Copyright © 1994, American Society for Microbiology
Orientation-Specific cis Complementation by Bulge-
and
Loop-Mutated
Human
Immunodeficiency Virus
Type 1 TAR RNAs
MARTIN BRADDOCK, ROBERT POWELL, JULIASUTTON, ALAN J. KINGSMAN, AND SUSAN M. KINGSMAN*
RetrovirusMolecularBiology Group, Department of Biochemistry, University of Oxord,
Oxford
OX13QU,
UnitedKingdom
Received 31 May1994/Accepted 19August1994Tat activates human immunodeficiency type 1gene expression by bindingto TAR RNA.TARcomprises a partiallybasepairedstemandhexanucleotide loop withatripyrimidinebulge in theupperstem. Invitro, Tat binds tothebulge and upper stem, with no requirementfor the loop. However, in vivo, loop sequences are critical foractivation,implyingthat aloop bindingcellular factormaybeinvolved intheactivationpathway. Giventhatactivation appearstobe atwo-componentsystemcomprisingaTat-bulgeinteraction and a cellular factor-loop interaction,weconsidered thatitmight bepossible tospatiallyseparate the twocomponents and retainactivation. Wehaveconstructed aseries of doubleTAR elements comprisingvarious combinations of mutated TAR structures. Defective TARs with nucleotide substitutions in either the bulge or the loop complemented eachother togivewild-typeactivation.However, thecomplementationwasorientation specific, requiring the intact Tat binding sitetoresideonthe5'-proximalTAR. These data suggestthatprovidedthe wild-typeorientation ofthebulge andloopelements isretained,there is norequirementfor them tocoexiston the same TAR structure.
Humanimmunodeficiency virus type 1 (HIV-1) replication
is critically dependent upon the virally encoded Tat protein.
Tat binds to the transactivation response (TAR) element, which is localized between +14 and +44 atthe 5' end of all HIV-1RNAs(7).TARisapartially base paired RNAstructure
comprisingatripyrimidinebulge andanunpaired hexanucleotide
loop (10,23).Theposition of TAR relativetothetranscription
startsite is critical foractivation, andTARisfunctional only when placed in the correct orientation with respect to the HIV-1long terminal repeat (15, 17, 24, 27). The predominant mode of action of Tat istostimulate transcription, and Tat is envisaged asbeing introducedto the transcription machinery inafeedback process as aprotein-bound TAR RNA complex
(8).
Invitro, Tat binds specifically at the bulge, with no require-ment for the loop sequence. However, in contrast to the in vitro binding data,mutations in the loop sequence abolish both Tatactivation of transcription and translation (4, 5, 11). This
finding implies arequirement for cellular factors to facilitate
thebinding and/or the activation process. A number of TAR RNA-binding proteins have been identified (12, 14, 19, 22, 25, 29, 37), some of which bind to the loop sequence, but the function of these cellular factors that bind the loop is contro-versial. The finding that Tat activates transcription
indepen-dently of TAR when tethered to RNA via a heterologous
RNA-protein interaction (28, 31) or when located upstream of theRNAstart in a TAR-less configuration (30) suggests that
loop-bindingfactors cannot be central to the activation
path-way. This isconsistent with the fact that Tat directly contacts components of the transcription initiation complex (18, 20). Thereis, however, some genetic evidence that implies that in the normal TAR configuration, Tat can bind to TAR only when complexed with a loop-binding protein (21). It has been
*Corresponding author. Mailing address: Retrovirus Molecular
Biology Group, Department of Biochemistry, South Parks Road, Oxford OX1 3QU, United Kingdom. Phone: 865 27548. Fax: 865 275259.
suggested that the loop-binding protein also mediates the
activation. Analternative view is that theloop-bindingfactors areessentialtofacilitateaccessof Tattothebulge byexcluding
competing cellularbulge-bindingfactors (4).
To investigate further the relationship between the Tat-bulge interaction and the cellular factor-loop interaction, we
separated the twobinding sitesby constructing tandem TAR
elements downstream of the HIV-1 promoter. The TAR elementswere mutated such that they lacked either the Tat
bindingsiteatthebulgeorthecellular factorloopbindingsite.
