Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Characterization of a Nerve Growth Factor-Inducible Cellular Activity
That Enhances Herpes Simplex Virus Type 1 Gene Expression and
Replication of an ICP0 Null Mutant in Cells of Neural Lineage
ROBERT JORDAN, JOSH PEPE,ANDPRISCILLA A. SCHAFFER*Division of Molecular Genetics, Dana-Farber Cancer Institute, and Department of Microbiology and Molecular Genetics, Harvard Medical School, Boston, Massachusetts 02115
Received 23 December 1997/Accepted 20 March 1998
Herpes simplex virus type 1 (HSV-1) ICP0 is required for efficient viral gene expression during lytic infection, especially at low multiplicities. A series of cellular activities that can substitute for ICP0 has been identified, suggesting that when the activity of ICP0 is limiting, these activities can substitute for ICP0 to activate viral gene expression. The cellular activities may be especially important during reactivation of HSV from neuronal latency when viral gene expression is initiated in the absence of prior viral protein synthesis. Consistent with this hypothesis, we have identified an inducible activity in cells of neural lineage (PC12) that can complement the low-multiplicity growth phenotype of an ICP0 null mutant, n212. Pretreatment of PC12 cells with nerve growth factor (NGF) or fibroblast growth factor (FGF) prior to infection produced a 10- to 20-fold increase in the 24-h yield of n212 but only a 2- to 4-fold increase in the yield of wild-type virus relative to mock treatment. Slot blot analysis of nuclear DNA isolated from infected cells treated or mock treated with NGF indicated that NGF treatment does not significantly affect viral entry. The NGF-induced activity in PC12 cells was expressed transiently, with peak complementing activity observed when cells were treated with NGF 12 h prior to infection. Addition of NGF 3 h after infection had little effect on virus yield. The NGF-induced cellular activity was inhibited by pretreatment of PC12 cells with kinase inhibitors that have high specificity for kinases involved in NGF/FGF-dependent signal transduction. RNase protection assays demonstrated that the NGF-inducible PC12 cell activity, like that of ICP0, functions to increase the level of viral mRNA during low-multiplicity infection. These results suggest that activation of viral transcription by ICP0 and transcrip-tional activation of cellular genes by NGF and FGF utilize common signal transduction pathways in PC12 cells.
Herpes simplex virus type I (HSV-1) establishes life-long infections of the human host characterized by productive and latent phases. During productive infection, nearly all of the more than 75 genes encoded by the virus are expressed. These genes have been categorized into three major kinetic classes, immediate-early (IE), early (E), and late (L), depending on the time of their peak expression and the sensitivity of their ex-pression to inhibitors of DNA, RNA, and protein synthesis (33, 34). The cascade of productive viral gene expression is regu-lated by virus-encoded proteins present in virions and five IE proteins which, in combination with host transcriptional ma-chinery, direct the expression of viral E and L genes (78).
HSV-1 initiates productive infection in epithelial cells of the skin and mucosal membranes at the site of infection (reviewed in references 25 and 60). Virus particles produced in epithelial cells during productive infection enter neuronal termini that innervate the skin in the area surrounding infection and then travel via retrograde axonal transport to neuronal cell bodies located in sensory ganglia (14). Viral DNA enters the neuronal nucleus, where limited genome amplification ensues and la-tency is ultimately established (14).
During latency, productive viral gene expression is almost completely repressed, with transcription being limited to a small region of the genome encoding the latency-associated transcripts (LATs) (17, 24). In the absence of viral transcrip-tion, viral DNA replication ceases, and viral genomes are
con-densed into chromatin-like structures containing nucleosomes (18). Months or years later, viral or cellular molecules induced in response to stress stimulate the resumption of viral gene expression (reactivation), resulting in the synthesis of new virus particles (25). These particles travel back to the initial site of infection via the axonal route and initiate lytic infection of epithelial cells, resulting in recurrent clinical disease (14). Al-though the general course of events involved in the establish-ment and reactivation of latency is clear, the molecular pro-cesses that underlie establishment and reactivation are poorly understood.
Mutant viruses have been widely used to identify viral genes involved in the establishment and reactivation of latent infec-tion (13, 36, 42, 50, 73). These studies have demonstrated that infected-cell polypeptide 0 (ICP0), a viral IE regulatory pro-tein, is important for efficient reactivation in mouse and rabbit models of HSV-1 latency (6, 13). ICP0 stimulates transcription of viral IE, E, and L genes when productive infection is initi-ated at low multiplicities (9, 10, 37). ICP0 activates a broad spectrum of viral and cellular promoters without apparent sequence specificity in transient expression assays and stimu-lates de novo gene expression after transfection of infectious DNA into permissive cells (7, 20, 21, 54, 55). In an in vitro cell culture model of HSV latency, ICP0 is both necessary and sufficient for reactivation (29, 61, 85). In addition, ICP0 may also play an important role in establishment of latent infection (80).
Cellular proteins likely play an important part in ICP0’s broad transactivating activity. In support of this hypothesis, ICP0 transiently colocalizes with antigens implicated in cell growth control in nuclear substructures called nuclear domain * Corresponding author. Present address: Department of
Microbi-ology, University of Pennsylvania School of Medicine, Philadelphia, PA 19104. Phone: (215) 573-9863. Fax: (215) 573-5344. E-mail: pschfr @mail.med.upenn.edu.
5373
on November 9, 2019 by guest
http://jvi.asm.org/
10 (ND10). Colocalization of ICP0 with ND10 antigens corre-lates with their redistribution in the infected cell (48, 49). In addition, ICP0 coimmunoprecipitates with a 135-kDa protein homologous to members of the ubiquitin-specific protease family of proteins (22, 52). Redistribution of ND10 antigens and coimmunoprecipitation with the 135-kDa protein requires intact transcriptional activating domains of ICP0, suggesting that these phenomena are functionally related (48, 51). ICP0 also destabilizes the catalytic subunit of a DNA-dependent protein kinase (DNA-PK) following viral infection (41). Thus, ICP0’s broad transactivating activity likely involves interactions with cellular proteins.
In studies of ICP0 null mutant viruses, several cell cycle-regulated and cell-type-specific cellular activities that are able to substitute for the transactivating activity of ICP0 have been identified (8, 84). Vero cells growth arrested in G0/G1by
iso-leucine deprivation express an activity after release from growth arrest that enhances the plating efficiency of an ICP0 null mutant virus (8). Similarly, Vero cells and cells of neural origin express an activity after release from growth arrest that activates HSV-1 gene expression in transient expression assays (57). Finally, U2OS cells, an osteosarcoma cell line, constitu-tively express high levels of an activity that stimulates the growth of an ICP0 null mutant virus (84). These observations demonstrate that cellular functions can substitute for the trans-activating activity of ICP0.
Initiation of viral gene expression at the onset of reactivation occurs in the absence of ICP0 and other viral transcriptional activators. Since ICP0 is an important activator of viral gene expression, the existence of cellular activities able to substitute for ICP0 suggests a possible mechanism by which viral gene expression is activated during reactivation. In this study, we describe an activity in cells of neural lineage (PC12) induced by treatment with two physiologically relevant growth factors, nerve growth factor (NGF) and fibroblast growth factor (FGF), that enhances gene expression and replication of an ICP0 null mutant, n212. The ICP0-like, n212-complementing activity is expressed transiently and can be blocked by inhibi-tors of NGF-dependent signal transduction. Moreover, like ICP0, the cellular complementing activity functions at the level of mRNA accumulation. We suggest that the NGF/FGF-in-duced n212-complementing activity may be responsible for the initiation of viral gene expression during reactivation from neuronal latency when no ICP0 is present.
MATERIALS AND METHODS
Cells and viruses.PC12 cells were the generous gift of John Wagner (Cornell University Medical College, New York, N.Y.) and were cultured in Dulbecco’s modification of Eagle’s minimal essential medium supplemented with 10% fetal bovine serum and 5% horse serum as described previously (26). MM17-26 cells were kindly provided by Geoffrey Cooper (Dana-Farber Cancer Institute, Bos-ton, Mass.) and were cultured as described for PC12 cells. Vero cells, L7 cells, which contain a stabily integrated copy of ICP0, and the osteosarcoma line U2OS were cultured as previously described (63, 65, 84).
