Prolactin Regulatory Element Binding Protein Is Involved in Hepatitis
C Virus Replication by Interaction with NS4B
Lingbao Kong,aAkira Fujimoto,aMariko Nakamura,aHaruyo Aoyagi,aMami Matsuda,aKoichi Watashi,aRyosuke Suzuki,a Minetaro Arita,aSatoshi Yamagoe,bNaoshi Dohmae,cTakehiro Suzuki,cYuriko Sakamaki,dShizuko Ichinose,dTetsuro Suzuki,e Takaji Wakita,aHideki Aizakia
Department of Virology II, National Institute of Infectious Diseases, Shinjuku-ku, Tokyo, Japana
; Department of Chemotherapy and Mycoses, National Institute of Infectious Diseases, Shinjuku-ku, Tokyo, Japanb
; RIKEN Center for Sustainable Resource Science, Saitama, Japanc
; Research Center for Medical and Dental Sciences, Tokyo Medical and Dental University, Bunkyo-ku, Tokyo, Japand
; Department of Infectious Diseases, Hamamatsu University School of Medicine, Hamamatsu, Shizuoka, Japane
ABSTRACT
It has been proposed that the hepatitis C virus (HCV) NS4B protein triggers the membranous HCV replication compartment,
but the underlying molecular mechanism is not fully understood. Here, we screened for NS4B-associated membrane proteins by
tandem affinity purification and proteome analysis and identified 202 host proteins. Subsequent screening of replicon cells with
small interfering RNA identified prolactin regulatory element binding (PREB) to be a novel HCV host cofactor. The interaction
between PREB and NS4B was confirmed by immunoprecipitation, immunofluorescence, and proximity ligation assays. PREB
colocalized with double-stranded RNA and the newly synthesized HCV RNA labeled with bromouridine triphosphate in HCV
replicon cells. Furthermore, PREB shifted to detergent-resistant membranes (DRMs), where HCV replication complexes reside,
in the presence of NS4B expression in Huh7 cells. However, a PREB mutant lacking the NS4B-binding region (PREBd3) could
not colocalize with double-stranded RNA and did not shift to the DRM in the presence of NS4B. These results indicate that PREB
locates at the HCV replication complex by interacting with NS4B. PREB silencing inhibited the formation of the membranous
HCV replication compartment and increased the protease and nuclease sensitivity of HCV replicase proteins and RNA in DRMs,
respectively. Collectively, these data indicate that PREB promotes HCV RNA replication by participating in the formation of the
membranous replication compartment and by maintaining its proper structure by interacting with NS4B. Furthermore, PREB
was induced by HCV infection
in vitro
and
in vivo
. Our findings provide new insights into HCV host cofactors.
IMPORTANCE
The hepatitis C virus (HCV) protein NS4B can induce alteration of the endoplasmic reticulum and the formation of a
membra-nous web structure, which provides a platform for the HCV replication complex. The molecular mechanism by which NS4B
in-duces the membranous HCV replication compartment is not understood. We screened for NS4B-associated membrane proteins
by tandem affinity purification and proteome analysis, followed by screening with small interfering RNA. We identified
prolac-tin regulatory element binding (PREB) to be a novel HCV host cofactor. PREB is induced by HCV infection and recruited into
the replication complex by interaction with NS4B. Recruited PREB promotes HCV RNA replication by participating in the
for-mation of the membranous HCV replication compartment. To our knowledge, the effect of NS4B-binding protein on the
forma-tion of the membranous HCV replicaforma-tion compartment is newly described in this report. Our findings are expected to provide
new insights into HCV host cofactors.
H
epatitis C virus (HCV) infects about 2% of the world’s
pop-ulation and leads to several diseases, including chronic
hep-atitis, liver cirrhosis, and hepatocellular carcinoma (HCC). HCV
is the most common reason for patients requiring a liver
trans-plant in developed countries and is thus a significant health
bur-den worldwide (1). To date, there are no efficacious vaccines
against HCV infection (2). The standard-of-care therapy against
HCV consists of pegylated alpha interferon (peg-IFN-
␣
) and
riba-virin, which can cure approximately 50% of HCV-infected
pa-tients. Direct-acting antivirals (DAAs) have recently been used
and have increased the cure rate to about 90% (3). Therefore, the
development of an HCV vaccine and the identification of host
factors for new anti-HCV targets remain significant challenges.
HCV cell culture systems containing replicon systems,
pseu-dotyped virus, and virus propagation systems have recently been
established. These systems are helpful for dissecting the viral life
cycle, identifying promising targets, and developing antiviral
compounds (4). Major challenges in research toward cures for
HCV infection, such as virus diversity, virus resistance, and host
genetics, are prompting considerable study of HCV-host
interac-tions. A number of host factors influencing the translation,
repli-cation, assembly, and release of HCV have recently been identified
using RNA interference screening and mass spectrometry (MS)
Received23 June 2015 Accepted30 December 2015
Accepted manuscript posted online6 January 2016
CitationKong L, Fujimoto A, Nakamura M, Aoyagi H, Matsuda M, Watashi K, Suzuki
R, Arita M, Yamagoe S, Dohmae N, Suzuki T, Sakamaki Y, Ichinose S, Suzuki T, Wakita T, Aizaki H. 2016. Prolactin regulatory element binding protein is involved in hepatitis C virus replication by interaction with NS4B. J Virol 90:3093–3111. doi:10.1128/JVI.01540-15.
Editor:J.-H. J. Ou
Address correspondence to Hideki Aizaki, [email protected].
Copyright © 2016, American Society for Microbiology. All Rights Reserved.
crossmark
on November 7, 2019 by guest
http://jvi.asm.org/
approaches. Unfortunately, the significance of most of these
fac-tors with respect to the HCV life cycle is currently unknown.
Positive-strand RNA virus infection may alter the host cell
membrane, resulting in the formation of distinct vesicle
struc-tures. These vesicle structures provide a platform for the virus
replication complex. The molecular mechanism of the
virus-in-duced membrane replication compartment is poorly understood.
HCV NS4B is a 27-kDa endoplasmic reticulum (ER)
membrane-associated protein. It induces alteration of the ER membrane and
the formation of a membranous web structure, which provides a
platform for the HCV replication complex (5). Increasing
evi-dence indicates that HCV NS4B can mediate virus-host
interac-tions by direct or secondary interacinterac-tions with cellular proteins,
and these interactions play important roles in the HCV life cycle
and pathogenesis (6).
The goal of this study was to identify new NS4B-associated host
membrane proteins involved in the HCV life cycle. We identified
prolactin regulatory element binding (PREB), also known as
Sec12, which has a function as the regulatory factor promoting
COPII vesicle budding (7), to be a novel HCV host cofactor. PREB
is induced by HCV infection and recruited into the replication
complex to promote viral RNA replication and the formation of
the membranous replication compartment. To our knowledge,
the effect of NS4B-binding protein on the formation of the
mem-branous HCV replication compartment is newly described in this
report. Consequently, the findings presented here provide new
insights into HCV host cofactors.
MATERIALS AND METHODS
Cell culture.Huh7 human hepatoma cells and HeLa cervical cancer cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) con-taining 10% fetal bovine serum (FBS). Huh7 cells harboring the genotype 1b subgenomic replicon (LucNeo#2; here designated SGR1b) or the ge-notype 2a JFH1 subgenomic replicon (SGRlucneo; here designated SGR2a) were maintained in complete DMEM supplemented with 0.5 mg/ml G418 (8,9).
Plasmid construction.Plasmids pJFH1 (containing the full-length HCV genotype2a JFH1 strain cDNA), pSGRlucneo (containing the geno-type 2a JFH1 subgenomic replicon), and pSGRlucneoGND (containing an inactivating mutation within NS5B) were previously described (10). The poliovirus replicon encoding firefly luciferase (Fluci) in place of the capsid genes was previously described (11).
The transmembrane domains (amino acids [aa] 82 to 104, aa 114 to 133, aa 140 to 159, and aa 169 to 191), the LIG_EH1_1 domain (aa 117 to 125), and the TRG_ER_diArg_1 domains (aa 191 to 193 and aa 246 to 248) of NS4B were predicted to interact with PREB domains, such as the WD40 domains (aa 71 to 110, aa 113 to 151, aa 217 to 256, and aa 259 to 310) or the transmembrane domains (aa 244 to 266 and aa 319 to 341) using basicELM software. Therefore, we constructed NS4B and PREB with the following deletions. To generate dually tagged NS4B expression plasmids for screening for NS4B-associated cellular proteins, the NS4B DNA fragment amplified by PCR was inserted into the NotI and KpnI sites of the pCNX2-Flag-HA vector, and the resultant plasmid was desig-nated pCXN2-Flag-HA-NS4B. To generate myc-tagged full-length NS4B and NS4B deletion constructs, including NS4Bd1 lacking aa 192 to 261, NS4Bd2 lacking aa 140 to 261, and NS4Bd3 lacking aa 1 to 133, the PCR-amplified full-length NS4B or NS4B deletion mutant sequences were in-serted into the pcDNA3-MEF vector, which includes the myc tag. The resultant full-length NS4B fragment and NS4B fragments with deletions with the myc tag at the N terminus were separately cloned into pCAGGS. To generate the full-length PREB pCAG-PREB construct, PREB cDNA was amplified using total RNA from Huh7 cells as a template and inserted into pCAGGS. To generate V5-tagged PREB and PREB deletion
constructs, including PREBd1 (lacking aa 259 to 345), PREBd2 (lacking aa 218 to 345), and PREBd3 (lacking aa 1 to 110), cDNAs encoding full-length PREB or PREB deletion mutant sequences with a V5 tag at the N terminus were amplified using pCAG-PREB as a template. The resultant fragments were cloned into pCAGGS.
pSilencer-shPREB expressing a short hairpin RNA (shRNA) targeting PREB (shPREB) under the control of the U6 promoter was constructed by cloning the oligonucleotide pair 5=-GATCCGGCTTATTATTGTGACC
ATTTCAAGAGAATGGTCACAATAATAAGCTTTTTTGGAAA-3= and
5=-AGCTTTTCCAAAAAAGCTTATTATTGTGACCATTCTCTTGAA
ATGGTCACAATAATAAGCCG-3= between the BamHI and HindIII
sites of pSilencer 2.1-U6 hygro (Ambion, Austin, TX). To generate the pCAG-PREBshr construct expressing shRNA-resistant PREB (PREBshr), the cDNA fragment encoding PREB, in which the shRNA-targeting re-gion (5=-GGGCTTATTATTGTGACCAT-3=) was replaced with GGTTT GATCATAGTAACGATT (resulting in no change in the amino acid se-quence), was amplified by PCR using pCAG-PREB as a template. The resulting fragment was confirmed by sequencing and then cloned into pCAGGS.