The bulge mutation was a substitution of residue U-23 for
C-23, and the loop mutationwas athree-base substitutionof the sequence30/CUGGG/34for30/AGGGU/34. Both of these classesof mutationseriously impairthetat activation process
(4, 26). Thegeneralconfigurations of single and double TAR
mutants areshown inFig. 1. Single TAR mutants were derived from the wild-type (WT) long terminal repeat-CAT plasmid
pOGS210 (1) and retained TAR in the precise WT position
but with the base changes indicated (Fig. 1A). Plasmids
containing tandem TARelements were constructed by
insert-ingasynthetic oligonucleotide, comprising alinker containing anSstII site andresidues +1 to + 62 of TAR as aWT,bulge mutant (BM), orloop mutant(LM) sequence into the unique
NheIsite of plasmid pOGS210, pPE511, or pPE682. The TAR elements studied were either WT, BM, or LM, and the double TARelements were combinations of these as indicated in Fig.
iC.
To ensure that theanalysis was quantitative, we established
limiting conditions for Tat activation. The activation of BM
and LM TARs in pPE682 and pPE511, respectively, was compared with that of WT (pOGS210) at a range of Tat concentrationsprovided by expression from plasmid pOGS213
(1). Plasmids were transfected into subconfluent HeLa cells,
andproteinextractswereassayed for chloramphenicol
acetyl-transferase (CAT) activity after 48 h. At a low input of Tat (1
ng of pOGS213), neither the BM nor LM TAR supported
activation, whereasWTgave about a 60-fold activation, (Fig.
2). Consistent with other studies (3, 26), the mutations were
8396
on November 9, 2019 by guest
http://jvi.asm.org/
Nh
HIV-1 LT AT I
+20 TAR
pOGS210
pPE682
pPE511
+20 bulge loop +38
AGAUCUGAGCCUGGGAGCU wildtype(WT)
AGACCUGAGCCUGGGAGCU bulgemutant(T1) AGAUCUGAGCAGGGJAGCU bopmutant(T2)
WT BM
LM c
CD
B
+1 +80
Nti SS Nh pA
TAR1 TAR2
Key: x
-X- wild type (WT)
_ -0- bulgemutant(T1)
-0_- loopmutant(T2)
0 1 1 0 100 1000
TAR1 TAR2
WT WT DT
WT LM T3
BM LM T4
BM WT T5
LM LM T6
LM WT T7
EM BM T8
LM BM T9
LM-10o-EM T10
BM-101-LM Ti1
FIG. 1. Structures of the expression plasmids used for WT (pOGS210), BM, and LM sequences. LTR, long terminal repeat; CAT, bacterialCATcodingregion; I, simianvirus 40 small t intron; Nh,NheI restriction site;pA,polyadenylation site. The TAR stem loop isrepresented byarrows.TheLMis plasmidpPE511(5), and the BM, which containsapointmutation of U23 to C23, was generated by PCR mutagenesis using standard techniques. The primerused for primer extension analysis of RNA was as described previously (3) and corresponded to nucleotides +33 to +13 in the CAT gene. (B) Structures of the HIV-1 LTR constructs harboring double TAR elements. Ss, SstII restrictionsite. PlasmidspOGS210, pPE682(T1) andplasmid pPE511(T2)werelinearizedattheuniqueNhel site, and
a synthetic oligonucleotide which comprised a linker containing an SstII restrictionsite and either theWT, LM,orBM TAR sequence (panelA)wasinserted. TAR1 is eitherWT,BM,orLM,and TAR2is
asyntheticoligonucleotide consisting ofalinkerandresidues +1 to +
62ofeither theWT,BM, or LM TAR sequence.(C)Configurations anddesignations of themutants.MutantsT10 andTllareT9andT4 in which the TARelementsareseparated bya101-nucleotidespacer.
lessdeleteriousat ahigh(500-ng) input ofTat.Allsubsequent
experimentswereconducted with 500 ng of TAR DNA and 1
ngofTat-expressing plasmid.