Wild-type HSV-1, strain KOS, and the ICP0 nonsense mutant, n212, derived from KOS were propagated as described previously (7, 67). Viral titers were measured by standard plaque assay on Vero cell monolayers for strain KOS and on U20S cells or L7 cells for n212. Genome numbers were determined for all virus stocks by slot blot analysis of viral DNA, and the number of biologically active genomes was determined by quantitating the number of ICP4-expressing Vero cells visualized by indirect immunofluorescence after low-multiplicity in-fection. The numbers of viral genomes and the number of ICP4-expressing cells correlated well with the titers of KOS as measured by plaque assay on Vero cell monolayers and of n212 as measured on U2OS or L7 cell monolayers.
Growth factors and kinase inhibitors.Viral infections were conducted in PC12 cells in the presence and absence of NGF or other growth factors with and without serine/threonine kinase inhibitors. Briefly, PC12 cell monolayers (106) were seeded in 35-mm-diameter dishes and incubated at 37°C in 10% CO2for 12 to 18 h. Three hours prior to infection at 0.01 PFU/cell, culture medium was removed and replaced with fresh medium containing NGF 2.5S (100 ng/ml;
Collaborative Biochemical, Bedford, Mass.) and FGF 100 ng/ml; Collaborative Biochemical). Serine/threonine kinase inhibitors K252a, KT5720, PD98059, and calphostin C were purchased from Calbiochem (La Jolla, Calif.) and were added 30 min prior to NGF addition. Inhibitors were added at concentrations 10-fold higher than the published 50% inhibitory concentration for each inhibitor (5, 38, 56). The concentrations of the inhibitors were as follows: K252a, 0.25mM; KT5720, 0.5mM; PD98059, 20mM; and calphostin C, 0.5mM. The specificity of each inhibitor for cellular kinases has been described elsewhere (5, 38, 40, 56, 71, 76). Replicate cultures were harvested at 24 h postinfection (hpi), and virus yields were determined by standard plaque assays on Vero (KOS) or L7 or U2OS (n212) cell monolayers as described previously (37).
PMA and second messenger analogs. PC12 cells were treated with 16 nM phorbol 12-myristate 13-acetate (PMA), 50mM dibutyryl cyclic AMP (dbcAMP), 1mM ionomycin, or 50mM forskolin, alone or in combinations, in the presence and absence of NGF for 3 h prior to infection. Following the 3-h incubation, the cultures were infected with 0.01 PFU of n212 or KOS per cell, and viral yields were measured at 24 hpi as described above.
Viral entry.PC12 cells (53106) in 60-mm-diameter dishes were treated with 100 ng of NGF per ml or mock treated 3 h prior to infection with 1.0 PFU of KOS or n212 per cell. At 3 hpi, the cells were scraped into the medium and pelleted by centrifugation at 1,0003g for 4 min at 4°C. The cell pellet was
washed once with 13Tris-buffered saline (25 mM Tris-HCl [pH 7.4], 140 mM sodium chloride, 5 mM potassium chloride), and the cells were repelleted by centrifugation at 1,0003g for 4 min at 4°C. The cell pellet was resuspended in
0.375 ml of lysis buffer (50 mM Tris-HCl [pH 7.6], 150 mM potassium acetate, 5 mM magnesium acetate, 0.5% Nonidet P-40, 1 mM dithiothreitol) and incubated on ice for 5 min to lyse the cells. The nuclei were centrifuged at 14,0003g for
5 min at 4°C through a 0.5-ml 20% sucrose cushion prepared in lysis buffer lacking Nonidet P-40. The nuclear DNA from this preparation was isolated by using minicolumns as recommended by the manufacturer (Qiagen, Chatsworth, Calif.).
Nuclear DNA was denatured in 0.3 N NaOH for 30 min at 65°C, neutralized with 0.2 ml of 3 M sodium acetate (pH 4.8), and applied to nylon membranes (Schleicher & Schuell, Keene, N.H.) by slot blot hybridization according to the manufacturer’s recommendations. The membrane was probed with 250 ng of 32P-labeled, nick-translated KOS DNA (8 3104cpM/ng). The radiolabeled probe was removed by heating the blot in a boiling water bath for 15 min. No detectable signal was observed by PhosphorImager analysis using a Storm 860 (Molecular Dynamics, Sunnyvale, Calif.) after a 4-h exposure at a sensitivity of 0.0 to 100 counts. The blot was then reprobed with a 140 ng of a32P-labeled antisense RNA probe (33105cpM/ng) specific for the rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, and the radiolabeled band was visu-alized by PhosphorImager analysis. The range values for the image display were set at 0 to 791 counts for the KOS probe and 0 to 258 counts for the GAPDH probe.
Northern blot analysis.PC12 cell cultures (53106) in 60-mm-diameter dishes were treated with 100 ng of NGF per ml for 0, 15, 30, and 60 min or infected with KOS and n212 at 5.0 PFU/cell for 30, 60, 90, or 120 min. Cytoplasmic RNA was isolated at each time point as described previously (37). The RNA was size fractionated by denaturing gel electrophoresis and transferred to nylon mem-branes by Northern blotting. The Northern blot was probed with 100 ng of 32P-labeled antisense RNA probe (83105cpM/ng) specific for the mouse c-fos message. The blot was reprobed with 150 ng of32P-labeled antisense RNA probe (8.83105cpM/ng) specific for the rat GAPDH message. The blot was visualized by PhosphorImager analysis. The range values for the image display were set at 0 to 500 counts.
RNA isolation and RNase protection.Cytoplasmic RNA isolation and quan-titative RNase protection assays were performed as described previously (37).
RESULTS
NGF and FGF stimulate replication of the ICP0 null mu-tant, n212, in PC12 cells.Reactivation of latent HSV-1 corre-lates with changes in levels of stress-induced growth factors and neurotrophins (32, 79). NGF, FGF, and epidermal growth factor (EGF) are among the many growth factors induced by stress that have a wide range of biological effects in vivo and in cell culture. In PC12 cells, physiological concentrations of NGF and FGF activate cellular signaling cascades leading to morphological and biochemical differentiation (28). Differen-tiated PC12 cells resemble sympathetic neurons in that they develop neurites, become electrically excitable, and express a number of neuron-specific genes (27, 28). In contrast, physio-logical concentrations of EGF stimulate mitogenesis in PC12 cells, even though many of the same signaling molecules are activated by all three factors (47). To determine the effects of growth factor-induced cellular activities on viral replication,
on November 9, 2019 by guest
http://jvi.asm.org/
PC12 cells were treated with NGF, FGF, or EGF or were mock treated for 3 h prior to infection with 0.01 PFU of wild-type HSV-1 strain KOS or the ICP0 null mutant n212 per cell. At 3 and 24 hpi, virus yields were measured by plaque assay.
Treatment of PC12 cells with NGF increased the 24-h yields of n212 and KOS;14- and 3-fold, respectively (Fig. 1). Sim-ilarly, FGF stimulated n212 and KOS replication nine- and fourfold, respectively. These treatments had little effect on the levels of infectious virus at 3 hpi (data not shown). EGF had only minor but reproducible effects on 24-h yields of n212 (1.5-fold) and KOS (2.3-fold). The results of these tests indi-cate that NGF and FGF, but not EGF, induce activities in PC12 cells that complement n212 and, to a lesser extent, en-hance replication of KOS.
One of the most dramatic morphological changes associated
with differentiation of PC12 cells is the formation of neurite-like processes. Not only is neurite formation induced by NGF and FGF, but neurite formation can also be induced by com-bined treatment with EGF and KCl (31, 46). EGF activates cellular signaling pathways, while KCl activates voltage-sensi-tive calcium channels, leading to elevated intracellular calcium levels (3). The combined effects of stimulating EGF-dependent signal transduction, while increasing intracellular calcium lev-els leads to neurite formation (31, 46). Notably, the extent of neurite formation following EGF and KCl treatment is signif-icantly less than that of NGF or FGF treatment (31, 46). To test whether neurite formation correlates with induction of the n212-complementing activity, PC12 cells were treated with EGF or EGF in combination with 50 mM KCl for 3 h prior to infection with 0.01 PFU of KOS or n212 per ml. Again, virus yields were measured at 24 hpi. In these tests, KCl alone and the combination of EGF and KCl (which produced moderate neurite outgrowth) had only modest effects (,2-fold) on rep-lication of n212 or KOS (Fig. 1). Taken together, these results suggest that the n212-complementing activity does not corre-late with neurite formation.