Antibodies.Mouse monoclonal antibodies against actin, bromode-oxyuridine, Flag, and CD81 were obtained from Sigma-Aldrich (St. Louis, MO). Mouse monoclonal double-stranded RNA (dsRNA) antibody was obtained from Biocenter Ltd. (Szirak, Hungary). Mouse monoclonal an-tibody against caveolin was obtained from BD Transduction Laboratories (San Jose, CA). Mouse monoclonal antibody against the HCV core pro-tein and rabbit polyclonal antibody against NS5A were described else-where (12). Rabbit polyclonal antibody against calnexin was obtained from Stressgen Bioreagents (Victoria, BC, Canada). Polyclonal antibodies against V5, myc, and PREB were obtained from Sigma-Aldrich. Goat polyclonal antibody against 24-dehydrocholesterol reductase (DHCR) was obtained from Santa Cruz Biotechnology (Dallas, TX). Anti-NS4B antibody was a gift from M. Kohara (13).
Screening of NS4B-associated cellular membrane fraction proteins.
pCNX2-Flag-HA-NS4B was transfected into HeLa cells to establish a sta-ble cell line expressing epitope-tagged NS4B under G418 selection. The cells were lysed in HB buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 1.5 mM MgCl2, 1⫻Complete protease inhibitor cocktail [Roche, Tokyo, Ja-pan]) using a Dounce homogenizer, and the nuclei were removed by centrifugation at 3,300⫻gfor 10 min. After addition of glycerol at a final concentration of 20% (vol/vol), the cell lysate was ultracentrifuged at 100,000⫻gfor 1 h. The resultant pellet was resuspended in 7 volumes of buffer (20 mM Tris-HCl [pH 7.5], 1.5 mM MgCl2, 0.2 mM EDTA, 0.02 mM KCl, 25% glycerol, 1⫻Complete, 2% Triton X-100 [TX100], 100 mM NaCl) and incubated at 4°C for 1 h. An anti-Flag M2 agarose affinity gel (Sigma-Aldrich) was added to the membrane fraction obtained after ultracentrifugation at 100,000⫻gfor 1 h, and the mixture was incubated at 4°C overnight and then loaded onto a empty Poly-Prep column (Bio-Rad, Hercules, CA). The column was washed with wash buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 10% glycerol, 0.1% Tween 20), and the immunocomplex was eluted two times with 300g/ml of 3⫻Flag peptide (Sigma-Aldrich) in wash buffer. Ana nti-hemagglutinin (anti-HA) affin-ity matrix (Roche) was added to the eluates, and the mixture was incu-bated at 4°C overnight and then washed with wash buffer. The immuno-complex was eluted with buffer containing 100 mM glycine-HCl (pH 2.5) and 10% glycerol, and the eluates were incubated with 10% trichloro-acetic acid on ice for 30 min. After centrifugation, the pellet was washed two times with acetone, dissolved in SDS sample buffer, and then sepa-rated on an SDS-polyacrylamide gel and silver stained using a Silver Stain MS kit (Wako, Osaka, Japan). The excised gel bands were reduced with dithiothreitol and carboxymethylated with iodoacetic acid. Then, the gel bands were treated with tosylsulfonyl phenylalanyl chloromethyl ketone-treated trypsin at 37°C overnight. The resultant peptides were analyzed by nano-liquid chromatography (LC)-MS/MS using an LCQ Deca XP ion-trap mass spectrometer (Thermo Scientific, Waltham, MA). The MS/MS spectra were searched against those in the nonredundant NCBI (NCBInr)
on November 7, 2019 by guest
http://jvi.asm.org/
database using an in-house MASCOT server (version 2.2.1; Matrix Sci-ences, Boston, MA).
RNA interference, DNA transfection, and cell viability.The small interfering RNAs (siRNAs) were purchased from Sigma-Aldrich and were introduced into the cells using the Lipofectamine RNAiMAX reagent (In-vitrogen, Tokyo, Japan). siRNAs targeting PREB (siPREB) included siPREB (5=-GGCUUAUUAUUGUGACCAU-3=), siPREB2 (5=-CUGACA
AGAUGAAUGCGCA-3=), and siPREB3 (5=-GAAGAAAUGUGGAGCG
GAA-3=). Silencing of DHCR has been reported to inhibit HCV replica-tion (14) and was used as a positive control. Nontargeting siRNA (siNT) was used as a negative control. DNA transfection was performed using the Trans LT1 transfection reagent (Mirus, Madison, WI) following the man-ufacturer’s instructions. Cell viability was analyzed using a Cell Titer-Glo luminescent cell viability assay (Promega, Madison, WI) according to the manufacturers’ protocol.
Establishment of stable cells expressing shRNA.Huh7 cells were transfected with pSilencer-shPREB or the negative-control pSilencer hy-gro vector (shNC), which expresses a hairpin siRNA with limited homol-ogy to any known sequences in the human, mouse, and rat genomes. Drug-resistant clones were selected by treatment with hygromycin B (Wako, Tokyo, Japan) at a final concentration of 300 mg/ml for 4 weeks.
HCV replication assay.For the HCV replication assay, cells in which HCV was replicating were harvested and luciferase activity was measured using a luciferase reporter assay system kit (Promega) according to the manufacturer’s protocol. The HCV RNA level was measured by real-time reverse transcription-PCR (RT-PCR) as described previously (10).
Measurement of PREB mRNA levels.The PREB mRNA level was measured by real-time RT-PCR (Applied Biosystems, Grand Island, NY) according to the manufacturer’s protocol.
HCV propagation assay.Plasmid pJFH1 was used to generate infec-tious JFH1 virus in Huh7 cells, as described previously (10). Naive Huh7 cells were infected with cell culture-produced JFH1 virus and then treated with siRNAs. After 3 days, the concentration of the HCV core antigen in filtered culture medium and in the infected cell lysate fraction was deter-mined by enzyme-linked immunosorbent assay (ELISA) using a Lumi-pulse Ortho HCV antigen kit (Ortho Clinical Diagnostics, Tokyo, Japan) as described previously (9). To analyze the infectivity of the HCV particles in the cells, JFH1-infected Huh7 cells were subjected to four cycles of freezing and thawing using dry ice and a 37°C water bath and centrifuged at 1,200 rpm for 5 min at 4°C to remove the cell debris, and then the supernatants were collected and used to infect Huh7 cells. At 3 days postinfection, the concentration of HCV core antigen in the cell lysate fraction of the infected cells was determined by ELISA using the Lumi-pulse Ortho HCV antigen kit. Using an immunoassay that also provided results indicative of HCV infectivity, we confirmed a good correlation between the levels of core antigen and infectious titers (8). The intracel-lular specific infectivity was determined by evaluation of the infectious HCV core antigen level relative to the total HCV core antigen level as described previously (8).
HCVpp systems.An HCV pseudoparticle (HCVpp) harboring the E1 and E2 glycoproteins of the TH clone (genotype 1b) (HCVpp1b), an HCVpp harboring the E1 and E2 glycoproteins of the JFH1 clone (geno-type 2a) (HCVpp2a), and a vesicular stomatitis virus (VSV) pseudopar-ticle containing VSV glycoprotein G (VSVpp) were produced as previ-ously described (12,15). Huh7 cells were seeded into 24-well plates, incubated overnight at 37°C, treated with the siRNAs indicated below for 3 days, and infected with untreated HCVpp or HCVpp that had been pretreated with CD81 antibody (0.5 mg/ml) for 2 h. At 4 h postinfection, the medium was replaced with DMEM with 10% FBS, and the cells were harvested 3 days later to determine intracellular luciferase activity.
Infection with dengue virus.Naive Huh7 cells were infected with dengue virus and then treated with siRNAs. After 3 days, the amount of dengue virus RNA in the infected cell was determined by the TaqMan RT-PCR method as described previously (16).
Expression of T7 RNA polymerase in Huh7 cells.Huh7 cells were infected with AdexCAT7, a recombinant adenovirus containing the bac-teriophage T7 RNA polymerase, and AxCAwt, a control virus containing only the promoter sequence, as described previously (17).
Translation and replication of HCV RNA.SGR2a RNAs and renilla luciferase (Rluci) RNA (Promega) were synthesized byin vitro transcrip-tion as described previously (10). Huh7 cells were electroporated with 1
g of SGR2a RNAs and 0.1g of Rluci RNA as an internal control by use of a single pulse of 260 V and 950F with a Bio-Rad Gene Pulser II apparatus (Bio-Rad, Hercules, CA). For analysis of RNA translation, transfected cells were harvested to determine Fluci and Rluci activities at 4 h posttransfection by use of a dual-luciferase reporter assay system (Pro-mega) following the manufacturer’s instructions. The numbers of relative light units (RLU) were measured with a luminometer (Berthod, Wildbad, Germany), and Fluci activity was normalized to Rluci activity. For analysis of RNA replication, transfected cells were harvested to determine the SGR2a RNA level at 3 days posttransfection.