Although RNA polymerase is poorly processive in the
absence ofcorrect Tatactivation (28), itseems unlikely that there is an accumulation oftranscripts truncated afterTAR,
as,forexample,aribozymeplaceddownstreamofTARis still
functional(18).Wethereforeanticipatedthat the second TAR element should besynthesized. However, to confirm this,we determined the level of activation fromasingleTARelement
compared with tandem TAR elements in which the first
[image:2.612.320.561.73.248.2] [image:2.612.65.303.74.386.2]elementwaseither aBM
(T5)
or anLM(T7).
Inaddition,
to control for the possibility that asuper-secondary
structure, deleterioustoWTactivation,mightarise from theduplication
[pOGS213]ng
FIG. 2. Effectsof titrating Tat-expressing (pOGS213)onthe
acti-vation of WT andmutantTARsequences.Transient transfectionwere
carried outby calcium phosphate coprecipitation onto subconfluent HeLa cells. Extractswereassayed after 48h for CAT activity by using
Quan-T-CAT, ascintillation counting-based assay(Amersham
Inter-national plc). RNAwas prepared as described previously (6) and
analyzed by primer extension. Transfection efficiencywasdetermined
by cotransfection of human cytomegalovirus promoter-driven lucif-erase plasmid (pMW41 [35]), and luciferase assays carried out as
describedpreviously (36). Plasmids encoding WT bulge BM Ti,and LM T2TAR RNAs (500ngof each)weretransfected into HeLa cells
with 0to500ngof pOGS213 and 250ngofpMW41. Proteinextracts
containing thesamenumberof luciferase unitswereassayed for CAT
activity.
of TAR elements,we analyzed the activation of tandem WT
TARs(DT)andatandem element in which the first TARwas
WT and thesecond TARwasmutated (T3). The double TAR
plasmids were cotransfected with pOGS213 under limiting
conditions, and after 48 h, extracts were analyzed for the
presence of CAT transcripts and CAT enzyme activity (Fig.
3A).
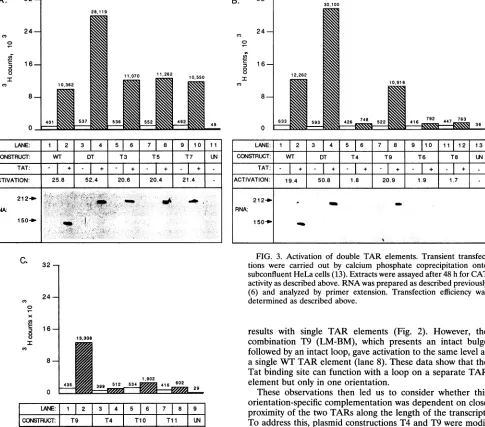
Interestingly, the tandem WT TAR gave twice the level of activation ofthe single TAR (Fig. 3A, lane 4compared with lane 2). AdefectiveTARplaced downstreamofWT had no
deleterious effect, as activation levels were the same as
ob-tained witha singleTAR (compare lanes 2 and6). Placinga
defectiveTAR upstream ofa WTTAR element also had no
effecton activation,whether itwas a BM (lane 8) or an LM (lane 10).Inthese latterconfigurations, activation levelswere
thesame asobtained with thesingleTAR(lane 2).These data indicated thataWT TARelementwasfullyfunctional whether placed upstream or downstream of a second defective TAR element, indicatingthatTARfoldingandproteininteractions
wereunperturbed bythe tandemstructure.Thisfindingis also consistent with reports that multiple tandem TAR elements functioneffectivelyasTARdecoys(32, 33).Thereasonfor the increased activation by the tandem WT TARs is not clear, although similar observations were made when HIV-2 TAR elementswereduplicated (2).ThefindingthataWTTARcan
function when placed downstream of a defective element about 90 nucleotidesawayfrom the mRNA startappears to be
inconsistent with thepreviouslydescribedposition dependence of TAR(15, 17, 24, 27).The result couldbe explained if the
compactsecondarystructureof thefirst TARservesto shorten
the distance between the initiation site and the active TAR.
Alternatively,an asyet uncharacterizedTAR-specific protein
A
on November 9, 2019 by guest
http://jvi.asm.org/
A.
32-28,119
24-0
0
B. 32
24
0
C;
-r
, 16 CO
10,916
C.