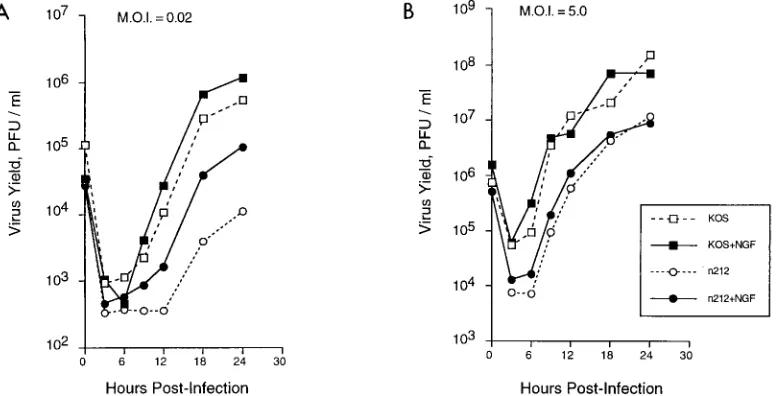
To examine more carefully the effects of NGF on growth of KOS and n212, PC12 cells were treated with NGF or mock treated 3 h prior to infection with 0.02 or 5.0 PFU of either virus per cell. Infected cultures were harvested at 3, 6, 9, 12, 18, and 24 hpi, and virus titers were measured by plaque assay. As shown in Fig. 2, at a low multiplicity of infection (0.02 PFU/ cell), NGF treatment caused an increase in n212 replication from 6 to 24 hpi compared to mock treatment. In contrast, KOS replication increased only slightly in response to NGF relative to mock-treated samples at all times postinfection. At a high multiplicity of infection (5.0 PFU/cell), NGF treatment did not affect replication of n212 or KOS. These results suggest that NGF treatment of PC12 cells stimulates replication of n212 after low-multiplicity infection and that ICP0 and high-multiplicity infection are dominant with regard to the NGF-dependent, n212-complementing activity.
[image:3.612.62.279.67.240.2]NGF treatment does not affect viral entry.It is conceivable that NGF treatment of PC12 cells may affect viral entry, even though the dramatic morphological changes associated with
FIG. 1. Effects of growth factors on replication of n212 and KOS. PC12 cells (106/35-mm-diameter dish) were treated with 100 ng of NGF, FGF, or EGF per
ml or 50 mM KCl in the presence and absence of EGF for 3 h prior to infection with 0.01 PFU of KOS or n212 per cell. At 24 hpi, the cultures were harvested and viral yields were measured. Titers of KOS and n212 were determined on Vero cells and U2OS cells, respectively. The data are expressed as fold difference in titer relative to mock treatment.
FIG. 2. Growth of KOS and n212 in PC12 cells in the presence and absence of NGF. PC12 cells (106/35-mm-diameter dish) were mock treated or treated with NGF
at 100 ng/ml for 3 h prior to infection with KOS or n212 at 0.02 or at 2.5 PFU/cell. At the times indicated, cultures were harvested and viral yields were measured by plaque assay. Titers of KOS and n212 were determined on Vero cells and U2OS cells, respectively.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.108.497.502.700.2]NGF-dependent differentiation require 24 h or more to de-velop. To test whether NGF treatment affects viral infectivity, PC12 cells were treated with NGF 3 h prior to infection with KOS or n212. At 3 hpi, nuclear DNA was isolated and immo-bilized on a nylon membrane by slot blot hybridization. The membrane was probed with radiolabeled KOS DNA. The blot was then stripped and reprobed with a radiolabeled RNA probe specific for the cellular gene, GAPDH. The results of these tests show that NGF treatment had no significant effect on the amount of nuclear KOS or n212 DNA recovered from PC12 cell nuclei (Fig. 3). These results indicate that ICP0 and NGF treatment do not effect viral entry or transport of viral DNA to the nucleus. Similar results were obtained when entry was measured by indirect immunofluorescence (i.e., by count-ing the number of ICP4-expresscount-ing cells in NGF-treated or mock-treated cells infected with KOS or n212 [data not shown]).
The NGF-induced n212-complementing activity requires ac-tivation of multiple NGF-dependent signal transduction path-ways. (i) Inhibitor studies.NGF activates multiple signal trans-duction pathways, including ras-dependent signaling pathways, whose downstream endpoint is the expression of differentia-tion-specific genes (16). The PC12-derived cell line MM17-26 constitutively expresses a dominant negative ras allele that blocks downstream functions of ras (69). Consequently, MM17-26 cells fail to differentiate in response to NGF or FGF treat-ment. To test whether ras is required for the NGF- or FGF-dependent stimulation of n212 replication, MM17-26 cells or PC12 cells were pretreated with NGF or FGF 3 h prior to infection with 0.01 PFU of n212 per cell. At 24 hpi, n212 yields were measured and compared to those on NGF-treated PC12 cells. As shown in Fig. 4, the replication efficiency of n212 in NGF- and FGF-treated MM17-26 cells was only 18% of that observed in NGF- and FGF-treated PC12 cells. These results indicate that the NGF/FGF-dependent stimulation of n212 replication is partially ras dependent.
NGF-dependent differentiation requires the activities of multiple serine/threonine kinases which function sequentially to regulate downstream differentiation-specific activities (16, 69). Serine/threonine kinase inhibitors that block NGF-depen-dent differentiation have been used to iNGF-depen-dentify specific kinases involved in the differentiation process (56, 76). To test whether
selected kinase inhibitors block the NGF-dependent comple-mentation of n212 replication, PC12 cells were treated for 30 min with K252a (a broad-spectrum serine/threonine kinase inhibitor), K5720 (a protein kinase A [PKA]-specific inhibitor), calphostin C (a PKC-specific inhibitor), or PD98059 (an inhib-itor specific for mitogen-activated protein kinase). After the 30-min incubation, NGF was added to cultures containing in-hibitors for 3 h prior to infection. The treated cultures were infected with 0.01 PFU of n212 per cell in the presence and absence of NGF and inhibitors. Virus yields were measured at 24 hpi. In control experiments, treatment of PC12 cells with each inhibitor blocked NGF-dependent differentiation (data not shown). The results of these tests show that the broad-spectrum kinase inhibitor K252a, which blocks multiple NGF-dependent signaling pathways, had the greatest inhibitory ef-fect on virus yield, almost completely blocking the ability of NGF to stimulate replication of n212 (Fig. 4). Calphostin C blocked NGF-dependent replication of n212 by 44%, whereas PD98059 reduced the NGF-dependent stimulation of n212 replication by ;50%. Treatment with K5720 had very little effect on the ability of NGF to stimulate replication of n212 (Fig. 4). Taken together, these results show that kinase inhib-itors able to block multiple NGF-dependent signaling path-ways were able to inhibit NGF-dependent replication of n212 more effectively than inhibitors that blocked fewer pendent signaling pathways, indicating that multiple NGF-de-pendent signaling pathways must be activated to complement n212 replication. Neither the inhibitors used in this study nor the presence of the dominant negative ras allele in MM17-26 cells had a significant effect (,2-fold) on replication of KOS or n212 in the absence of NGF (data not shown). These obser-vations suggest either that HSV-1 does not require these en-zymes for productive infection or that multiple redundant sig-naling pathways are used by the virus.
(ii) Activator studies. Many of the intracellular signaling pathways activated by NGF can be stimulated indirectly by activators of cellular protein kinases or second messenger an-alogs. To test whether direct stimulation of protein kinases or
FIG. 3. NGF does not affect viral entry. PC12 cells were mock treated or treated with NGF (100 ng/ml) for 3 h prior to infection with KOS or n212. Nuclear DNA was harvested prior to the onset of viral DNA replication at 3 hpi and applied to a nylon membrane by slot blot hybridization (inf.). Cesium chloride-purified KOS DNA was applied to the membrane as indicated (std.). The blot was probed for viral DNA by using32P-labeled nick-translated KOS DNA. The blot was stripped and reprobed for cellular DNA by using a32 P-labeled antisense RNA probe specific for the cellular gene GAPDH. The image was visualized by PhosphorImager analysis. The range values for the image display were set at 0 to 791 counts for the KOS probe and 0 to 258 counts for the
GAPDH probe.