Immunoprecipitation.Cells were washed with ice-cold phosphate-buffered saline (PBS) and suspended in lysis buffer (20 mM Tris-HCl [pH 7.4] containing 135 mM NaCl, 1% Triton X-100, and 10% glycerol) sup-plemented with 50 mM NaF, 5 mM Na3VO4, and EDTA-free Complete protease inhibitor cocktail (Roche). Cell lysates were sonicated for 10 min and then incubated for 30 min at 4°C, followed by centrifugation at 14,000⫻gfor 10 min. The supernatants were immunoprecipitated with anti-NS4B, anti-PREB, anti-myc, or anti-V5 antibody in the pres-ence of protein G Dynabeads (Invitrogen). The immunocomplexes with the beads were collected by centrifugation at 800⫻gfor 30 s and were then washed four times with lysis buffer. The proteins binding to the beads were boiled with SDS sample buffer and then subjected to SDS-PAGE.
Immunoblots.Cells were washed with PBS and lysed with 50 mM Tris-HCl (pH 7.4), 300 mM NaCl, 1% Triton X-100. The lysates were sonicated for 10 min and added to the same volume of SDS sample buffer. The protein samples were boiled for 10 min, separated by SDS-PAGE, and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA). After the membranes were blocked, they were probed with the pri-mary antibodies, followed by incubation with peroxidase-conjugated sec-ondary antibody. Antigen-antibody complexes were visualized by use of an enhanced chemiluminescence detection system (Super Signal West Pico chemiluminescent substrate; Pierce, Rockford, IL) according to the manufacturer’s protocol and were detected using an LAS-3000 image an-alyzer system (Fujifilm, Tokyo, Japan).
Indirect immunofluorescence assay.Cells were fixed in acetone-methanol (1:1) for 10 min at⫺20°C. Cells were blocked and then incu-bated with the primary antibodies. After they were washed with PBS, the cells were incubated with Alexa Fluor 555 or 488 secondary antibodies. Cell nuclei were stained with DAPI (4=,6-diamidino-2-phenylindole). Stained samples were then examined with a Leica TCS SPE confocal mi-croscope (Leica Microsystems, Tokyo, Japan). The surface area of each color was calculated using MetaMorph software (Molecular Devices Cor-poration, Sunnyvale, CA).
PLA.The proximity ligation assay (PLA) was performed according to the manufacturer’s instructions (Olink Bioscience, Uppsala, Sweden). Huh7 cells were transfected with an expression plasmid, NS4B tagged with myc (NS4Bmyc) and/or PREB tagged with V5 (PREBv5). Transfected cells were grown on glass coverslips. At 2 days posttransfection, cells were fixed with 4% paraformaldehyde in PBS for 20 min and then blocked and permeabilized with 0.3% Triton X-100 in a nonfat milk solution (Block Ace; Snow Brand Milk Products Co., Sapporo, Japan) for 60 min at room temperature. The samples were incubated with a mixture of mouse an-ti-V5 monoclonal antibody and rabbit anti-myc polyclonal antibody for 60 min, washed three times, and incubated with PLA probes. After the samples were washed, the ligation mixture containing connector oligonu-cleotide was added for 30 min. The washing step was repeated, and an amplification mixture containing fluorescently labeled DNA probe was
PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
added for 100 min. Finally, the samples were washed and mounted with DAPI mounting medium. The samples were analyzed for a signal repre-senting an interaction using a Leica TCS SPE confocal microscope.
Labeling and immunofluorescence staining ofde novo-synthesized viral RNA.The procedures for labeling and immunofluorescence staining ofde novo-synthesized viral RNA were modified from previously de-scribed methods (18,19). Briefly, cells were plated on 8-well chamber slides at a density of 5⫻104cells per well. At 1 day after seeding, the cells were treated with actinomycin D (10g/l) for 1 h and then transfected with 2 mM bromouridine triphosphate (BrUTP) using the Lipofectamine RNAiMAX reagent according to the manufacturer’s instructions. After 1 h, the cells were fixed and processed for immunofluorescence staining.
Membrane flotation assay.The membrane flotation assay was per-formed as previously described (20). The cells were first lysed in 0.5 ml of hypotonic buffer (10 mM Tris-HCl [pH 7.5], 10 mM KCl, 5 mM MgCl2) and passed through a 25-gauge needle 20 times. The nuclei and unbroken cells were removed by centrifugation at 1,000⫻gfor 5 min in a micro-centrifuge at 4°C. The cell lysates were then mixed with 3 ml of 75% sucrose in low-salt buffer (LSB; 50 mM Tris-HCl [pH 7.5], 25 mM KCl, 5 mM MgCl2) and overlaid with 4 ml of 55% sucrose in LSB, followed by 1.5 ml of 10% sucrose in LSB. To isolate the detergent-resistant membrane (DRM) fractions of the cells, the cell lysates were treated with 1% TX100 at 4°C or 37°C for 1 h before being loaded onto a sucrose gradient. The sucrose gradient was centrifuged at 38,000 rpm in a Beckman SW41Ti rotor for 14 h at 4°C. One-milliliter fractions were collected from the top (10% sucrose fraction) and bottom (75% sucrose fraction) of the gradi-ent, and each was concentrated by passage through a Centricon YM-30 filter unit (Millipore).
Resistance assays of RNase and protease.The DRM from cells was used to analyze RNase and protease resistance. For analysis of RNase resistance, the sample was treated with RNase A (500 ng/ml) at 37°C for 5 min, and then the HCV RNA level was determined by real-time RT-PCR and the protein level was determined by immunoblot analysis. For anal-ysis of protease resistance, the sample was treated with trypsin (25g/ml) at 37°C for 10 min, and then the protein level was determined by immu-noblot analysis.
Ultrastructural analysis.Cells or the cellular membrane fraction iso-lated by a membrane flotation assay was fixed with 1.5% glutaraldehyde in 1.0% cacodylate buffer (pH 7.4) for 5 min, postfixed with 2% OsO4in phosphate buffer (pH 7.4) for 1 h, dehydrated in ethanol, and embedded in Epon. Ultrathin sections were double stained and examined using a JEM-ARM200F transmission electron microscope (TEM; JEOL, Tokyo, Japan).
HCV infection and immunoblot analysis of SCID mice with human-ized liver.SCID mice transgenic for the urokinase-type plasminogen ac-tivator gene and in which the liver was repopulated with human hepato-cytes (chimeric mice) were purchased from Phoenix Bio Co., Ltd. (Hiroshima, Japan). The chimeric mice were infected with serum con-taining HCV isolate S310, which has a high infectivity for primary human hepatocytes (21). Mock-infected mice were used as controls. Lysates of mouse liver tissues were analyzed by immunoblot analysis. Allin vivo experiments and protocols for animal experiments were approved by the Laboratory Animal Ethics Committee at PhoenixBio Co., Ltd.
Statistical analysis.Statistical analysis was performed by one-way analysis of variance using the Statistical Package for the Social Sciences (SPSS) program (version 11.5), and significant differences among groups were determined by use of the least significant difference (LSD). The ac-cepted level of statistical significance was aPvalue of⬍0.05.
RESULTS
Identification of NS4B-associated host cofactors by tandem
af-finity purification, proteomic analysis, and siRNA silencing.
As
an intracellular parasite, HCV depends heavily on host proteins to
complete its life cycle. HCV NS4B rearranges the cellular
mem-brane to form a membranous web that contains the HCV
replica-tion complex (5). Accumulating evidence shows that NS4B plays
an important role in the viral life cycle and pathogenesis by
inter-acting with host proteins (6). HeLa cells support only the
replica-tion step in the HCV life cycle (22). Therefore, we started with
HeLa cells to screen for NS4B-associated host membrane proteins,
which are involved in HCV replication, by the use of established
NS4B-expressing cells dually tagged with Flag and HA. The
cellu-lar membrane fraction containing NS4B was isolated by
differen-tial centrifugation, and then NS4B-associated host proteins in the
membrane fraction were obtained by dual-affinity purification of
Flag-HA. Some proteins were reproductively detected, but none
were recovered from lysed control cells transfected with empty
vector alone (Fig. 1A). They were analyzed by liquid
chromatog-raphy-tandem mass spectrometry (LC-MS/MS), leading to the
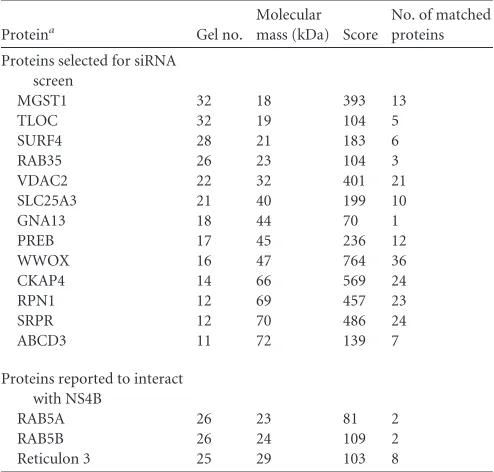
identification of 202 host proteins associated with NS4B,
includ-ing 9 chaperones, 14 lipoproteins, and 2 glycosylated proteins.