0
CO
x
0
am
I
mayrecognizethedefective TAR andfacilitate the activity of thesecondelement despite its distancefromthe initiationsite. Given that tandem HIV-1 TAR elements were functional, we then examined whether different mutated TAR elements could be combinedin tandem to allowactivation by comple-mentationbetween the Tatbinding site and the loop factor bindingsite (Fig. 3B). The combination T4 (BM-LM), which effectively presentsaWTloop in the firstTARfollowed bya
WT bulge in the second TAR, showed no activation by Tat
(lane 6). Similarly,doublebulgemutants (T8)ordouble loop
mutants(T6)weredefective (lanes12and10), paralleling the
LANE: 1
12
3 4 5 6 71
8 9 10 11 12 13CONSTRUCT: WT DT |T4 TT9 T6 T8 IN
TAT:
l
+
|
+
T
+
r
+
-
+
ACTIVATION: 19.4 50.8 1.8 20.9 1.9 1.7
212W "a
RNA:
150_ _
FIG. 3. Activation ofdouble TAR elements. Transient transfec-tions were carried out by calcium phosphate coprecipitation onto subconfluent HeLacells(13). Extractswereassayed after 48 h forCAT activityasdescribedabove.RNA waspreparedasdescribedpreviously (6) and analyzed by primer extension. Transfection efficiency was determinedasdescribedabove.
results with single TAR elements (Fig. 2). However, the combination T9 (LM-BM), which presents an intact bulge
followed by an intactloop, gave activationtothesamelevel as asingle WT TAR element (lane 8).These data show that the Tatbinding sitecan function witha loopon aseparate TAR element but only inoneorientation.
These observations then led us to consider whether this orientation-specific complementationwas dependentonclose proximity of the twoTARsalongthelengthof thetranscript.
Toaddressthis, plasmidconstructions T4 and T9were modi-fied to produce plasmids Ti1 and T10, respectively, by the insertion of a 101-nucleotide oligonucleotide (5'-GGTAA GACACGACTTATCGCCACTGGCAGCAGCCACTGGTA
ACAGGATTAGCAGAGCGAGGT'lTGTAGGCGGTGCTA
CAGAGTTCTAGAAGTGGTGGCCGC-3')
at the unique SstII site, thus spacing the two TAR elements. Plasmid con-structions Tll and T10were tested in a Tat activation assay(Fig. 3C). As formutant T4, Tat was unable to activate the
TARconfiguration inmutantTll(Fig.3C, lane8).Activation ofmutantT10,however,wasmuch reducedcomparedwiththe parental construct T9 (compare lanes 2 and 6). Taken
to-gether,these datashow that while there issurprisingflexibility
intherelationshipof the Tatbinding site to the TAR loop, that
flexibilitydoesnotextendto aseparation of 101 nucleotides.
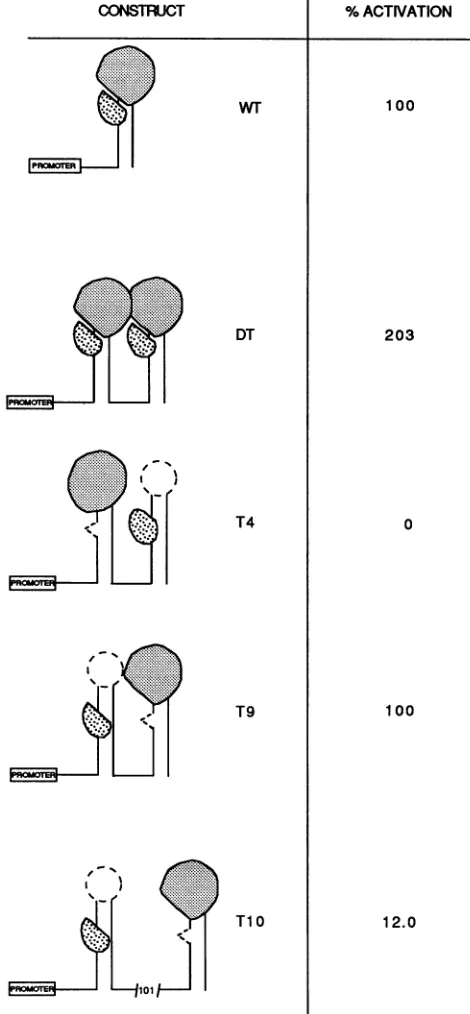
The resultsaredepictedschematically in Fig.4.Wepropose
thatTatandtheloop-bindingproteinareasymmetric and that
theypresent interaction surfaces in the WTconfiguration. In
T4,althoughTatand theloopfunctionaredisplayingthesame
facetothe promoter, theyare notdisplayingthesamefaceto each other. Thisconfiguration isinactive. IfTatand theloop factor bind in thisconfiguration,thenclearlytheirindependent
presentation to the initiation complex is not sufficient for
36
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.66.551.80.507.2]CONSTRJCT
IER I
rpp LJ0iR
WT
DT
T4
T9
T10
FRMTP-- - L- ~o-
~-%ACTIVATION
100
203
0
100
12.0
FIG. 4. Activationconfigurationsof doubleTAR elements. Shown
isasimplecartoonrepresentingTARsequence,indicatingthebinding
oftwoasymmetric factors,Tat andaloop-bindingfactor. Thepercent
activation of thesequence configurations by Tatas determined from
data inFig. 3 isindicated.