FIG. 4. Serine/threonine kinase inhibitors block NGF-dependent replication of n212. PC12 cells (106/35-mm-diameter dish) were incubated for 30 min prior
to NGF addition with serine/threonine kinase inhibitors at the following con-centrations: K252a, 0.25mM; KT5720, 0.5mM; PD98059, 20mM; and calphostin C, 0.5mM. At 3 h posttreatment, the cultures were infected with n212 (0.01 PFU/cell) in the presence and absence of inhibitors and NGF. At 24 hpi, the cultures were harvested and viral yields were measured. The data are expressed as percentage of NGF-induced n212 replication, which was set at 100% to show all of the data in a single figure. MAPKinase, mitogen-activated kinase.
on November 9, 2019 by guest
http://jvi.asm.org/
second messenger pathways result in n212-complementing ac-tivity, and to test whether NGF can synergize with these activ-ities, PC12 cells were treated alone or in combination with PMA, forskolin, dbcAMP, and ionomycin in the presence and absence of NGF for 3 h prior to infection with 0.01 PFU of KOS or n212 per cell. At 24 hpi, virus yields were measured (Table 1).
PMA activates PKC and stimulates mitogenesis in many cell types, including PC12 cells (75). PMA had only minor effects on replication of both KOS and n212. PMA did not affect NGF-induced replication of n212 since PC12 cells treated with both PMA and NGF produced a 10.5-fold increase in n212 yield relative to mock-treated cultures. This increase in repli-cation efficiency was consistent with the 12-fold increase in n212 replication observed in NGF-treated cultures in the ab-sence of PMA (Fig. 1). The combination of PMA and dbcAMP
treatment in the presence and absence of NGF also had little effect on virus replication (Table 1).
Forskolin increases intracellular cAMP levels, which in turn activate cAMP-dependent enzymes (43). In a similar manner, dbcAMP, a membrane-permeable analog of cAMP, directly activates cAMP-dependent enzymes (43). Treatment of PC12 cells with either forskolin or dbcAMP stimulated n212 repli-cation (1.6- or 2.6-fold, respectively) and KOS replirepli-cation (1.4-or 0.4-fold, respectively) only slightly (Table 1). Treatment of PC12 cells with forskolin and NGF increased replication of n212 4.8-fold and that of KOS 3.3-fold. While we do not have a satisfactory explanation for the increased replication effi-ciency of KOS in the presence of forskolin and NGF, it is a real and reproducible effect. The results of these tests indicate that activation of cAMP-dependent kinases did not induce the n212-complementing activity.
Ionomycin, a calcium-specific ionophore, increases intracel-lular calcium levels, thereby activating calcium-dependent en-zymes (44). Treatment of PC12 cells with ionomycin alone or in combination with dbcAMP in the presence and absence of NGF had little effect on replication of both KOS or n212 (Table 1). Dimethyl sulfoxide, which was used as the vehicle for ionomycin delivery, had little effect on replication of KOS and n212 (data not shown). Taken together, the results of these tests indicate that activators of cellular protein kinases and second messenger analogs cannot substitute for NGF- or FGF-dependent stimulation of n212 replication in PC12 cells.
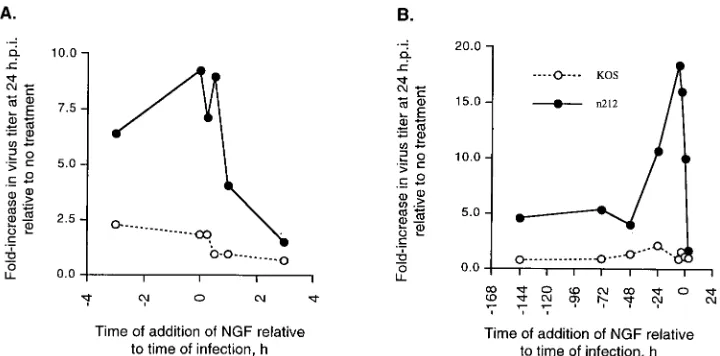
The NGF-induced ICP0-complementing activity is expressed transiently. To determine the optimal time of addition and length of NGF treatment relative to the time of infection required to produce maximal expression of the NGF-induced n212-complementing activity, PC12 cells were treated with NGF 3 h prior to infection, at the time of infection, or 15 min, 30 min, 1 h, and 3 h after infection with 0.01 PFU of KOS or n212 per cell. Twenty-four hours after infection, virus yields were measured by plaque assay. Addition of NGF to cultures 3 h prior to infection and at the time of infection produced six-and ninefold increases, respectively, in the 24-h yield of n212 (Fig. 5A). However, addition of NGF 15 min, 1 h, or 3 h after infection produced a progressive decrease in the 24-h yield of n212 (Fig. 5A). These treatments had only minor effects
(ap-FIG. 5. Time course of the NGF-induced n212-complementing activity. (A) PC12 cells (106/35-mm-diameter dish) were treated with NGF (100 ng/ml) 3 h prior to
infection, at the time of infection, or 15 min, 30 min, 1 h, and 3 h after infection with 0.01 PFU of KOS and n212 per cell. Virus yields were measured at 24 hpi and expressed as fold increase in viral replication relative to mock-treated samples. (B) PC12 cells (106/35-mm-diameter dish) were treated with NGF (100 ng/ml) for 144,
[image:5.612.50.290.89.252.2]72, 48, 24, 12, 6, and 3 h prior to infection, at the time of infection, or 3 h after infection with 0.01 PFU of n212 or KOS per cell. Virus yields were measured at 24 hpi. NGF-induced n212-complementing activity is expressed as fold increase in viral titer relative to mock-treated samples.
TABLE 1. Effects of PMA and second messenger analogs on replication of n212 and KOSa
Treatment Fold (SD)
n212 KOS
Mock 1 1
NGF 10.0 (3.1) 1.7 (1.2)
PMA 1.9 (1.2) 1.1 (0.8)
PMA1NGF 10.5 (2.1) 1.6 (1.4)
PMA1dbcAMP 1.8 (0.1) 0.6 (0.4)
PMA1dbcAMP1NGF 10.8 (7.8) 1.0 (0.1)
dbcAMP 2.6 (2.0) 0.4 (0.2)
dbcAMP1NGF 9.4 (4.0) 1.1 (0.5)
Forskolin 1.6 (0.4) 1.4 (0.7)
Forskolin1NGF 4.3 (0.6) 3.3 (0.4)
Ionomycin 1.0 (0.8) 0.4 (0.1)
Ionomycin1NGF 9.0 (0.1) 1.6 (0.3)
Ionomycin1dbcAMP 0.7 (0.6) 1.0 (0.9)
Ionomycin1dbcAMP1NGF 10.4 (3.9) 2.1 (0.2)
aPC12 cells were treated with compounds as indicated for 3 h prior to
infec-tion with 0.01 PFU of n212 or KOS per cell. At 24 hpi, the cultures were harvested and viral yields were measured. The data are expressed as fold increase in virus titer at 24 hpi relative to no treatment. Each value represents the average of at least three independent experiments.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.120.480.505.683.2]proximately twofold) on the 24-h yield of KOS (Fig. 5A). The results of these tests suggest either that (i) the NGF-induced activity which is able to substitute for ICP0 is required within the first 3 h prior to infection and the 30 min immediately after infection or (ii) by 3 hpi, infected PC12 cells no longer respond to stimulation by NGF.
