Copyright © 1997, American Society for Microbiology
Distinct Functions and Requirements for the Cys-His Boxes of
the Human Immunodeficiency Virus Type 1 Nucleocapsid
Protein during RNA Encapsidation and Replication
MICHAEL D. SCHWARTZ, DICCON FIORE,
ANDANTONITO T. PANGANIBAN*
McArdle Laboratory for Cancer Research, University of Wisconsin
Medical School, Madison, Wisconsin 53706
Received 13 June 1997/Accepted 22 August 1997
The process of retroviral RNA encapsidation involves interaction between trans-acting viral proteins and
cis-acting RNA elements. The encapsidation signal on human immunodeficiency virus type 1 (HIV-1) RNA is
a multipartite structure composed of functional stem-loop structures. The nucleocapsid (NC) domain of the
Gag polyprotein precursor contains two copies of a Cys-His box motif that have been demonstrated to be
important in RNA encapsidation. To further characterize the role of the Cys-His boxes of the HIV-1 NC protein
in RNA encapsidation, the relative efficiency of RNA encapsidation for virus particles that contained mutations
within the Cys-His boxes was measured. Mutations that disrupted the first Cys-His box of the NC protein
resulted in virus particles that encapsidated genomic RNA less efficiently and subgenomic RNA more efficiently
than did wild-type virus. Mutations within the second Cys-His box did not significantly affect RNA
encapsi-dation. In addition, a full complement of wild-type NC protein in virus particles is not required for efficient
RNA encapsidation or virus replication. Finally, both Cys-His boxes of the NC protein play additional roles in
virus replication.
The core of human immunodeficiency virus type 1 (HIV-1)
particles is composed mainly of proteins expressed from the
gag and pol genes. The Gag precursor, Pr55
gag, and the
Gag-Pol precursor, Pr160
gag-pol, are initially synthesized as
polypro-teins (77), and assembly and release of HIV-1 particles occur
at the plasma membrane where Pr55
gagand Pr160
gag-polare
associated with the plasma membrane by way of N-terminal
hydrophobic amino acids (74, 80) and an N-terminal myristic
acid residue (11, 28). Pr55
gagis the only viral protein required
for virus particle formation (24, 30). During assembly or
bud-ding of the virus particle, a virally encoded protease
proteo-lytically processes the Gag polyprotein into the matrix (MA,
p17), capsid (CA, p24), p2, nucleocapsid (NC, p7), p1, and
p6
gagproteins (28, 45, 60, 65). Proteolytic processing is not
required for efficient HIV-1 particle assembly or release (28,
45, 65) but is required for the production of mature, infectious
particles (35, 45, 65).
An essential component of the HIV-1 particle is dimeric,
unspliced, genomic-length viral RNA (51). The process by
which this RNA becomes associated with the virus particle is
termed RNA encapsidation or packaging. RNA encapsidation
is highly specific, since greater than 95% of RNA found in
particles is viral RNA, although viral RNA makes up only
about 1% of the total cytoplasmic mRNA found in cells.
En-capsidation also results in the preferential enEn-capsidation of
unspliced, genomic-length viral RNA; more than 95% of viral
RNA found in virus particles is unspliced, genomic-length
RNA, even though subgenomic RNA makes up about half of
the viral RNA found in the cytoplasm of virus-producing
cells.
Encapsidation requires interaction between cis-acting
se-quences on viral RNA and proteins of the assembling virus
particle. cis-acting sequences involved in HIV-1 RNA
encap-sidation are located near the 5
9
ends of the viral genome (3, 13,
32, 42, 50, 54, 56, 64, 72). In vitro biochemical probing,
com-puter modeling, and phylogenetic comparisons suggest that the
secondary structure at the 5
9
end of HIV-1 RNA consists of
seven stem-loop structures (4, 14, 31, 69). The importance of
some of these stem-loop structures in RNA encapsidation in
vivo has been confirmed (46, 56, 57, 78). It is likely that
higher-order RNA structure, rather than primary nucleotide
se-quence, is involved in RNA encapsidation (57, 78). Some of
the cis-acting sequences that have been implicated in HIV-1
RNA encapsidation are contained on both genomic and
sub-genomic RNA (3, 6, 7, 13, 14, 42, 50, 54, 56, 68). Because
retroviruses contain both intronic and exonic encapsidation
signals, it is not yet clear how preferential encapsidation of
genomic RNA over subgenomic RNA occurs.