activation. Alternatively,neither of themmaybind. InT9,Tat
and the loop-binding protein display the same face to each
otherand to the initiationcomplexas in theWTsingleTAR, and thisisnowactive. InT10,althoughTat andaloop-binding
cellular factor are in the correct orientation with respect to
each other, we would suggest that the spacing of the two
protein binding sites does not permit efficient interaction to
allowactivation to occur.
If, as proposed previously, Tat must form a stable
het-erodimer beforebindingto TAR(21),then thiscomplex must
beabletobridgetwo TARelements. Given the in vitro binding
propertiesof Tat (9, 10, 34),itisperhaps more likely that Tat
and the loop-binding factor bind independently but upon contact,facilitatedbytheflexibilityof the RNA, theremightbe aconformationalchangeinTat tocreate anactivation surface. Thenotion of conformationalchangein Tat is consistent with
thefindingthat isolated activation domainscomprisingamino
acids 1 to 48 bind different proteins from full-length Tat in
vitro
(16).
Itis interestingthat separating thebulge and loopondifferent TAR elements isfunctional,whereasspacingthem
by increasingthe upper stemresults in anonfunctional TAR
(2, 29).
Either there is insufficient flexibility in this latter structuretoallowproteininteractionorthestretchedstructure binds a competing factor such as TRBP1 (12), which may inhibit Tat binding. Irrespective of whether Tat binds TARindependentlyor as aheterodimer withaloop factor,it is clear
that the bulge region in the first TAR is available for Tat
binding.
Thisis notconsistent with the notionproposedforasingle
TAR(4)
that in the absence ofloop-binding factors,
competing
cellular bulge-binding factors block Tatbinding.
However, it is possible that the loop-binding factors on the
second TARalso
effectively
exclude otherbulge-binding
pro-teins.
Taken
together,
these resultsmeanthatanydetailed model of the Tat-TARphenomenon
would needtotakeaccountofa greaterflexibility
intherelationship
of thebulge
and theloop
than has been
appreciated
todate.MartinBraddock isaRoyal SocietyResearchFellow;Robert Powell isaSERCpostgraduate.This researchwasfundedbyGLAXO andthe SERC.
We thank our colleagues in the Retrovirus Molecular Biology Groupforstimulating discussions.
REFERENCES
1. Adams,S.E.,
L.
D.Johnson,M.Braddock,A.J.Kingsman,and R. Edwards. 1988. Synthesis of a gene for the HIV transactivator proteinTatbyanovelsinglestrandedapproachinvolvingin vivo gaprepair.Nucleic AcidsRes.16:4287-4298.2. Berkhout, B.,A.Gatignol, J.Silver,andD.Jeang.1990.Efficient transactivation bythe HIV-2 Tat protein requires a
duplicated
TAR RNAstructure.Nucleic AcidsRes. 18:1839-1846. 3. Blanchard,A.D., R.
Pow4l1,
M.Braddock,A.J.Kingsman,andS. M.Kingsman.1992. An adenosineatposition27in the human immunodeficiencyvirustype 1trans-activationresponse element is
notcriticalfortranscriptionalortranslationalactivationbyTat.J. Virol.66:6769-6772.