To measure the duration of the NGF-induced complement-ing activity, PC12 cells were treated with NGF for 144, 72, 48, 24, 12, 6, and 3 h prior to infection, at the time of infection, or 3 h after infection with 0.01 PFU of KOS or n212 per cell. Virus yields were measured 24 h later. Pretreatment of PC12 cells with NGF from 0 to 12 h prior to infection produced a 10-to nearly 20-fold increase in the yield of n212, while only minor (,2-fold) effects on the yield of KOS were observed (Fig. 5B). The NGF-induced stimulation of n212 replication was less apparent in PC12 cells treated with NGF for 48 h or longer prior to infection. Thus, treatment of PC12 cells from 48 to 144 h prior to infection had only modest (;5-fold) effects on the replication of n212 and very little effect on the replication of KOS (Fig. 5B). Consistent with the findings presented in Fig. 5B, by 3 hpi NGF had little effect on replication of either n212 or KOS (Fig. 5B). The results of these tests indicate that the NGF-induced stimulation of n212 replication is transient and is maximal if initiated 12 h prior to infection.
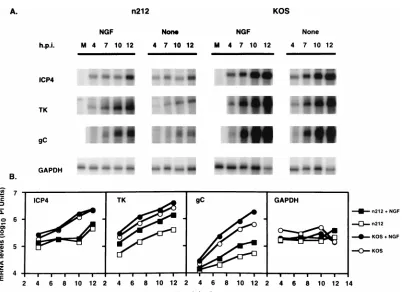
NGF treatment of PC12 cells increases the steady-state lev-els of viral mRNA.ICP0 increases the steady-state levels of viral E and L mRNAs during low-multiplicity infection by increasing the rate of initiation of mRNA synthesis (37). To test whether the NGF-induced n212-complementing activity functions at the same level as ICP0 to increase the steady-state level of viral mRNA, PC12 cells were treated with NGF or
mock treated for 12 h prior to infection with 0.1 PFU of KOS or n212 per cell. At 0, 4, 7, and 10 hpi, cytoplasmic RNA was isolated and levels of ICP4, thymidine kinase (TK), and gC mRNAs were measured by quantitative RNase protection as-say. The results of these tests showed that at all times tested after infection, the levels of TK and gC but not ICP4 mRNAs in n212-infected cells were markedly higher in the presence than in the absence of NGF, whereas the level of cellular GAPDH mRNA remained relatively constant in the presence and absence of NGF (Fig. 6). A similar result was observed for these viral mRNAs in KOS-infected cells; however, the NGF-induced enhancement of viral mRNA accumulation was great-er in n212-infected cells. This result indicates that the NGF-induced n212-complementing activity functions to increase the accumulation of E and L but not IE viral mRNAs. Notably, although levels of viral mRNAs in KOS-infected cells were also enhanced, this NGF-induced enhancement was less evident at the level of viral replication (Fig. 2A and 5). The results of these tests indicate that the NGF-induced activity that com-plements n212 functions at the level of mRNA accumulation and, like ICP0, serves to increase E and L gene expression.
[image:6.612.99.503.63.355.2]ICP0 does not affect the induction of mRNAs of cellular pri-mary response genes. Activation of growth factor-dependent signal transduction pathways leads to the transient expression of cellular primary response genes such as c-fos, c-jun, junD, and krox24 (30). These genes encode transcription factors which regulate secondary response genes. In PC12 cells, the second-ary response genes induced by NGF treatment are responsible for the morphological and biochemical changes associated with differentiation (66). In addition, herpesvirus infection also in-duces selected cellular primary response genes (1, 4). The
FIG. 6. NGF induces viral mRNA accumulation in infected PC12 cells. PC12 cells (53106/60-mm-diameter dish) were mock treated or treated with NGF (100 ng/ml) for 12 h prior to infection with 0.1 PFU of KOS or n212 per cell. At 0, 4, 7, and 10 hpi, cytoplasmic RNA was isolated and levels of ICP4, TK, and gC mRNAs were measured by quantitative RNase protection assay. The range values for the image display were set at 0 to 2,000 (ICP4), 0 to 1,000 (TK), and 0 to 200 (gC).
on November 9, 2019 by guest
http://jvi.asm.org/
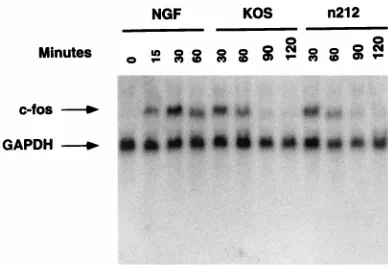
biological consequences of this induction are not well under-stood. To determine whether ICP0, like the NGF-induced n212-complementing activity, is involved in the herpesvirus infection-specific induction of cellular primary response genes, PC12 cells were infected with KOS or n212 for 60, 90, or 120 min or treated with NGF for 0, 30, 60, 90 or 120 min. At the times indicated (Fig. 7) cytoplasmic RNA was isolated. The RNA was separated according to molecular weight by dena-turing gel electrophoresis and transferred to a nylon mem-brane by Northern blotting. The memmem-brane was probed with radiolabeled RNA probes specific for the human c-fos message and the rat GAPDH mRNA. As shown in Fig. 7, the kinetics and extent of activation of c-fos mRNA were similar in cells treated with NGF or infected with KOS or n212, suggesting that ICP0 is not involved in the herpesvirus-induced activation of cellular primary response genes. Similar results were ob-served when northern blots were probed with radiolabeled c-jun, junD, and krox24 probes (data not shown). Moreover, as a control, addition of media in the absence of NGF did not induce cellular primary response gene expression. The results of these tests indicate that ICP0 is not involved in the herpes-virus infection-specific induction of cellular primary response gene expression.
DISCUSSION
The balance between productive and latent infection is ul-timately determined by the transcriptional permissivity of the infected cell. Growth factors and extracellular signals influence the transcriptional permissivity of the cell by activating intra-cellular signaling cascades. Viral IE proteins circumvent the need for extracellular signaling by increasing the transcrip-tional permissivity of the infected cell, thereby promoting viral gene expression. During reactivation, in the absence of viral regulatory proteins, activation of cellular signaling cascades likely changes the transcriptional permissivity of the infected cell and stimulates viral gene expression. In support of this hypothesis, Tal-Singer et al. have reported that viral E and L genes may be induced prior to IE gene expression during reactivation in mice, suggesting that cellular functions can sub-stitute for viral IE genes during reactivation (70). In addition,
the levels of cyclic nucleotides, which function as second mes-sengers during signal transduction, influence the maintenance and reactivation of latent virus in mice (23, 64). Moreover, activators of PKA and PKC as well as second messenger ana-logs stimulate reactivation of latent HSV-1 in primary rat neu-rons latently infected in vitro (68). While activators of PKA and PKC induce reactivation in this system, they fail to induce the ICP0-like activity, suggesting that activation of these en-zymes alone is not sufficient to complement replication of n212. Taken together, these observations suggest that intracel-lular signaling plays a major role in regulating viral gene ex-pression during reactivation.
NGF and FGF are two of the many neurotropic factors whose concentrations change during the host stress response and may contribute to the signals that induce HSV reactivation (45). NGF induces expression of the LAT promoter in a ras-dependent manner in PC12 cells, suggesting that during la-tency, LAT expression may be regulated by NGF or NGF-like extracellular signals (26). Changes in the levels of stress-in-duced growth factors like NGF may well lead to activation of cellular signaling pathways which in turn induce activities that stimulate viral gene expression.
We have identified and characterized an NGF/FGF-induc-ible cellular activity that stimulates replication of the ICP0 null mutant, n212. The NGF-induced cellular activity is partially ras dependent and requires activation of multiple NGF-dependent signaling pathways. This activity is transiently expressed, with peak complementing activity observed within the first 12 h of NGF treatment. Like ICP0, the NGF-dependent n212-comple-menting activity functions to stimulate viral E and L mRNA accumulation.
The NGF-induced complementing activity is cell type spe-cific. The NGF/FGF-induced n212-complementing activity is specific to PC12 cells. NGF treatment of a human neuroblas-toma cell line, SY5Y, had little effect on viral growth (data not shown). Notably, even clonal isolates of SY5Y constitutively expressing the trkA gene, which encodes the NGF receptor, failed to induce the n212-complementing activity in response to NGF. Furthermore, treatment of Vero cells with NGF, FGF, or EGF produced little difference in virus yields at 3 and 24 hpi relative to mock treatment. Although Vero cells do not express NGF receptors, they do express FGF and EGF recep-tors (12, 19). Notably, FGF and EGF treatment of Vero cells stimulates mitogenesis, indicating that these receptors are bi-ologically active (12). These data imply that NGF/FGF signal-ing in conjunction with some other factor(s) in PC12 cells is required for complementation of n212 and that NGF/FGF signaling alone is not sufficient for induction of the n212-complementing activity.
