immune response. Future studies
will utilize vIRF-ko RRV to focus on the impact of vIRFs during
in
vivo
RRV infection in the rhesus macaque to further our
under-standing of their role(s) in immune evasion and pathogenesis.
REFERENCES
1.Albert TJ, et al.2005. Mutation discovery in bacterial genomes: metro-nidazole resistance in Helicobacter pylori. Nat. Methods2:951–953. 2.Alexander L, et al.2000. The primary sequence of rhesus monkey
rhadi-novirus isolate 26-95: sequence similarities to Kaposi’s sarcoma-associated herpesvirus and rhesus monkey rhadinovirus isolate 17577. J. Virol.74:3388 –3398.
3.Areste C, Blackbourn DJ.2009. Modulation of the immune system by Kaposi’s sarcoma-associated herpesvirus. Trends Microbiol.17:119 –129. 4.Bergquam EP, Avery N, Shiigi SM, Axthelm MK, Wong SW. 1999. Rhesus rhadinovirus establishes a latent infection in B lymphocytes in vivo. J. Virol.73:7874 –7876.
5.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP.1999. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol.17:189 –220.
6. Bowie AG, Unterholzner L. 2008. Viral evasion and subversion of pattern-recognition receptor signalling. Nat. Rev. Immunol.8:911–922. 7. Boyne JR, Whitehouse A. 2006. Gamma-2 herpes virus
post-transcriptional gene regulation. Clin. Microbiol. Infect.12:110 –117. 8.Brown KN, Barratt-Boyes SM.2009. Surface phenotype and rapid
quan-tification of blood dendritic cell subsets in the rhesus macaque. J. Med. Primatol.38:272–278.
9. Burysek L, et al. 1999. Functional analysis of human herpesvirus 8-encoded viral interferon regulatory factor 1 and its association with cellular interferon regulatory factors and p300. J. Virol.73:7334 –7342. 10. Burysek L, Yeow WS, Pitha PM.1999. Unique properties of a second
human herpesvirus 8-encoded interferon regulatory factor (vIRF-2). J. Hum. Virol.2:19 –32.
11. Chung E, et al.2005. Characterization of virus-responsive plasmacytoid dendritic cells in the rhesus macaque. Clin. Diagn. Lab. Immunol.12:426 – 435.
12. Dai J, Megjugorac NJ, Amrute SB, Fitzgerald-Bocarsly P.2004. Regu-lation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J. Immunol.173:1535–1548.
13. Dittmer DP.2003. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res.63:2010 –2015.
14. Dittmer DP, et al.2005. Whole-genome transcription profiling of rhesus monkey rhadinovirus. J. Virol.79:8637– 8650.
15. Donnelly RP, Kotenko SV.2010. Interferon-lambda: a new addition to an old family. J. Interferon Cytokine Res.30:555–564.
16. Estep RD, Powers MF, Yen BK, Li H, Wong SW.2007. Construction of an infectious rhesus rhadinovirus bacterial artificial chromosome for the analysis of Kaposi’s sarcoma-associated herpesvirus-related disease devel-opment. J. Virol.81:2957–2969.
17. Feldman SB, et al.1994. Viral induction of low frequency interferon-alpha producing cells. Virology204:1–7.
18. Fensterl V, Sen GC.2009. Interferons and viral infections. Biofactors 35:14 –20.
19. Fitzgerald-Bocarsly P, Dai J, Singh S.2008. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev.19:3–19.
20. Flowers CC, Flowers SP, Nabel GJ.1998. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor confers resistance to the an-tiproliferative effect of interferon-alpha. Mol. Med.4:402– 412. 21. Fuld S, Cunningham C, Klucher K, Davison AJ, Blackbourn DJ.2006.
Inhibition of interferon signaling by the Kaposi’s sarcoma-associated her-pesvirus full-length viral interferon regulatory factor 2 protein. J. Virol. 80:3092–3097.
22. Gao SJ, et al.1997. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene15:1979 –1985.
23. Genin P, Vaccaro A, Civas A.2009. The role of differential expression of human interferon-a genes in antiviral immunity. Cytokine Growth Factor Rev.20:283–295.
24. Honda K, Taniguchi T.2006. IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol.6:644 – 658.
25. Huber JP, Farrar JD.2011. Regulation of effector and memory T-cell functions by type I interferon. Immunology132:466 – 474.
26. Isaacs A, Lindenmann J.1957. Virus interference. I. The interferon. Proc. R. Soc. Lond. B Biol. Sci.147:258 –267.
27. Ito T, et al.2001. Differential regulation of human blood dendritic cell subsets by IFNs. J. Immunol.166:2961–2969.
28. Iwamura T, et al.2001. Induction of IRF-3/-7 kinase and NF-kappaB in response to double-stranded RNA and virus infection: common and unique pathways. Genes Cells6:375–388.
29. Joo CH, et al.2007. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J. Virol.81:8282– 8292. 30. Juang YT, et al.1998. Primary activation of interferon A and interferon B
gene transcription by interferon regulatory factor 3. Proc. Natl. Acad. Sci. U. S. A.95:9837–9842.
31. Ketloy C, et al.2008. Expression and function of Toll-like receptors on dendritic cells and other antigen presenting cells from non-human pri-mates. Vet. Immunol. Immunopathol.125:18 –30.
32. Kirchoff V, Wong S, St Jeor S, Pari GS. 2002. Generation of a life-expanded rhesus monkey fibroblast cell line for the growth of rhesus rhadinovirus (RRV). Arch. Virol.147:321–333.
33. Koyama S, Ishii KJ, Coban C, Akira S.2008. Innate immune response to viral infection. Cytokine43:336 –341.
34. Lagos D, et al.2007. Kaposi sarcoma herpesvirus-encoded vFLIP and vIRF1 regulate antigen presentation in lymphatic endothelial cells. Blood 109:1550 –1558.
35. Lee EC, et al.2001. A highly efficient Escherichia coli-based chromosome engineering system adapted for recombinogenic targeting and subcloning of BAC DNA. Genomics73:56 – 65.
36. Li M, et al.2000. Inhibition of p300 histone acetyltransferase by viral interferon regulatory factor. Mol. Cell. Biol.20:8254 – 8263.
37. Li M, et al.1998. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor. J. Virol.72:5433–5440.
38. Lin R, Genin P, Mamane Y, Hiscott J.2000. Selective DNA binding and association with the CREB binding protein coactivator contribute to dif-ferential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Mol. Cell. Biol.20:6342– 6353.
39. Lin R, et al.2001. HHV-8 encoded vIRF-1 represses the interferon