Previously reported NS4B-binding proteins, such as Ras-related
protein Rab 5A (RAB5A), RAB5B, and reticulon 3 (23,
24), were
among these identified proteins (Table 1), suggesting that our
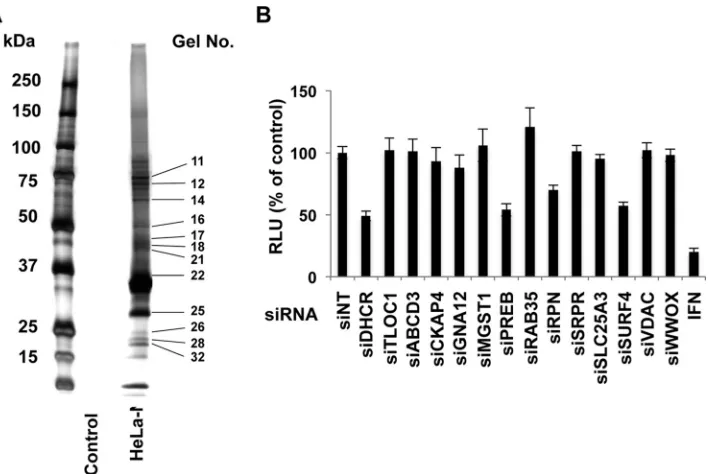
screening method is appropriate. PREB was identified (Fig. 1A, gel
no. 17) on the basis of 8 independent MS/MS spectra (Table 2). To
identify NS4B-associated HCV host cofactors involved in viral
replication, SGR2a was transfected with small interfering RNAs
(siRNAs) targeting 13 NS4B-associated host proteins providing
high scores in the above-described screening test (Table 1). Scores
were calculated as described in Materials and Methods. At 3 days
posttransfection, cells were harvested and luciferase activity, an
indicator of replicon replication, was measured. As shown in
Fig.
1B, treatment with siRNAs targeting PREB, ribophorin I (RPN),
and surfeit 4 (SURF4) inhibited SGR2a replication. Treatment
with siRNA targeting 24-dehydrocholesterol reductase (siDHCR)
and interferon (IFN), used as positive controls, reduced the level
of SGR2a replication, while treatment with siNT, used as a
nega-tive control, provided no inhibition. None of the siRNAs tested
exhibited cytotoxicity against the replicon cells (data not shown).
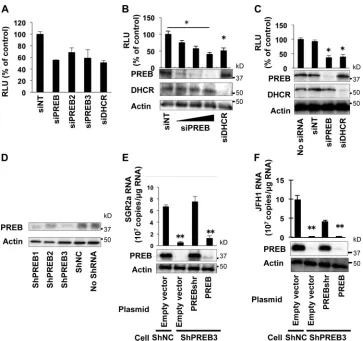
PREB participates in HCV replication.
To confirm the
inhib-itory effect of siRNA targeting PREB on SGR2a replication, cells
harboring SGR2a (SGR2a cells) were treated with the three unique
siRNAs targeting different sites of PREB. siPREB (used in the assay
whose results are presented in
Fig. 1B), siPREB2, and siPREB3
inhibited SGR2a replication, albeit to slightly different degrees.
siDHCR, used as a positive control, inhibited SGR2a replication,
while siNT, used as a negative control, provided no inhibition
(Fig. 2A). None of the siRNAs tested exhibited cytotoxicity against
the replicon cells (data not shown), ruling out cytotoxicity as a
mechanism for the inhibition of SGR2a replication. These results
suggest that the inhibition of SGR2a replication by siRNAs
target-ing PREB is likely PREB target specific and not an off-target effect.
To investigate whether PREB silencing inhibits SGR2a
replica-tion in a dose-dependent manner, different concentrareplica-tions of
siPREB were transfected into SGR2a cells. siPREB decreased the
levels of PREB expression (Fig. 2B, bottom) and luciferase activity
in a dose-dependent manner (Fig. 2B, top) but had no effect on
cell viability (data not shown). Similarly, siPREB decreased the
levels of PREB expression (Fig. 2C, bottom) and luciferase activity
(Fig. 2C, top), an indicator of the replication level of SGR1b,
with-out affecting cell viability (data not shown). Collectively, these
results indicate that PREB is involved in HCV replication in a
genotype-independent manner.
To further confirm the function of endogenous PREB in HCV
on November 7, 2019 by guest
http://jvi.asm.org/
replication, we constructed Huh7 cells with the stable knockdown
of PREB. Huh7 cells were transfected with a plasmid carrying
shRNA targeting PREB (shPREB) and selected with hygromycin
B, resulting in cell clones shPREB1, shPREB2, and shPREB3.
Among the three clones, shPREB3 showed the lowest level of
PREB expression, whereas Huh7 cells stably transfected with
neg-ative-control shRNA (shNC) showed a normal level of PREB
ex-pression (Fig. 2D). Cells transfected with shPREB3 or shNC were
electroporated with SGR2a RNA in the presence or absence of an
expression plasmid for shRNA-resistant PREB (PREBshr). An
im-munoblotting assay showed that transfection of the PREBshr
ex-pression plasmid produced significant exex-pression of PREB, while
transfection with the empty vector resulted in no PREB expression
(Fig. 2E, bottom). As expected, transfection of shPREB3 cells with
PREBshr increased the level of SGR2a replication compared to the
level achieved by transfection of the empty vector (Fig. 2E, top).
However, transfection of wild-type PREB did not cause a
signifi-cant increase in the levels of PREB expression and SGR2a
replica-tion compared to those achieved by transfecreplica-tion of the empty
vector into shPREB3 cells (Fig. 2E). We also examined the
func-tion of endogenous PREB in HCV replicafunc-tion in the context of
virus infection. shPREB3 cells were electroporated with JFH1
ge-nome RNA in the presence or absence of an expression plasmid
for PREBshr. An immunoblotting assay showed the expression
levels of PREB in transfected cells (Fig. 2F, bottom). Transfection
of the PREBshr expression plasmid increased the levels of PREB
expression and JFH1 genome replication in shPREB3 cells
com-pared to those achieved by transfection of the empty vector and
the wild-type PREB expression plasmid (Fig. 2F, top). The empty
vector and the wild-type PREB expression plasmid were used as
negative controls. These results together further indicate that
PREB is involved in HCV replication.
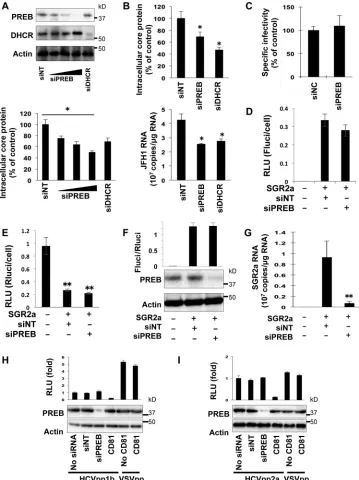
PREB participates in the propagation of HCV.
To investigate
the role of endogenous PREB in HCV propagation, JFH1-infected
Huh7 cells were treated at 20 days postinfection with siPREB,
siDHCR, or siNT for 3 days. siPREB decreased the level of PREB
expression (Fig. 3A, top) and the intracellular level of the HCV
core protein in a dose-dependent manner (Fig. 3A, bottom) but
had no effect on cell viability (data not shown). As a positive
con-trol, siDHCR decreased the intracellular level of the HCV core
protein (Fig. 3A). These results indicate that PREB participates in
the propagation of HCV. To further investigate the effect of
silenc-ing of endogenous PREB on the infectivity of HCV, the virus
par-ticles from the siRNA-treated cells described above were harvested
and used to infect naive Huh7 cells. At 3 days postinfection, the
intracellular levels of the HCV core protein were determined by
ELISA (Fig. 3B, top). Intracellular HCV RNA levels were analyzed
by real-time RT-PCR (Fig. 3B, bottom). siPREB and the positive
control, siDHCR, resulted in reduced intracellular HCV core
pro-tein and RNA levels in infected cells. On the basis of the results
presented in
Fig. 3A
and
B, we determined the specific infectivity
of HCV in cells treated with siPREB or control siNT. Specific
infectivity was determined as the ratio of the infectious virus titer
to the HCV core antigen level in general. We confirmed a good
correlation between the levels of core antigen and infectious titers
(8). siPREB did not alter the specific infectivity (Fig. 3C),
suggest-ing that PREB has no influence on viral assembly.
To investigate the effect of PREB silencing on the replication
FIG 1Identification of NS4B-associated host cofactors by tandem affinity purification, proteomic analysis, and siRNA silencing. (A) pCNX2-Flag-HA-NS4B was transfected into HeLa cells to establish a stable cell line expressing epitope-tagged NS4B under G418 selection. The cellular membrane fraction containing NS4B was isolated by differential centrifugation, and then NS4B-associated host proteins in the membrane fraction were obtained by dual-affinity purification of Flag-HA. They were separated on an SDS-polyacrylamide gel and silver stained using a Silver Stain MS kit. Control experiments were performed using vector alone. (B) SGR2a cells were treated with 25 nM siRNA specific for each of the 13 indicated NS4B-associated host proteins, siDHCR, nontargeting siRNA (siNT), or IFN (100 IU/ml). Three days later, HCV replication was determined by a luciferase assay. siDHCR and IFN were used as positive controls, while siNT was used as a negative control. Relative luciferase activity (RLU) is indicated. Values were obtained from quadruplicate wells in two independent experiments and are means⫾SDs.PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.119.474.65.302.2]and translation of HCV RNA, Huh7 cells were treated with
siPREB for 3 days and then electroporated with SGR2a RNA and
Rluci RNA. Fluci activity at 4 h after HCV RNA transfection has
been reported to reflect HCV RNA translation levels but not
rep-lication levels (25–27). Fluci activity (Fig. 3D) was normalized to
Rluci activity (Fig. 3E), indicating the transfection efficiency.