4. Braddock, M.,R Powell,A. D.Blanchard,A.J. Kingsman,and S. M.Kingsman.1993. HIV-1 TARRNA-binding
proteins
control Tat activation of translation in Xenopus oocytes. FASEB J. 7:214-222.5. Braddock, M., A. Thorburn, A. Chambers, G. D. Elliott, G.J. Anderson,A.J.Kingsman,and S. M.Kingsman. 1990. Anuclear translational blockimposed bytheHIV-1 3
region
is relievedby
theTat/TARinteraction.Cell 62:1123-1133.
6. Braddock, M., A. M. Thorburn, A. J.
Kingsman,
and S. M. Kingsman. 1991. BlockingofTat-dependentHIV-1 RNA modi-ficationbyaninhibitorof RNApolymerase
IIprocessivity.
Nature(London)350:439-441.
7. Cullen,B. R. 1992. Mechanism of action of
complex regulatory
proteinsencodedbycomplexretroviuses. Microbiol.Rev. 56:375-394.8. Cullen, B. R. 1993. Does HIV-1 tat induce a
change
in viral initiationrights?Cell73:417-420.9. Dingwall,C.,I.Ernberg,M.J. Gait,S.M.Green, S.Heaphy,J. Karn,A. D.Lowe,M. Singh,and M. A.Skinner. 1990. HIV-1tat
,-1% I I
1% ol
-1I
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.69.304.81.588.2]protein stimulatestranscriptionbybindingto aU-richbulgeinthe stemof theTARRNA structure. EMBOJ.9:4145-4153. 10. Dingwall, C., I. Ernberg, M. J.Gait, S. M.Green, S.Heaphy,J.
Karn, A. D. Lowe, N. Singh, and M. A. Skinner. 1989. Human immunodeficiency tat protein binds transactivation responsive region(TAR)RNA in vitro. Proc.Natl.Acad.Sci.USA 86:6925-6929.
11. Feng, S., and E. C. Holland. 1988. HIV-1 tat transactivation requires theloop sequences withinTAR. Nature (London) 334: 165-167.
12. Gatignol, A., A. Buckler-White, B. Berkhout and K.-T. Jeang. 1991. Characterisation of a human TAR-RNA binding protein that activates the HIV-1 LTR.Science 251:1597-1600.
13. Gorman, C. M., L. F.Moffat,and B. H.Howard.1982. Recombi-nant genomeswhichexpresschloramphenicol acetyltransferasein mammalian cells.Mol.Cell. Biol.2:1044-1051.
14. Gunnery, S., S. R. Greeen, and M. B. Mathews. 1992. Tat responsive region of humanimmunodeficiency virustype 1 stim-ulatesproteinsynthesis in vivoand in vitro: relationship between structure and function. Proc. Natl. Acad. Sci. USA 89:11556-11561.
15. Hauber, J., and B. R. Cullen. 1988. Mutational analysis ofthe transactivation responsiveregion of thehumanimmunodeficiency virusTatprotein. J.Virol.62:673-679.
16. Hermann, C. H., and A. Rice. 1993. Specific interaction ofthe humanimmunodeficiency virustatproteins withacellular protein kinase.Virology 197:601-608.
17. Jakobovits,A., D. H. Smith, E. B. Jakobovits, and D. J. Capon. 1988. A discrete element 3' of human immunodeficiency virus1 (HIV-1) and HIV-2 mRNA initiation sites mediates transcrip-tional activation by an HIV trans activator. Mol. Cell. Biol. 8:2555-2561.
18. Jeang, K.-T.,R.Chun, N. H. Lin, A.Gatignol, C. G. Glabe, and H. Fan. 1993. In vitro and invivo binding of human immunodefi-ciency virus type 1 Tat protein and Spl transcription factor. J. Virol.67:6224-6233.
19. Kaczmarski, W., and S. A. Khan. 1993. Lupus autoantigen ku proteinbindsHIV-1 TAR RNAinvitro. Biochem. Biophys. Res. Commun. 196:935-942.