[image:7.612.71.265.68.202.2]Cellular signaling, phosphorylation, and transcription fac-tor activation.How do NGF and FGF complement n212? A well-recognized endpoint of NGF/FGF-induced signal trans-duction is the phosphorylation of nuclear transcription factors (58, 66, 82). Activation of transcription factors by phosphory-lation is a common mechanism by which growth factors initiate changes in cellular transcription (35). NGF and FGF activate multiple serine/threonine kinase cascades including the ras/ raf-1/MEK/ERK pathway and cyclin-dependent kinase activi-ties (16, 69). Once activated, these kinases phosphorylate tran-scription factors such as cAMP response element binding protein and serum response factor, thereby stimulating their transcriptional activating activities (43, 58, 66). NGF also in-duces phosphorylation of SP1 and c-Fos and stimulates NF-kB DNA binding activity (72, 81, 83). The NGF/FGF-induced n212-complementing activity may require several of these ac-tivated kinase signaling pathways to phosphorylate and activate
FIG. 7. ICP0 does not induce c-fos expression. PC12 cells (53106 /60-mm-diameter dish) were treated with NGF (100 ng/ml) for 0, 15, 30, and 60 min or infected with 5.0 PFU of KOS and n212 per cell for 30, 60, 90, and 120 min. Cytoplasmic RNA was isolated at each time point as described in the text. The RNA was size fractionated by denaturing gel electrophoresis and transferred to a nylon membrane by Northern blotting. The Northern blot was probed with 100 ng of32P-labeled antisense RNA probe (83105cpM/ng) specific for the mouse c-fos message. The blot was reprobed with 150 ng of32P-labeled antisense RNA probe (8.83105cpM/ng) specific for the rat GAPDH message. The image was visualized by PhosphorImager analysis. The range values for the image display were set at 0 to 500 counts.
on November 9, 2019 by guest
http://jvi.asm.org/
transcription factors which are then used by the virus in the absence of ICP0 to stimulate viral gene expression. The obser-vation that serine/threonine kinase inhibitors partially block the NGF-dependent stimulation of n212 replication (Fig. 4) is consistent with this hypothesis.
Like the cellular ICP0-like activity, ICP0 may indirectly reg-ulate the phosphorylation state of viral and cellular proteins during the course of infection. ICP0 physically interacts with HUASP, a component of the ubiquitin-dependent proteolysis system (22). Ubiquitin-dependent proteolysis of phosphory-lated transcription factors is one mechanism by which cells down regulate cellular transcription induced by extracellular signaling (11, 39, 53, 77). The interaction of ICP0 with HUASP may serve to modify cellular enzymes that directly phosphor-ylate transcription factors. Indeed, ICP0 is required to desta-bilize the catalytic subunit of a host DNA-PK (41). In the absence of DNA-PK, the intranuclear phosphorylation state of numerous proteins may be altered, potentially affecting the ability of these proteins to interact with DNA. Thus, ICP0 and the NGF-induced n212-complementing activity may function to stimulate transcription indirectly by altering the levels of phosphorylation of transcription factors which activate viral transcription. Consistent with this hypothesis, two-dimensional gel electrophoresis of infected-cell nuclear proteins from KOS-and n212-infected PC12 cells shows significant differences in the pattern of phosphorylation of multiple nuclear phospho-proteins (37a).
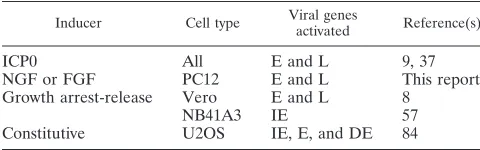
A comparison of ICP0-like cellular activities.A comparison of the known ICP0-like cellular activities is shown in Table 2. Like ICP0, both the NGF-induced activity in PC12 cells and the cell cycle-regulated activity in Vero cells stimulate viral E and L but not IE gene expression. In contrast, the activity expressed constitutively in U2OS cells and the cell cycle-regu-lated activity in NB41A3 cells preferentially stimulate basal IE gene expression in transient expression assays. These differ-ences may be due to the differential effects of the cellular activity on DNA delivered by transfection versus infection. In addition, it is unknown whether increased basal IE gene ex-pression is sufficient to complement ICP0 null mutant replica-tion. The U20S cell activity also stimulates IE gene expression during infection; however, it is unknown whether this effect occurs throughout infection or only at later times postinfec-tion.
It is conceivable that all of the activities that complement ICP0 null mutants require activation of cellular kinases and phosphorylation of transcription factors. Quiescent cells enter-ing the cell cycle induce cell cycle-regulated kinases that phos-phorylate cellular transcription factors (2, 15, 59, 62, 74). Like-wise, NGF induces many of these same activities in PC12 cells (15, 81, 83). Although it is unknown whether U2OS cells ex-press elevated levels of kinase activity, transcription factors that regulate viral IE gene expression are activated (84). Thus, in at least two instances (Vero cells entering the cell cycle from G0and NGF treatment of PC12 cells), induction of the
ICP0-like cellular activity correlates with activation of cellular ki-nases.
Cellular kinase activation may be only one part of the ICP0-like activity. Peak expression of the NGF-induced n212-com-plementing activity in PC12 cells and the cell cycle-regulated ICP0-like activity in Vero cells occur after the initial activation of cellular primary response genes (8) (Fig. 5B). Moreover, ICP0 does not affect the kinetics or level of expression of c-fos mRNA following infection of PC12 cells (Fig. 7). These obser-vations suggest that the cellular functions that complement ICP0 null mutants require activities expressed downstream of initial signaling events.
ICP0 activates viral gene expression during productive in-fection and promotes efficient reactivation from latency both in vitro and in vivo. Thus, ICP0 regulates viral gene expression during all phases of the HSV-1 life cycle. Cellular activities that functionally substitute for ICP0 and complement replication of ICP0 null viruses have been described (8, 84). The existence of these activities may provide insight into the mechanisms that regulate viral gene expression during productive infection and reactivation from latency. We have shown that cellular activi-ties induced by NGF and FGF in a neurally derived cell line can substitute for ICP0 and stimulate viral gene expression. We suggest that these activities may be similar to the activities that regulate viral gene expression during reactivation from latency.
ACKNOWLEDGMENTS
We thank Anh Nguyen-Huynh, Lily Yeh, David Fraser, and Luis Schang for helpful discussions of this work.
This research was supported by Public Health Service grants R37CA20260 and POINS35138-10 (P.A.S.) and F32 AI09127 (R.J.).
REFERENCES
1. Albrecht, T., I. Boldogh, and M. Fons. 1996. Receptor-initiated activation of cells and their oncogenes by herpes-family viruses. J. Invest. Dermatol. 98: 29s–35s.
2. Baldwin, A. S., J. C. Azizkhan, D. E. Jensen, A. A. Beg, and L. R. Coodly. 1991. Induction of NF-kB DNA-binding activity during the G0-to-G1 tran-sition in mouse fibroblasts. Mol. Cell. Biol. 11:4943–4951.
3. Bartel, D. P., M. Sheng, L. F. Lau, and M. E. Greenberg. 1989. Growth factors and membrane depolarization activate distinct programs of early response gene expression: dissociation of fos and jun induction. Genes Dev. 3:304–313.
4. Boldogh, I., S. AbubBakar, and T. Albrecht. 1990. Activation of proto-oncogenes: an immediate early event in human cytomegalovirus infection. Science 247:561–564.
5. Bruns, R. F., F. D. Miller, R. L. Merriman, J. J. Howbert, W. F. Heath, E. Kobayashi, I. Takahashi, T. Tamaokin, and H. Nakano.1991. Inhibition of protein kinase C by calphostin C is light-dependent. Biochem. Biophys. Res. Commun. 176:288–293.
6. Cai, W., T. L. Astor, L. M. Liptak, C. Cho, D. M. Coen, and P. A. Schaffer. 1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67: 7501–7512.