PREB silencing had no effect on the Fluci/Rluci ratio at 4 h
post-electroporation (Fig. 3F). However, PREB silencing significantly
decreased the SGR2a RNA level at 3 days postelectroporation (Fig.
3G). We next examined the effect of PREB silencing on HCV entry
using HCVpp systems. Huh7 cells were treated with siPREB and
infected with HCVpp1b, harboring the E1 and E2 glycoproteins of
the TH clone (genotype 1b); HCVpp2a, harboring the E1 and E2
glycoproteins of the JFH1 clone (genotype 2a); or VSVpp,
con-taining the VSV G glycoprotein. PREB expression was determined
by an immunoblotting assay (Fig. 3H
andI, bottom). PREB
silenc-ing did not inhibit the entry of HCVpp1b, HCVpp2a, or VSVpp
(Fig. 3H
and
I, top). As a control, pretreatment with CD81
anti-body inhibited the entry of HCVpp1b and HCVpp2a but had no
effect on VSVpp entry. Taken together, these results show that
PREB participates in the replication of HCV RNA but not in the
translation of viral proteins or in viral entry.
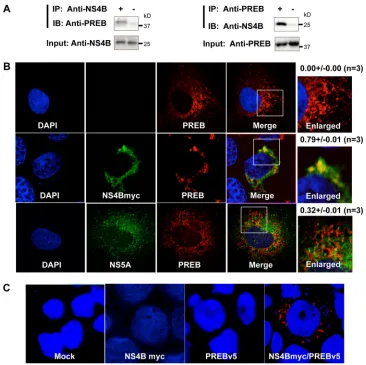
PREB interacts with NS4B.
As described above, our screen
identified that PREB is a novel NS4B-associated host protein. To
confirm the interaction between PREB and NS4B, we performed
coimmunoprecipitation, immunofluorescence staining, and PLA.
First, coimmunoprecipitation of PREB and NS4B in SGR2a cells
using antibodies against the two proteins was analyzed.
Endoge-nous PREB was coimmunoprecipitated with NS4B by using NS4B
antibody (Fig. 4A, left). NS4B was also coimmunoprecipitated
with PREB by using PREB antibody (Fig. 4A, right). Second,
indi-rect immunofluorescence staining of the cellular PREB and NS4B
proteins was performed. Several attempts to perform
immunoflu-orescence staining of NS4B using NS4B antibody were not
suc-cessful. Therefore, we used myc-tagged NS4B for
immunofluores-cence staining. Huh7 cells were transfected with the plasmid
expressing full-length NS4B with a myc tag (NS4Bmyc) and then
used for immunofluorescence staining. NS4Bmyc and PREB were
shown to colocalize in the perinuclear area (Fig. 4B, middle). We
further performed immunofluorescence staining of PREB with
NS5A, which is a membrane-associated protein. Huh7 cells were
transfected with the plasmid expressing full-length NS5A and
then used for immunofluorescence staining. NS5A and PREB
were not shown to colocalize (Fig. 4B, bottom). PREB with NS5A
was stained diffusely in the same way that PREB with the empty
vector was stained (Fig. 4B, top), suggesting that the NS4B-PREB
interaction is not a simple membrane association.
To further confirm the intracellular colocalization of PREB
and NS4B, we performed PLA, which used antibodies tagged with
circular DNA probes. If the antibodies are in close proximity, the
probes can be ligated together and subsequently amplified by a
polymerase. We were able to detect a red PLA signal in cells
coex-pressing NS4Bmyc and PREB tagged with V5 (PREBv5). In
con-trast, the PLA signal was not observed in cells mock transfected or
transfected with plasmids expressing only NS4B or PREB (Fig.
4C). Taken together, these results strongly indicate that PREB
interacts with NS4B.
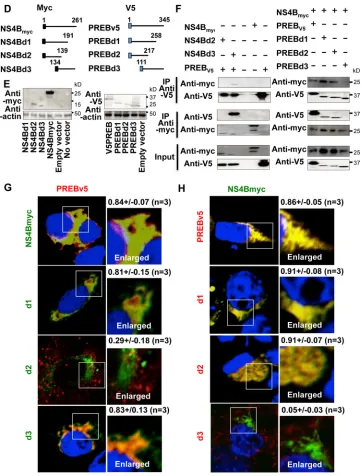
[image:6.585.300.545.77.175.2]To determine what region of NS4B interacts with PREB,
dele-tion mutants of myc-tagged NS4B were constructed (Fig. 4D, left).
Full-length tagged NS4B and deletion mutants of
myc-tagged NS4B were coexpressed with PREBv5 in Huh7 cells and
subjected to immunoprecipitation and immunofluorescence
staining. The expression of NS4B deletion mutant NS4Bd1 was
not detected by the immunoblotting assay (Fig. 4E, left, first lane),
presumably due to its misfolding and/or instability. Deletion
mu-tants NS4Bd2 and NS4Bd3, as well as full-length NS4B, were
sub-jected to immunoprecipitation. NS4B and NS4Bd3
coimmuno-precipitated with PREBv5, whereas NS4Bd2 did not (Fig. 4F, left).
Similarly, immunofluorescence staining showed no colocalization
between NS4Bd2 and PREBv5, whereas NS4B, NS4Bd1, and
NS4Bd3 strongly colocalized in the cytoplasm with PREBv5 (Fig.
4G). These results raise the possibility that the NS4B region
(amino acids 140 to 191), which is deleted in NS4Bd2, is
respon-sible for the interaction with PREB. To determine what region of
PREB interacts with NS4B, deletion mutants of V5-tagged PREB
TABLE 1Selected NS4B-associated host proteins identified by tandemaffinity purification and proteome analysis
Proteina Gel no.
Molecular mass (kDa) Score
No. of matched proteins
Proteins selected for siRNA screen
MGST1 32 18 393 13
TLOC 32 19 104 5
SURF4 28 21 183 6
RAB35 26 23 104 3
VDAC2 22 32 401 21
SLC25A3 21 40 199 10
GNA13 18 44 70 1
PREB 17 45 236 12
WWOX 16 47 764 36
CKAP4 14 66 569 24
RPN1 12 69 457 23
SRPR 12 70 486 24
ABCD3 11 72 139 7
Proteins reported to interact with NS4B
RAB5A 26 23 81 2
RAB5B 26 24 109 2
Reticulon 3 25 29 103 8
aMGST1, microsomal glutathioneS-transferase 1; TLOC, translocation protein;
SURF4, surfeit 4; RAB35, member of the RAS oncogene family; VDAC2, voltage-dependent anion-selective channel protein 2; SLC25A3, solute carrier family 25-3; GNA13, guanine nucleotide-binding protein subunit alpha-13; WWOX, WW domain-containing oxidoreductase; CKAP4, cytoskeleton-associated protein 4; RPN1, ribophorin I; SRPR, signal recognition particle receptor; ABCD3, ATP-binding cassette subfamily D member 3.
TABLE 2Identification of PREB by tandem mass spectrometrya
Sequence determined Residues
APFPLYALQVDPSTGLLIAAGGGGAAK 12–38 NGVHFLQLELINGR 43–56 LSASLLHSHDTETR 57–70 ATMNLALAGDILAAGQDAHCQLLR 71–94 VENLQAVQTDFSSDPLQK 141–158 AHEGEIEDLALGPDGK 194–209 FGQVPDQPAGLR 252–263 QPPPCYLTAWDGSNFLPLR 276–294
aThe protein was PREB (GenBank accession no.AAH16906).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.40.287.87.323.2]were constructed (Fig. 4D, right). The expression of PREB
con-structs in Huh7 cells was confirmed by an immunoblotting assay
(Fig. 4E, right). We next coexpressed V5-tagged PREB constructs
and NS4Bmyc in Huh7 cells. The interaction of PREB and NS4B
was analyzed by immunoprecipitation and
immunofluores-cence staining. PREBv5 and the deletion mutants (PREBd1 and
PREBd2) coimmunoprecipitated with NS4Bmyc, whereas PREB
deletion mutant PREBd3 did not (Fig. 4F, right). Consistent with
this finding, immunofluorescence staining found no
colocaliza-tion between PREBd3 and NS4Bmyc, whereas PREBv5, PREBd1,
and PREBd2 strongly colocalized with NS4Bmyc in the cytoplasm
(Fig. 4H). Taken together, these results indicate that the PREB
region (amino acids 1 to 110) deleted in PREBd3 is important for
binding NS4B. Further experiments are needed to determine the
amino acids involved in the interaction between NS4B and PREB.
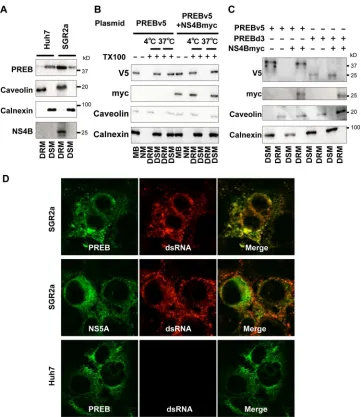
PREB is recruited into the HCV RNA replication complex by
NS4B.