20. Kashanchi, F., G. Piras, M. F. Radonovich, J. F. Duvall, A. Fattaey, C.-M.Chiang, R. G. Roeder, and J. N. Brady. 1994. Direct interaction of human TFIIDwith the HIV-1 transactivator Tat. Nature(London)367:295-296.
21. Madore, S. J., and B. R. Cullen. 1993. Genetic analysis of the cofactor requirement for human immunodeficiency virus type 1 Tatfunction.J. Virol. 67:3703-3711.
22. Marciniak, R. A., M. A. Garcia-Blanco, and P. A. Sharp. 1990. Identification and characterisationof aHeLa nuclear protein that specificallybinds to the transactivation response element of
hu-manimmunodeficiencyvirus. Proc. Natl. Acad. Sci. USA 87:3624-3628.
23. Muesing,M.A.,D. Smith,and D.J. Capon. 1987.Regulationof mRNAaccumulationbya humanimmunodeficiencyvirus trans-activatorprotein.Cell48:691-701.
24. Peterlin, B.N.,P.J. Luciw,P.J. Barr, and M. D.Walker. 1986. Elevated levels ofmRNA can account for thetransactivation of human immunodeficiency virus. Proc.Natl. Acad. Sci. USA 83: 9734-9738.
25. Rounsville, M. P., and A. Kumar. 1992. Binding of a host cell nuclear proteinto the stemregion ofhuman immunodeficiency virus type 1 transactivation-responsive RNA. J. Virol. 66:1688-1694.
26. Roy, S., N. T.Parkin, C.Rosen, J.Itovitch, and N. Sonenberg. 1990.Structuralrequirementsof transactivation of human immu-nodeficiency virus type 1 long terminal repeat-directed gene expressionbytat:importanceofbasepairing, loop sequences,and bulges in thetat-responsive sequence region. J.Virol. 64:1402-1406.
27. Selby, M. J., E. S. Bain, P. A. Luciw, and B. M.Peterlin. 1989. Structure, sequence, and position of the stem-loop in TAR determine transcriptional elongation by Tat through the HIV-1 longterminal repeat.Genes Dev.3:547-558.
28. Selby,M.J., and M. B. Peterlin. 1990.TransactivationbyHIV-1 tatviaaheterologousRNA-binding protein. Cell62:769-776. 29. Sheline, T., L. H.Milocco, and K. A.Jones. 1991. Two distinct
nucleartranscriptionfactorsrecognize loopandbulge residuesof theHIV-1 TAR RNAhairpin.Genes Dev. 5:2508-2520. 30. Southgate, C., L. L. Zapp, and M. R. Green. 1990. Activation of
transcription by HIV-1 Tat protein tethered to nascent RNA through another protein. Nature(London)345:640-642. 31. Southgate, C. M., and M. R. Green. 1991.Tat proteinactivates
transcriptionfrom an upstreamDNAbinding site: implicationsfor tatfunction. Genes Dev. 5:2496-2507.
32. Sullenger,B.A., H. F.Gallardo,G. E. Ungers, and E. Gilboa. 1990. Overexpression of TAR sequences renders cells resistant to humanimmunodeficiency virusreplication. Cell63:601-608. 33. Sullenger,B.A., H. F. Gallardo, G. E. Ungers, and E. Gilboa.1991.
Analysisoftransactingresponse decoyRNA-mediatedinhibition of humanimmunodeficiencyvirus type 1transactivation.J.Virol. 65:6811-6816.
34. Weeks, K. M., and D. M.Crothers. 1991. RNA recognitionby Tat-derivedpeptides: interaction in the majorgroove.Cell 66:577-588.
35. White,M., andS. M.Kingman. Unpublisheddata.
36. White, M. R. H., J. Morse, Z. A. M. Boniszewski, C. R. Mundy, M. A. W. Brady, and D. J. Chiswell. 1990. Imaging of firefly luciferase expressioninsinglemammalian cells using high sensi-tivitycharge-coupled devicecameras.Technique 2:194-201. 37. Wu, F. J., D. Garcia, D. Sigman, and R Gaynor. 1991. Tat
regulatesbinding ofthehumanimmunodeficiencyvirus transacti-vating region RNA loop-binding protein TRP-185. Genes -Dev. 5:2128-2140.