7. Cai, W., and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4579–4589.
8. Cai, W., and P. A. Schaffer. 1991. A cellular function can enhance gene expression and plating efficiency of a mutant defective in the gene for ICP0, a transactivating protein of herpes simplex virus type 1. J. Virol. 65:4078– 4090.
9. Cai, W., and P. A. Schaffer. 1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 66:2904–2915.
10. Chen, J., and S. Silverstein. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 66: 2916–2927.
11. Chen, Z., J. Hagler, V. J. Palombella, F. Melandri, D. Scherer, D. Ballard, and T. Maniatis.1995. Signal-induced site-specific phosphorylation targets IkBato the ubiquitin-proteasome pathway. Genes Dev. 9:1586–1597. 12. Clark, J. M., C. Gebb, and M. D. Hirtenstein. 1981. Serum supplements and
[image:8.612.49.289.90.165.2]serum-free media: applicability for microcarrier culture of animal cells. Dev. Biol. Stand. 50:81–91.
TABLE 2. Comparison of ICP0-like cellular activities expressed in different cell types
Inducer Cell type Viral genesactivated Reference(s)
ICP0 All E and L 9, 37
NGF or FGF PC12 E and L This report
Growth arrest-release Vero E and L 8
NB41A3 IE 57
Constitutive U2OS IE, E, and DE 84
on November 9, 2019 by guest
http://jvi.asm.org/
13. Clements, J. B., and N. D. Stow. 1989. A herpes simplex virus type 1 mutant containing a deletion within immediate early gene 1 is latency-competent in mice. J. Gen. Virol. 70:2501–2506.
14. Corey, L., and P. G. Spear. 1986. Infections with herpes simplex viruses. N. Engl. J. Med. 314:686–691.
15. Cowley, S., H. Paterson, P. Kemp, and C. J. Marshall. 1994. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell 77:841–852.
16. D’Arcangelo, G., and S. Halegoua. 1993. A branched signaling pathway for nerve growth factor is revealed by Src-, Ras-, and Raf-mediated gene induc-tions. Mol. Cell. Biol. 13:3146–3155.
17. Deatly, A. M., J. G. Spivack, E. Lavi, D. R. O’Boyle, and N. W. Fraser. 1988. Latent herpes simplex virus type 1 transcripts in peripheral and central nervous system tissues of mice map to similar regions of the viral genome. J. Virol. 62:749–756.
18. Deshmane, S. L., and N. W. Fraser. 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 63.2:943–947.
19. Distefano, P. S., J. B. Schweitzer, M. Taniuchi, and E. M. Johnson. 1985. Selective destruction of nerve growth factor receptor-bearing cells in vitro using a hybrid toxin composed of ricin A chain and a monoclonal antibody against the nerve growth factor receptor. J. Cell Biol. 101:1107–1114. 20. Everett, R. D. 1984. Trans activation of transcription by herpes virus
prod-ucts: requirement for two HSV-1 immediate-early polypeptides for maxi-mum activity. EMBO J. 3:3135–3141.
21. Everett, R. D. 1986. The products of herpes simplex virus type-1 (HSV-1) immediate early genes 1, 2 and 3 can activate HSV-1 gene expression in trans. J. Gen. Virol. 67:2507–2513.
22. Everett, R. D., M. Meredith, A. Orr, A. Cross, M. Kathoria, and J. Parkin-son.1997. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 16:566–577.
23. Foster, C. S., E. M. Opremcak, and N. Tolchin. 1989. Evidence for the potential influence of cyclic nucleotides on maintenance of or reactivation from latency of herpes simplex virus in trigeminal ganglionic neurons. Acta Ophthalmol. 192:142–144.
24. Fraser, N. W., T. M. Block, and J. G. Spivack. 1992. The latency-associated transcripts of herpes simplex virus: RNA in search of function. Virology 191: 1–8.
25. Fraser, N. W., J. G. Spivack, Z. Wroblewska, T. Block, S. L. Deshmane, T. Valyi-nagy, and R. Natarajan.1990. A review of the molecular mechanism of HSV-1 latency. Curr. Eye Res. 10:1–13.
26. Frazier, D. P., D. Cox, E. M. Godshalk, and P. A. Schaffer. 1996. The herpes simplex virus type 1 latency-associated transcript promoter is activated through Ras and Raf by nerve growth factor and sodium butyrate in PC12 cells. J. Virol. 70:7424–7432.
27. Greene, L. A. 1978. Nerve growth factor prevents the death and stimulates the neuronal differentiation of clonal pc12 pheochromocytoma cells in se-rum-free medium. J. Cell Biol. 78:747–754.
28. Greene, L. A., and A. S. Tischler. 1976. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc. Natl. Acad. Sci. USA 73:2424–2428.
29. Harris, R. A., R. D. Everett, X. Zhu, S. Silverstein, and C. M. Preston. 1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro system. J. Virol. 63:3513– 3515.
30. Herschman, H. R. 1991. Primary response genes induced by growth factors and tumor promoters. Annu. Rev. Biochem. 60:281–319.
31. Hilborn, M. D., S. G. Rane, and J. D. Pollock. 1997. EGF in combination with depolarization or cAMP produces morphological but not physiological differentiation in PC12 cells. J. Neurosci. Res. 47:16–26.
32. Hill, T. J. 1985. Herpes simplex virus latency, p. 175–240. In B. Roizman (ed.), The herpesviruses. Plenum Publishing Corp., New York, N.Y. 33. Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus
macromo-lecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19.
34. Honess, R. W., and B. Roizman. 1975. Regulation of herpesvirus macromo-lecular synthesis: sequential transition of polypeptide synthesis requires functional viral polypeptides. Proc. Natl. Acad. Sci. USA 72:1276–1280. 35. Hunter, T., and M. Karin. 1992. The regulation of transcription by
phos-phorylation. Cell 70:375–387.
36. Jacobson, J. G., D. A. Leib, D. J. Goldstein, C. L. Bogard, P. A. Schaffer, S. K. Weller, and D. M. Coen.1989. A herpes simplex virus ribonucleotide reductase deletion mutant is defective for productive acute and reactivatable latent infections of mice and for replication in mouse cells. Virology 173: 278–283.
37. Jordan, R., and P. A. Schaffer. 1997. Activation of gene expression by herpes simplex virus type 1 ICP0 occurs at the level of mRNA synthesis. J. Virol. 71: 6859–6862.
37a.Jordan, R., and P. A. Schaffer. Unpublished data.
38. Kase, H., K. Iwahashi, S. Nakanishi, Y. Matsuda, K. Yamada, M. Taka-hashi, C. Murakata, A. Sato, and M. Kaneko.1987. K-252 compounds, novel
and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem. Biophys. Res. Commun. 142:436–440. 39. Kim, T. K., and T. Maniatis. 1996. Regulation of interferon-g-activated
STAT1 by the ubiquitin-proteasome pathway. Science 273:1717–1719. 40. Kobayashi, E., H. Nakano, M. Morimoto, and T. Tamaoki. 1989. Calphostin
C (ucn-1028c), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochem. Biophys. Res. Commun. 159:548– 553.
41. Lees-Miller, S. P., M. C. Long, M. A. Kilvert, V. Lam, S. A. Rice, and C. A. Spencer.1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70:7471–7477.
42. Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe, K. L. Tyler, and P. A. Schaffer.1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol. 63:759–768.
43. Leib, D. A., C. K. Nadeau, S. A. Rundle, and P. A. Schaffer. 1991. The promoter of the latency-associated transcripts of herpes simplex virus type-1 contains a functional cAMP-response element: role of the latency-associated transcripts and cAMP in reactivation of viral latency. Proc. Natl. Acad. Sci. USA 88:48–52.
44. Liu, C., and T. E. Hermann. 1978. Characterization of ionomycin as a calcium ionophore. J. Biol. Chem. 253:5892–5894.
45. Live-Montalcini, R., S. D. Skaper, R. D. Toso, L. Petrelli, and L. Leon. 1997. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 19: 514–520.
46. Mark, M. D., Y. Liu, S. T. Wong, T. R. Hinds, and D. R. Storm. 1995. Stimulation of neurite outgrowth in PC12 cells by EGF and KCl. J. Cell Biol. 130:701–710.