Since NS4B provides a platform for the formation of the
HCV RNA replication complex and PREB binds NS4B, it is
con-ceivable that PREB is involved in the HCV RNA replication
com-plex. The HCV RNA replication complex has been reported to be
located in a detergent-resistant membrane (DRM), and these
complexes are known as lipid rafts (20). We analyzed whether
endogenous PREB is located in DRM in HCV replicon cells. PREB
was detected in the detergent-soluble membrane (DSM) in Huh7
cells but in DRM in SGR2a cells (Fig. 5A). As expected, caveolin, a
DRM marker, was detected in the DRM fraction, and calnexin, a
DSM marker, was absent from the DRM fraction. To investigate
whether NS4B causes the retargeting of PREB to DRM, Huh7 cells
were transfected with plasmids expressing PREBv5and NS4Bmyc,
and then the membrane fraction from the transfected cells was
FIG 2Effect of PREB silencing on HCV replication. (A) Effect of different siRNAs targeting PREB on SGR2a replication. SGR2a cells were treated with siRNAs, including siPREB (used for the assay whose results are presented inFig. 1B), siPREB2, and siPREB3, which target different regions of PREB. Three days later, HCV replication was determined by a luciferase assay. siDHCR, used as a positive control, inhibited SGR2a replication, while siNT, used as a negative control, caused no inhibition. (B) Effect of different concentrations of siPREB on SGR2a replication. SGR2a cells were transfected with siPREB (10 nM, 25 nM, and 50 nM), siDHCR (25 nM), or siNT (25 nM). At 3 days posttransfection, silencing of proteins (bottom) and inhibition of HCV replication (top) were determined by immunoblotting and luciferase assays, respectively. (C) Effect of PREB silencing on SGR1b. SGR1b cells were transfected with siPREB (25 nM), siDHCR (25 nM), or siNT (25 nM). At 3 days posttransfection, silencing of proteins (bottom) and inhibition of HCV replication (top) were determined by immunoblotting and luciferase assays, respectively. (D) Huh7 cells were transfected with pSilencer-shPREB or negative-control shRNA (shNC) and selected using hygromycin B. The knockdown of PREB in stable transfectants, including shPREB1, shPREB2, and shPREB3, was determined by immunoblotting. (E) Cells that had been transfected with shPREB3 and shNC were transfected with plasmids expressing shRNA-resistant PREB (PREBshr), PREB, or the empty vector, as indicated. After 24 h, transfected cells were electroporated with SGR2a RNA. Three days later, cell lysates were used to determine PREB expression (bottom). Intracellular SGR2a RNA was quantified by real-time RT-PCR (top). (F) Cells that had been transfected with shPREB3 and shNC were transfected with plasmids expressing PREBshr, PREB, or the empty vector, as indicated. After 24 h, transfected cells were electroporated with JFH1 RNA. Three days later, PREB silencing was determined by an immunoblotting assay (bottom). Intracellular JFH1 RNA was quantified by real-time RT-PCR (top). Values were obtained from quadruplicate wells in two independent experiments and are means⫾SDs. *,P⬍0.05; **,P⬍0.01.PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.111.474.63.404.2]FIG 3Effect of PREB silencing on HCV propagation. (A) JFH1-infected cells were transfected with siPREB (10 nM, 25 nM, or 50 nM), siDHCR (25 nM), or siNT (25 nM). At 3 days posttransfection, silencing of proteins was determined by an immunoblotting assay (top). The amounts of core proteins in the cell lysate were determined by ELISA (bottom). (B) For the determination of intracellular infectivity, intracellular HCV particles were collected from the cells treated with siPREB (25 nM) and used to infect Huh7 cells. At 3 days postinfection, the amount of intracellular HCV core antigen was determined by ELISA (top). Intracellular JFH1 RNA was evaluated by real-time RT-PCR (bottom). (C) The intracellular specific infectivity of the cells treated with siPREB (25 nM) or siNT (25 nM) was determined by measurement of the infectious HCV core antigen level (B, top, second histogram) relative to the total HCV core antigen level (A, bottom, third histogram). The intracellular specific infectivity of cells treated with siNT (25 nM) was used as a negative control. (D and E) SGR2a RNA and Rluci RNA, used as an internal standard, were introduced into Huh7 cells pretreated with the indicated siRNA. For analysis of SGR2a translation, transfected cells were harvested to determine the Fluci activity (D) and Rluci activity (E) at 4 h posttransfection. (F and G) SGR2a RNA was introduced into Huh7 cells pretreated with the indicated siRNA. For analysis of SGR2a translation, transfected cells were harvested to determine luciferase activity (F, top) as the ratio of Fluci activity (D)/Rluci activity (E) and protein levels (F, bottom) at 4 h posttransfection (G). For analysis of SGR2a replication, transfected cells were harvested to determine the SGR2a RNA level by real-time RT-PCR at 3 days posttransfection. (H and I) Huh7 cells were seeded into 24-well plates, incubated overnight at 37°C, treated with the indicated siRNAs for 3 days, and infected with untreated HCVpp or HCVpp that had been pretreated with CD81 antibody (0.5 mg/ml) for 2 h. At 4 h postinfection, the medium was replaced with DMEM with 10% FBS, and the cells were harvested 3 days later to determine intracellular luciferase activity. Values were obtained from quadruplicate wells in two independent experiments and are means⫾SDs. *,P⬍0.05; **,P⬍0.01.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.112.471.67.547.2]subjected to immunoblot analysis (Fig. 5B). PREBv5 was detected
in the membrane fraction in the absence of detergent. When the
cell lysates were preincubated with cold 1% TX100 at 4°C for 1 h,
PREBv5 was recovered in the DSM fraction in the absence of
NS4Bmyc expression. However, PREBv5 shifted to the DRM
frac-tion in the presence of NS4Bmyc expression. NS4B, PREB, and
caveolin were solubilized by pretreatment with TX100 at 37°C for
1 h. This condition disrupted the interaction of PREB and caveolin
with the DRM structure, as observed for other raft-localized
pro-teins (28), suggesting that PREB was present in DRM in the cells
cotransfected with NS4B. To confirm whether the interaction of
PREB with NS4B is necessary for PREB localization in DRM,
Huh7 cells were transfected with plasmids expressing PREBv5,
PREBd3, or NS4Bmyc. PREBv5 was detected in the DRM fraction
FIG 4Interaction of PREB and NS4B. (A) Lysates of SGR2a cells were immunoprecipitated with anti-NS4B antibody (left) or anti-PREB antibody (right). The resulting immunoprecipitates (IP) and whole-cell lysates used for immunoprecipitation (Input) were examined by an immunoblotting (IB) assay using anti-NS4B antibody and anti-PREB antibody. The negative-control sample was generated by using control IgG antibody. (B) Huh7 cells were transfected with plasmid expressing full-length NS4B with a myc tag (NS4Bmyc) (middle), NS5A (bottom), or empty vector (top). At 2 days posttransfection, the cells were fixed, permeabilized with Triton X-100, and then subjected to immunofluorescence staining using anti-myc (green), NS5A (green), and anti-PREB antibodies (red). The boxed areas are shown as enlargements in the corresponding panels on the right. The surface area of each color was calculated using MetaMorph software. Quantities for the degree of colocalization (overlapping area/PREB area) are given at the top of the enlarged pictures and are indicated as means⫾SDs. (C) Huh7 cells were transfected with the indicated expression plasmids. At 2 days posttransfection, the cells were fixed, permeabilized with Triton X-100, and then subjected to PLA using anti-myc and anti-V5 antibodies. Nuclei were stained with DAPI. (D) Diagrams of the NS4B (left) and PREB (right) constructs. The myc and V5 tags are depicted by black and blue boxes, respectively. The positions of the amino acid residues are indicated above each line. (E) Expression of the NS4B (left) and PREB (right) constructs. Huh7 cells were transfected with the indicated plasmids. Cell lysates were examined by an immunoblotting assay using anti-myc, anti-V5, or antiactin antibody, as indicated. (F) Huh7 cells were transfected with the indicated plasmids. Cell lysates were immunoprecipitated with anti-myc antibody or anti-V5 antibody. The resulting precipitates and whole-cell lysates used for immunoprecipitation were examined by an immunoblotting assay using anti-myc or anti-V5 antibody. (G) Huh7 cells were cotransfected with PREB and NS4B deletion mutants. At 3 days posttransfection, the cells were subjected to immunofluorescence staining using anti-myc (green) and anti-V5 (red) antibody. Nuclei were stained with DAPI. The panels on the left present low-magnifi-cation overviews; the boxed areas are shown as enlargements in the corresponding panels on the right. Quantities for the degree of colocalization (overlapping area/PREB area) are given at the top of the enlarged pictures and are indicated as means⫾SDs. d1, d2, and d3 in green, NS4Bd1, NS4Bd2, and NS4Bd3, respectively. (H) Huh7 cells were cotransfected with NS4B and PREB deletion mutants. At 3 days posttransfection, the cells were subjected to immunofluores-cence staining using anti-myc (green) and anti-V5 (red) antibodies. The boxed areas are shown as enlargements in the corresponding panels on the right. Quantities for the degree of colocalization (overlapping area/NS4B area) were obtained from three samples and are indicated as means⫾SDs. d1, d2, and d3 in red, PREBd1, PREBd2, and PREBd3, respectively.PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.109.475.64.429.2]in the presence of NS4Bmyc expression. However, PREBd3, which
lacks the ability to bind NS4B, was not detected in the DRM
frac-tion in the presence of NS4Bmyc expression (Fig. 5C). These
re-sults together indicate that PREB is recruited into DRM via
inter-action with NS4B.
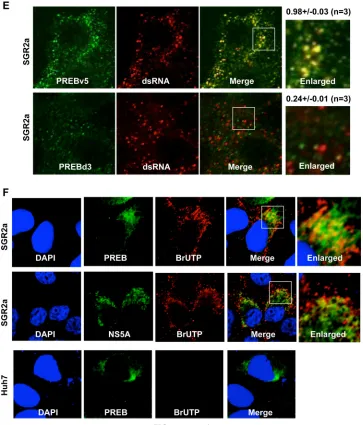
To visualize whether PREB localizes in the HCV RNA
replica-tion complex, we examined the colocalizareplica-tion of PREB and
dou-ble-stranded RNA (dsRNA), a marker of the HCV RNA
replica-tion complex. PREB colocalized with dsRNA in SGR2a cells (Fig.
