Interaction of the Influenza A Virus Nucleocapsid Protein with the Viral RNA Polymerase Potentiates Unprimed Viral RNA Replication
Full text
Figure
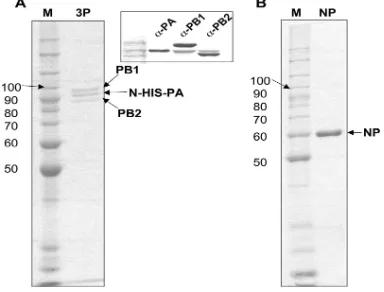
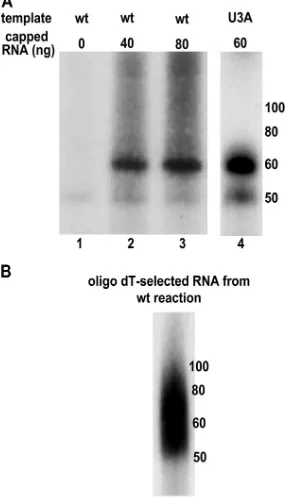

Related documents
Only a few viral gene products are expressed by the latent virus, and one of the best characterized is the latency-associated nuclear antigen (LANA), a nuclear protein required for
Importantly, neither of these 25- and 27-kDa tNef proteins was observed in similar immunoblot analysis of cells infected with a stock of SIVmac239⌬ 152 nef (Fig. 4A, lane 3) or in
This analysis demon- strated a clear relationship between pR78-like peptides and other GCRs; the predicted amino acid sequence of the UL78 gene was similar to sequences of GCRs such
mine whether MAV-1 is able to alter surface expression of class I MHC antigens other than those shown in Fig. 1, we obtained additional cultured cell lines expressing different
clearance of intestinal infection. Replication of reovirus in MuMT mice 7 to 11 days after p.o. 1 and 2), combined with the importance of B cells in the control of intestinal
We previously encountered a similar lack of correla- tion between infectivity titers and the number of cells labeled by in situ hybridization in neonatal C3H and C57BL/6 mice
For the particular case of an abstract linear control systems, we shall derive now a different conservation property based on our observation concerning evolutionary systems...
The aim of this study is to identify current Knowledge Management practice in Thai SMEs within the manufacturing sector and to examine how employees in the organization capture,