47. Marshall, C. J. 1995. Specificity of receptor tyrosine kinase signaling: tran-sient versus sustained extracellular signal-regulated kinase activation. Cell 80:179–185.
48. Maul, G. G., and R. D. Everett. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 75:1223–1233.
49. Maul, G. G., H. H. Guldner, and J. G. Spivack. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 74:2679–2690.
50. Meignier, B., R. Longnecker, P. Mavromara-Nazos, A. E. Sears, and B. Roizman.1988. Virulence of and establishment of latency by genetically engineered deletion mutants of herpes simplex virus 1. Virology 162:251– 254.
51. Meredith, M., A. Orr, M. Elliott, and R. D. Everett. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174–187.
52. Meredith, M., A. Orr, and R. D. Everett. 1994. Herpes simplex virus type-1 immediate-early protein vmw110 binds strongly and specifically to a 135-kDa cellular protein. Virology 200:457–469.
53. Musti, A. M., M. Treier, and D. Bohmann. 1997. Reduced ubiquitin-depen-dent degradation of c-jun after phosphorylation by MAP kinase. Science 275: 400–402.
54. O’Hare, P., and G. S. Hayward. 1985. Three trans-acting regulatory proteins of herpes simplex virus modulate immediate-early gene expression in a pathway involving positive and negative feedback regulation. J. Virol. 56: 723–733.
55. O’Hare, P., and G. S. Hayward. 1985. Evidence for a direct role for both 175,000- and 110,000-molecular-weight immediate-early proteins of herpes simplex virus in the transactivation of delayed-early promoters. J. Virol. 53: 751–760.
56. Pang, L., T. Sawada, S. J. Decker, and A. R. Saltiel. 1995. Inhibition of MAP kinase kinase blocks the differentiation of PC-12 cells induced by nerve growth factor. J. Biol. Chem. 270:13585–13588.
57. Ralph, W. M., JR., M. S. Cabatingan, and P. A. Schaffer. 1994. Induction of herpes simplex virus type 1 immediate-early gene expression by a cellular activity expressed in Vero and NB41a3 cells after growth arrest-release. J. Virol. 68:6871–6882.
58. Riccio, A., B. A. Pierchala, C. L. Ciarallo, and D. D. Ginty. 1997. An NGF-trkA-mediated retrograde signal to transcription factor CREB in sympa-thetic neurons. Science 277:1097–1100.
59. Roberts, S. B., N. Segil, and N. Heintz. 1991. Differential phosphorylation of the transcription factor oct-1 during the cell cycle. Science 253:1022–1026. 60. Roizman, B., and A. E. Sears. 1990. Herpes simplex viruses and their
repli-cation, p. 1795–1841. In B. N. Fields and D. M. Knipe (ed.), Fields virology. Raven Press, Ltd., New York, N.Y.
61. Russel, J., N. D. Stow, E. C. Stow, and C. M. Preston. 1987. Herpes simplex virus genes involved in latency in vitro. J. Gen. Virol. 68:3009–3018. 62. Ryseck, R. P., S. I. Hirai, and R. Bravo. 1988. Transcriptional activation of
c-jun during the G0/G1transition in mouse fibroblasts. Nature 334:535–537. 63. Sacks, W. R., C. C. Greene, D. P. Aschman, and P. A. Schaffer. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 55: 796–805.
on November 9, 2019 by guest
http://jvi.asm.org/
64. Sainz de la Maza, M., P. A. Wells, and C. S. Foster. 1989. Cyclic nucleotide modulation of herpes simplex virus latency and reactivation. Invest. Oph-thalmol. Visual Sci. 30:2154–2159.
65. Samaniego, L. A., N. Wu, and N. A. DeLuca. 1997. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 71: 4614–4625.
66. Sheng, M., and M. E. Greenberg. 1990. The regulation and function of c-fos and other immediate early genes in the nervous system. Neuron 4:477–485. 67. Smith, K. O. 1964. Relationship between the envelope and the infectivity of
herpes simplex virus. Proc. Soc. Exp. Biol. Med. 115:814–816.
68. Smith, R. L., L. I. Pizer, E. M. Johnson, and C. L. Wilcox. 1992. Activation of second-messenger pathways reactivates latent herpes simplex virus in neuronal cultures. Virology 188:311–318.
69. Szebereni, J., H. Cai, and G. M. Cooper. 1990. Effect of a dominant inhib-itory Ha-ras mutation on neuronal differentiation of PC12 cells. Mol. Cell. Biol. 10:5324–5332.
70. Tal-Singer, R., T. M. Lasner, W. Podrzucki, A. Skokotas, J. J. Leary, S. L. Berger, and N. W. Fraser.1997. Gene expression during reactivation of herpes simplex virus type 1 from latency in the peripheral nervous system is different from that during lytic infection of tissue cultures. J. Virol. 71: 5268–5276.
71. Tamaoki, T. 1991. Use and specificity of staurosporine, UCN-01, and cal-phostin C as protein kinase inhibitors. Methods Enzymol. 201:340–347. 72. Taylor, L. K., K. D. Swanson, and K. D. Mobley. 1994. Isolation and
char-acterization of a nerve growth factor-regulated fos kinase from PC12 cells. J. Biol. Chem. 269:308–318.
73. Tenser, R. B., and M. E. Dunstan. 1979. Herpes simplex virus thymidine kinase expression in infection of the trigeminal ganglion. Virology 99:417– 422.
74. Thomas, N. S. B., L. C. Burke, A. Bybee, and D. C. Linch. 1991. The phosphorylation state of the retinoblastoma (RB) protein in G0/G1is de-pendent on growth status. Oncogene 6:317–322.
75. Tischler, A. S., J. C. Riseberg, M. A. Hardenbrook, and V. Cherington. 1993.
Nerve growth factor is a potent inducer of proliferation and neuronal dif-ferentiation for adult rat chromaffin cell in vitro. J. Neurosci. 13:1533–1542. 76. Tischler, A. S., L. A. Ruzicka, and R. L. Perlman. 1990. Mimicry and inhi-bition of nerve growth factor effects: interactions of staurosporine, forskolin, and K252a in PC12 cells and normal rat chromaffin cells in vitro. J. Neuro-chem. 55:1159–1165.
77. Tsurumi, C., N. Ishida, T. Tomohiro, A. Kakizuka, E. Nishida, E. Okumura, T. Kishimoto, M. Inagaki, K. Okazaki, N. Sagat, A. Ichihara, and K. Tanaka.1995. Degradation of c-Fos by the 26S proteasome is accelerated by c-Jun and multiple protein kinases. Mol. Cell. Biol. 15:5682–5687. 78. Wagner, E. K. 1991. Herpesvirus transcription and its regulation. CRC Press,
Boston, Mass.
79. Wilcox, C. L., and E. M. Johnson. 1987. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol. 61: 2311–2315.
80. Wilcox, C. L., R. L. Smith, R. D. Everett, and D. Mysofski. 1997. The herpes simplex virus type 1 immediate-early protein ICP0 is necessary for the effi-cient establishment of latent infection. J. Virol. 71:6777–6785.
81. Wood, J. N. 1995. Regulation of NF-kB activity in rat dorsal root ganglia and PC12 cells by tumor necrosis factor and nerve growth factor. Neurosci. Lett. 192:41–44.
82. Xing, J., D. D. Ginty, and M. E. Greenberg. 1996. Coupling of the ras-MAPK pathway to gene activation by RSK2, a growth factor-regulated CREB ki-nase. Science 273:959–963.
83. Yan, G. Z., and E. B. Ziff. 1997. Nerve growth factor induces transcription of the p21 WAF1/CIP1/ and cyclin D1 genes in PC12 cell by activating the SP1 transcription factor. J. Neurosci. 17:6122–61332.
84. Yao, F., and P. A. Schaffer. 1995. An activity specified by the osteosarcoma line U2OS can substitute functionally for ICP0, a major regulatory protein of herpes simplex virus type 1. J. Virol. 69:6249–6258.
85. Zhu, X., J. Chen, C. S. H. Young, and S. Silverstein. 1990. Reactivation of latent herpes simplex virus by adenovirus recombinants encoding mutant IE-0 gene products. J. Virol. 64:4489–4498.