There is strong evidence that the Gag polyprotein precursor
is involved in RNA encapsidation. Moreover, virus particles
produced in the absence of the pol and env gene products
specifically encapsidate viral RNA (39, 61, 70), suggesting that
the Gag protein is sufficient for RNA encapsidation. One
do-main of Pr55
gagthat is involved in RNA encapsidation is the
nucleocapsid (NC) protein domain, since mutations within NC
have been demonstrated to affect RNA encapsidation in vivo
(3, 19, 25, 27, 62, 66) and RNA binding in vitro (7, 15, 18, 53,
71). During the initial interaction with viral RNA, the NC
protein most likely acts as part of Pr55
gag, since proteolytic
processing is not required for efficient RNA encapsidation
(39). The mature HIV-1 NC protein is 55 amino acid residues
in length and contains many basic amino acid residues.
Muta-tions that affect some of the basic amino acid residues of
HIV-1 NC reduce RNA encapsidation in vivo (19, 66) and
RNA binding in vitro (71). The mature NC protein is closely
associated with genomic RNA in mature virions (16) and has
been demonstrated to have both nonspecific (17, 37, 40, 49, 52,
59) and specific (7, 14, 15, 34, 53) RNA binding activities in
* Corresponding author. Mailing address: McArdle Laboratory for
Cancer Research, University of Wisconsin Medical School, 1400
Uni-versity Ave., Madison, WI 53706. Phone: (608) 263-7820. Fax: (608)
262-2824. E-mail: [email protected].
9295
on November 9, 2019 by guest
http://jvi.asm.org/
vitro. Analysis of chimeric Gag proteins has provided some
evidence that the NC protein provides at least some
speci-ficity for viral RNA during encapsidation in vivo (8, 21, 70,
79).
With the exception of spumaretroviruses, all known
retrovi-ruses contain at least one copy of the sequence Cys-X
2-Cys-X
4-His-X
4-Cys, termed a cysteine-histidine (Cys-His) box or
motif. Retrovirus Cys-His boxes resemble zinc finger domains
found in many nucleic acid binding proteins (5), and the HIV-1
NC protein contains two Cys-His boxes that are capable of
binding Zn
21(9). The Cys-His boxes of HIV-1 have been
shown to play a critical role in RNA encapsidation, since
mu-tations that alter the cysteine or histidine residues of NC
re-duce the efficiency of RNA encapsidation (3, 20, 25–27, 33, 79).
Mutational analysis of NC proteins that contain two Cys-His
boxes indicates that the boxes are not functionally equivalent
(10, 25) and that the first Cys-His box of HIV-1 NC appears to
be more important for HIV-1 RNA encapsidation (25).
Dele-tion of the first Cys-His box of HIV-1 NC and mutaDele-tions that
disrupt both Cys-His boxes of HIV-1 NC have been
demon-strated to increase the encapsidation efficiency of subgenomic
RNA (8, 79).
To further characterize the role of the HIV-1 NC protein in
efficient and specific RNA encapsidation, mutant virus
parti-cles that contained mutations within the Cys-His boxes were
analyzed. Results indicated that mutation of the first two
cys-teine residues of Cys-His box 1 leads to both a decrease in the
efficiency of genomic RNA encapsidation and an increase in
the efficiency of subgenomic RNA encapsidation. Analysis
of heterogeneous virus particles containing both wild-type
(WT) and NC mutant Gag molecules revealed that a full
complement of WT Gag molecules is not required for
effi-cient or specific genomic RNA encapsidation or for virus
replication.
MATERIALS AND METHODS
Maintenance of cell lines, transfections, and infections.Two hundred ninety-three cells were maintained in Dulbecco’s modified Eagle’s medium supple-mented with 10% fetal bovine serum (HyClone), 0.05 mg of gentamicin (Gibco BRL) per ml, and 10 mM HEPES (Gibco BRL) and incubated at 37°C at 5% CO2. Twenty-four hours prior to transfection, 293 cells were seeded at a density of 2.53106cells per 100-mm-diameter cell culture dish. Transfections were carried out by a modified calcium phosphate precipitation method (2). Briefly, 10 mg of plasmid DNA was suspended in 0.5 ml of a solution containing 125 mM CaCl2. An equal volume of 23BES buffer {50 mM BES [N,N-bis(2-hydroxy-ethyl)-2-aminosulfonic acid], 280 mM NaCl, 1.5 mM Na2HPO4z2H2O (pH 6.96)} was added, and the DNA was incubated at room temperature for 45 min. This precipitate solution was added to a 100-mm-diameter plate, and the cells were incubated at 37°C and 5% CO2for 16 to 20 h. The medium was then removed, fresh medium was added, and the cells were incubated for an addi-tional 48 h.
Infectivity assays were performed as previously described with CD4-long ter-minal repeat (LTR)/b-galactosidase (b-Gal) (23, 43). Briefly, 293 cells were transfected as described above, except that 15 mg of DNA/plate (7.5mg of pSV-A-MLV and 7.5mg of virus expression plasmid) was transfected. DEAE-dextran was added to a final concentration of 80mg/ml, and 500ml of media was used to infect 33104CD4-LTR/b-Gal cells per 24-well cell culture plate. CD4-LTR/b-Gal cells were assayed forb-Gal expression 2 days after infec-tion.