5D, top). As a positive control, NS5A residing in the HCV RNA
replication complex colocalized with dsRNA in SGR2a cells (Fig.
5D, middle). As a negative control, Huh7 cells without the HCV
replicon exhibited no dsRNA signal (Fig. 5D, bottom). To
inves-tigate whether the interaction of PREB with NS4B is necessary for
PREB colocalization with dsRNA, SGR2a cells were transfected
with PREBv5 or PREBd3, which lacks the ability to bind NS4B.
PREBv5 colocalized with dsRNA (Fig. 5E, top), but PREBd3
did not (Fig. 5E, bottom). These results together provided visual
evidence that PREB was recruited into the HCV RNA replication
complex via interaction with NS4B. To further visualize whether
PREB localizes in the active HCV RNA replication complex, we
examined the colocalization of PREB and bromouridine
triphos-phate (BrUTP) in actinomycin D-treated SGR2a cells. Under
these conditions, only
de novo
-synthesized viral RNA, which
de-pended on RNA-dependent RNA polymerase, was labeled. PREB
colocalized with BrUTP in SGR2a cells (Fig. 5F, top). As a positive
FIG 4continuedon November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.112.474.64.540.2]control, the colocalization of NS5A with BrUTP was detected
in SGR2a cells (Fig. 5F, middle). However, the BrUTP signal
was absent from Huh7 cells lacking the HCV replicon (Fig. 5F,
bottom). These results, together with the findings described
above, strongly indicate that PREB is recruited into the HCV
RNA replication complex by interaction with NS4B.
PREB is involved in the formation of the HCV-induced
membranous replication compartment.
Since PREB is recruited
into the HCV replication complex by NS4B, we investigated the
effect of PREB silencing on the formation of the membrane web.
SGR2a cells were treated with siPREB or siNT and analyzed by
transmission electron microscopy (TEM). siPREB treatment
de-creased the level of formation of membrane webs compared to
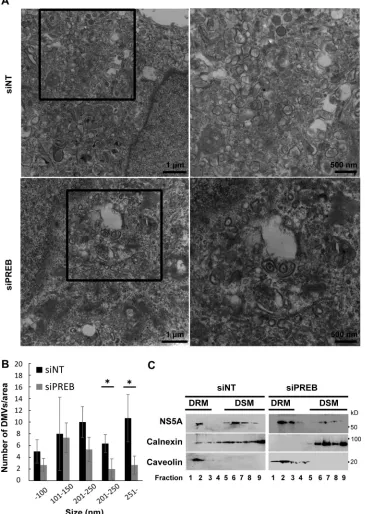
that achieved with siNT treatment (Fig. 6A). The sizes of the
mem-brane webs in boxed areas of siRNA-treated cells in
Fig. 6A
were
quantified. siPREB treatment decreased the sizes of the membrane
FIG 5PREB is recruited into the HCV RNA replication complex by NS4B. (A) PREB recruitment into DRM in SGR2a cells. DRM and DSM fractions from Huh7 and SGR2a cells were analyzed by an immunoblotting assay with anti-PREB, anti-NS4B, anticaveolin, and anticalnexin antibodies. (B) PREB recruitment into DRMs by NS4Bmyc in Huh7 cells. Huh7 cells were transfected with plasmids expressing PREBv5 and NS4Bmyc, as indicated. At 3 days posttransfection, cell lysates were treated with 1% TX100 at 4°C or 37°C as indicated. Membrane fractions were isolated by a membrane flotation assay and then analyzed by an immunoblotting assay with anti-V5, anti-myc, anticaveolin, and anticalnexin antibodies. MB, membrane fraction; NM, nonmembrane fraction. (C) Huh7 cells were transfected with plasmids expressing PREBv5, PREBd3, and NS4Bmyc, as indicated. The DRM and DSM fractions of transfected cells were analyzed by an immunoblotting assay with anti-V5, anti-myc, anticaveolin, and anticalnexin antibodies. (D) Colocalization of PREB with dsRNA. SGR2a and Huh7 cells were fixed and subjected to immunofluorescence staining using anti-PREB (green), anti-NS5A (green), and anti-dsRNA (red) antibodies. (E) SGR2a cells were transfected with plasmids expressing PREBv5 or PREBd3. At 3 days posttransfection, cells were subjected to immunofluorescence staining using anti-V5 (green) and anti-dsRNA (red) antibodies. The boxed areas are shown as enlargements in the corresponding panels on the right. The surface area of each color was calculated using the MetaMorph software. Quantities for the degree of colocalization (overlapping area/dsRNA area) are given at the top of the enlarged images. Values were obtained from three samples and are given as means⫾SDs. (F) Colocalization of PREB withde novo-synthesized viral RNA. SGR2a cells were treated with actinomycin D, transfected with BrUTP, and subjected to immunofluorescence staining using anti-NS5A (green), anti-PREB (green), and anti-BrUTP (red) antibodies. Nuclei were stained with DAPI. The boxed areas are shown as enlargements in the corresponding panels on the right.PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.112.473.63.480.2]webs compared to those obtained with siNT treatment (Fig. 6B).
Double-membrane vesicles (DMVs) and multimembrane vesicles
(MMVs) were recently suggested to be predominant HCV
repli-cation sites (29,
30). To determine the effect of PREB silencing on
the formation of DMVs and MMVs, we treated SGR2a cells with
siPREB or siNT; isolated the DRM fraction (Fig. 6C, fraction 2),
which is rich in replication complexes (20); and then quantified
the DMV and MMV percentages in whole-membrane structures.
We confirmed that the fractions contained HCV NS5A and
caveo-lin (Fig. 6C). A smaller number of DMVs and MMVs was detected
in siPREB-treated cells than siNT-treated cells (Fig. 6D, right),
suggesting that PREB is involved in the formation of
HCV-in-duced DMVs and MMVs. Furthermore, interesting structures,
presumably from DMVs whose formation was hindered or whose
structure was impaired, appeared in siPREB-treated cells (Fig. 6D,
left).
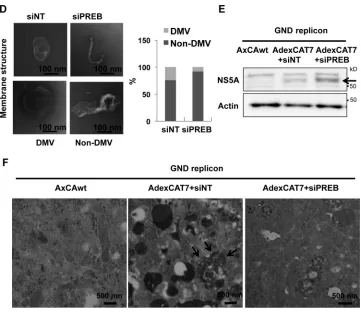
Furthermore, it would be important to know that a decrease in
DMV number is not linked to the overall decrease in replication
upon PREB knockdown. Thus, we used a replication-independent
system for the expression of HCV proteins. Huh7 cells were
infected with AdexCAT7 and AxCAwt, a control virus;
trans-fected with pSGRlucneoGND, containing an inactivating
muta-tion within NS5B; and analyzed by immunoblotting (Fig. 6E) and
TEM (Fig. 6F). HCV protein-induced membranous
compart-ments were observed in siNT-treated cells (Fig. 6F, middle), but
siPREB treatment decreased the sizes of the vesicle structures (Fig.
6F, right). These results together indicated that PREB is involved
in the formation of the HCV-induced membranous replication
compartment.
PREB silencing decreases the resistance of the HCV RNA
replication complex to proteases and nucleases.
The viral
pro-teins and genomic RNA in the HCV-induced membranous
repli-cation compartment have been reported to be protected from
degradation by cellular proteases and nucleases, respectively (29).
To investigate the role of PREB in the structure of the
HCV-in-duced membranous replication compartment, we examined the
FIG 5continuedon November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.111.474.67.492.2]FIG 6PREB is required for the formation of the HCV-induced membranous replication compartment. (A) SGR2a cells were transfected with 25 nM siPREB or siNT. At 3 days posttransfection, cells were examined by TEM. The right panels are magnified views of the squares in the left panels. (B) The sizes of the membrane webs in the boxed areas of siRNA-treated cells in panel A were quantified. Values were obtained from three samples and are means⫾SDs. *,P⬍0.05. (C) Detection of HCV proteins in DRM fractions of HCV replicon cells treated with siRNA. Cell lysates were treated with 1% TX100 for 30 min on ice and fractionated by discontinuous sucrose gradient centrifugation. Each fraction was concentrated in a Centricon YM-30 filter unit. Equal volumes of the recovered fractions were analyzed on a 12% SDS-polyacrylamide gel, followed by immunoblotting with antibodies against NS5A, calnexin, or caveolin. Fractions are numbered from 1 to 9 in order from top to bottom (light to heavy), respectively. (D) DRM fractions (C, fraction 2) from treated cells were examined by TEM. Representative DMV and non-DMV membranous structures are shown (left). The number of membrane structures in 10 randomly chosen squares (3,600
m2/square) was determined. The percentages of DMVs and non-DMVs in siPREB-treated or control cells were determined for more than 200 membrane
structures (right). (E and F) A replication-independent system of HCV protein expression. Huh7 cells were infected with AdexCAT7, a recombinant adenovirus containing the bacteriophage T7 RNA polymerase (middle and right), or AxCAwt, a control virus (left), and transfected with pSGRlucneoGND, which contains an inactivating mutation within NS5B. Then, the cells were transfected with 25 nM siPREB or siNT. At 3 days posttransfection, cells were examined by immunoblotting (E) and TEM (F). Arrows in panel F, HCV protein-induced membranous compartments.
PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
[image:13.585.111.476.72.586.2]effect of PREB silencing on the enzymatic resistance of the HCV
replicase complex and genomic RNA. SGR2a cells were treated
with siPREB or siNT, and then the cellular DRM fraction was
concentrated. The same amounts of proteins from the DRM
frac-tions were analyzed for their resistance to enzymatic degradation.
An immunoblotting assay confirmed PREB silencing in
siPREB-treated SGR2a cells (Fig. 7A, top). PREB silencing decreased the
RNase resistance of HCV RNA in DRMs (Fig. 7A, bottom). Next,
we investigated the effect of PREB silencing on the protease
resis-tance of viral replicase proteins, including NS4B and NS5A, in
DRMs. PREB silencing significantly decreased the protease
resis-tance of NS4B and NS5A in DRMs (Fig. 7B). Collectively, these
data provide evidence that PREB silencing may impair the
struc-ture of the HCV-induced membranous replication compartment
and, thus, increase the degradation of HCV RNA and proteins in
the replication complex.
HCV increases PREB expression both
in vitro
and
in vivo
.
Having demonstrated that PREB participates in HCV RNA
repli-cation and is recruited into the HCV RNA replirepli-cation complex by
its interaction with NS4B, we were interested in the role of the
PREB-NS4B interaction in HCV RNA replication. SGR2a cells
were transfected with plasmids expressing PREB, PREBv5, or
mu-tant PREBd3 lacking the region interacting with NS4B. SGR2a
replication was increased by full-length PREB (PREB and
PREBv5) expression, whereas the expression of PREBd3 had no
effect on HCV RNA replication (Fig. 8A). Likewise, replication of
SGR1b RNA was increased by elevated levels of full-length PREB
(PREB and PREBv5) expression, whereas this effect was not
ob-served in the presence of PREBd3 expression (Fig. 8B). Since
PREBd3 lost the ability to bind NS4B and go to the HCV
replica-tion complex, it is reasonable that expression of the mutant has no
dominant negative impact on HCV replication. Collectively, these
results indicate that the PREB-NS4B interaction is important for
the role of PREB in HCV RNA replication.
SGR2a cells have higher PREB levels than Huh7 cells (Fig. 5A),
suggesting that HCV may increase the level of PREB expression.
To confirm this, Huh7 cells were electroporated with SGR2a RNA
or the JFH1 genome. At 3 days postelectroporation, cellular PREB
protein and PREB mRNA were analyzed by immunoblotting
anal-ysis and the TaqMan PCR method, respectively. Huh7 cells
elec-troporated with the JFH1 replicon (SGR2a) or the JFH1 genome
exhibited elevated levels of PREB protein and mRNA expression
compared to control Huh7 cells (Fig. 8C, left and right).
Consis-tent with these results, SGR2a and SGR1b cells with persisConsis-tent
replicon replication also exhibited higher levels of PREB
ex-pression than control Huh7 cells (Fig. 8D). We further
inves-tigated by immunoblotting analysis the PREB levels in the liver
tissues of HCV strain S310 genotype3a-infected SCID mice
into which human hepatocytes had been transplanted (21).
HCV infection was confirmed by the presence of NS5A and
core proteins. Liver tissues from the HCV-infected mice
exhib-ited higher levels of PREB expression than tissues from the
mock-infected mice, suggesting that PREB expression was
in-creased by HCV infection
in vivo
(Fig. 8E). Taken together,
these results indicate that HCV increases the levels of PREB
expression both
in vitro
and
in vivo
.
Effect of PREB silencing on replication of poliovirus and
dengue virus.
The specificity of PREB for HCV replication was
FIG 6continued
on November 7, 2019 by guest
http://jvi.asm.org/
[image:14.585.112.473.63.377.2]further validated using other positive-stranded RNA viruses
(po-liovirus and dengue virus) to exclude the possibility of a
nonspe-cific effect associated with membrane integrity other than a direct
interaction between HCV NS4B and PREB. Naive Huh7 cells were
transfected with poliovirus replicon RNA or infected with dengue
virus and then treated with siRNAs. After 8 h, the replication of
poliovirus replicon RNA in the transfected cell was analyzed by
measurement of luciferase activity. After 3 days, the amount of
dengue virus RNA in the infected cell was determined by the
Taq-Man PCR method. siPREB decreased the level of poliovirus
repli-cation (Fig. 9A) but not the level of dengue virus replirepli-cation (Fig.
9B). Poliovirus and dengue virus are known to use DMV and
invaginated/sphere-type vesicles (31), respectively, specifically
suggesting the effects of PREB on the DMV structure. We need to
perform further experiments to analyze the mechanisms that the
viruses use to alter the membranes.
DISCUSSION
The host cellular membrane is tightly involved in each step of the
life cycle of viruses of the family
Flaviviridae
, including HCV.
HCV NS4B rearranges the cellular membrane to form a
membra-nous web where HCV replication takes place (6). However, the
roles of NS4B-associated host proteins in HCV replication,
par-ticularly in the formation of the membranous replication
com-partment, are largely unknown. Recent progress in protein
tech-niques, such as protein purification and proteomics, has provided
new avenues for high-throughput screening of host cofactors
in-teracting with viral proteins. In the present study, we used HeLa
cells expressing NS4B dually tagged with Flag and HA to perform
high-throughput screening of NS4B-associated host membrane
proteins by tandem affinity purification and proteome analysis.
Previously reported NS4B-binding proteins, such as RAB5A,
RAB5B, and reticulon 3 (23,
24), were among these identified
proteins (Table 1). Prior to the present study, Paul et al. (29)
isolated NS4B-containing membranes from stable HCV
repli-con cells using a single affinity purification method and found
that vesicle-associated membrane protein-associated protein
A (VAP-A) and VAP-B were enriched in NS4B-containing
DMVs, the presumed site of HCV RNA replication. VAP-A and
-B have been reported to interact with HCV replicase proteins
NS5A and NS5B (32,
33). Our screen was based on HeLa cells
without the expression of HCV replicase proteins NS5A and
NS5B. As expected, VAP-A and -B were not identified using our
screen. Taken together, these results strongly support the
appro-priate quality of our screen. Sequence-specific gene silencing with
siRNA is ideal for the screening of host cofactors involved in the
viral life cycle. Recently, limited and genome-wide siRNA screens
using HCV replication or infection models have been used to
identify new host cofactors, such as phosphatidylinositol 4-kinase
III alpha (PI4KIII
␣
), signal peptidase complex subunit 1 (SPCS1),
COPI, and hepcidin (34–38). We used siRNAs targeting 13
NS4B-associated host proteins with high scores in our above-described
screen to examine whether these proteins are involved in HCV
replication. A high correlation between PREB silencing and the
triggered of SGR2a/1b or virus inhibition by siPREB treatment
was observed (Fig. 1B
and
2A
to
C). The exogenous expression of
shRNA-resistant PREB rescued the replication of SGR2a and the
viral genome in shPREB3 cells with stable silencing of PREB (Fig.
2E
and
F). These results suggest that PREB is a common cofactor
of HCV genotype 2a and 1b replication. Similarly, PI4KIII
␣
has
been reported to act as a common cofactor of HCV genotype 1b
(Con1) and genotype 2a (JFH1). However, PI4KIII

is identified
to be a cofactor of Con1 replication but not JFH1 replication (34).
Interestingly, the impact of silencing on RNA replication was
dif-ferent in difdif-ferent assay systems. Presilencing of PREB produced
higher levels of inhibition (Fig. 2E
and
F
and
3G) than
postsilenc-ing of PREB (Fig. 1B,
2A
to
C, and
3A
and
B), possibly because
HCV DMVs have been shown to be relatively stable and
preexist-ing PREB proteins exert a role to promote HCV replication in the
case of postsilencing of PREB.
PREB is DNA-binding protein that specifically binds to a
Pit1-binding element of the prolactin (PRL) promoter and then acts as
a transcriptional regulator. PREB is mainly located in the ER and
is detected in a variety of tissues, including human liver (39).
PREB is also called the mammalian guanine nucleotide exchange
factor (Sec12) (39,
40). Sec12 is known to promote COPII vesicle
FIG 7PREB silencing decreases the enzymatic resistance of HCV RNA andproteins in DRMs. (A, B) SGR2a cells were treated with siPREB or siNT. Three days later, proteins from the DRM fractions were concentrated. The same amounts of proteins from the DRM fractions were analyzed by RNase A (A) or trypsin (B) digestion. (A) DRMs from treated cells were digested with RNase A (500 ng/ml) at 37°C for 5 min, as indicated, and were then used to determine the HCV RNA level by real-time RT-PCR (bottom) and the protein level by an immunoblotting assay using PREB, NS5A, and anticaveolin anti-bodies (top). For real-time RT-PCR, data are averages of triplicate values, with error bars representing standard deviations. (B) DRMs from transfected cells were treated with trypsin (25g/ml) at 37°C for 10 min, as indicated, and were then used to determine the protein level by an immunoblotting assay using anti-PREB, anti-NS4B, and anti-NS5A antibodies. Values were obtained from quadruplicate wells in two independent experiments and are means⫾SDs. **, P⬍0.01.
PREB Is Involved in HCV Replication by NS4B Interaction
on November 7, 2019 by guest
http://jvi.asm.org/
[image:15.585.101.226.63.368.2]FIG 8PREB expression bothin vitroandin vivo. (A and B) SGR2a (A) and SGR1b (B) cells were transfected with plasmids expressing PREB, PREBv5, PREBd3, or the empty control vector. At 3 days posttransfection, HCV replication and the expression of proteins were determined by luciferase (top) and immunoblotting (bottom) assays, respectively. (C) Huh7 cells were electroporated with the SGR2a replicon or JFH1 genome. At 3 days postelectropo-ration, cellular PREB protein (left) and PREB mRNA (right) were analyzed by immunoblotting analysis and the TaqMan RT-PCR method, respectively. Mock-electroporated Huh7 cells were used as controls. (D) SGR2a, SGR1