Construction of plasmid DNAs.Standard molecular cloning techniques (55) were used to generate plasmids. pMSMDEnv2 is a derivative of the infectious proviral clone pNL4-3 (1). The nucleotide designations refer to the DNA se-quence of pNL4-3. The construction of pMSMDEnv2 has been previously de-scribed (56), and this plasmid is identical to pMSMBA (also previously dede-scribed [56]), except that pMSMDEnv2 retains the naturally occurring BspEI site at base pair (bp) 9383 in the distal LTR.
pNC1 was created by PCR amplification of pMSMDEnv2 with the sense primer 1465–1485 (59-GGC CAG ATG AGA GAA CCA AGG) and the anti-sense mismatch primer 1977–1953 SspI (59-GCC ATA ATT GAA ATA TTT AAC AGT C). A second fragment was derived from the PCR amplification of pMSMDEnv2 with the antisense primer 2470–2450 (59-CC TAT AGC TTT ATG TCC GCA G) and the sense mismatch primer 1953–1977 SspI (59-G ACT GTT
AAA TAT TTC AAT TAT GGC). These fragments were digested with SspI, ligated, and then reamplified with the sense primer 1465–1485 and the antisense primer 2470–2450. The amplified fragment was digested with SpeI and BclI, and this 922-bp fragment containing the desired mutations was ligated into pMSMDEnv2 that had been digested with SpeI and BclI.
pNC2 was created by PCR amplification of pMSMDEnv2 with the sense primer 1953–1977 and the antisense mismatch primer 2048–2023 NdeI (59-CCT TCC TTT CCA TAT GTC CAA TAG CC). A second fragment was derived from the PCR amplification of pMSMDEnv2 with the antisense primer 2470– 2450 and the sense mismatch primer 2023–2048 NdeI (59-GGC TAT TGG ACA TAT GGA AAG GAA GG). These fragments were digested with NdeI and ligated and were then reamplified with the sense primer 1465–1485 and the antisense primer 2470–2450. The amplified fragment was digested with SpeI and
BclI, and this 922-bp fragment containing the desired mutations was ligated into
pMSMDEnv2 that had been digested with SpeI and BclI. pNC12 was created by the same method as pNC2, except that pNC1 was used as the PCR template.
pDCA was generated by creating an NsiI (nucleotide [nt] 1251) to PstI (nt 1419) deletion in pMSMDEnv2. This deletion is identical to the deletion in pdl.NsiPst (previously described [76]).
pGEM(600-900) was generated by PCR amplification of pMSMDEnv2 with the sense mismatch primer 587–610 BamHI (59-ACTAGAGATGGATCCGAC CCTTTT) and the antisense mismatch primer 911–888 XhoI (59-AGCTCCCT GCTCGAGCATACTATA). The amplified fragment was digested with BamHI and XhoI, and the resulting 300-bp fragment was ligated into pGEMllzf(2) that had been digested with BamHI and XhoI. pGEM(600-900) has been previously described (56).
Isolation of cytoplasmic RNA and protein. Cytoplasmic protein and RNA were isolated from a 100-mm-diameter plate of cells by first washing the cells with phosphate-buffered saline (PBS) and then scraping the cells into 1 ml of PBS. The cells were then pelleted by centrifugation for 5 min at 6,000 rpm (2,2003g). The supernatant was then removed, and the cells were lysed in 0.5
ml of Nonidet P-40 lysis buffer (10 mM Tris [pH 7.5]–10 mM NaCl–3 mM MgCl2–0.5% [wt/vol] Nonidet P-40). The lysate was incubated on ice for 10 min and then centrifuged for 5 min at 6,000 rpm in a microcentrifuge (2,2003g) to
pellet nuclei. The supernatant (cytoplasmic fraction) was removed and used for protein analysis and as a source of cytoplasmic RNA.
RNA was purified from the cytoplasmic fraction by incubation in the presence of proteinase K (Boehringer-Mannheim Biochemicals [BMB]) (500mg/ml) and 1% sodium dodecyl sulfate (SDS) for 30 min at 37°C. This mixture was then phenol-chloroform extracted, and the nucleic acid was ethyl alcohol (EtOH) precipitated in the presence of 0.3 M sodium acetate. The nucleic acid was then incubated in DNase treatment buffer (50 mM Tris [pH 7.5]–10 mM MgCl2–1 mM dithiothreitol–400 U of RNasin ribonuclease inhibitor (Promega, Madison, Wis.) per ml–100 U of RNase-free DNase I [BMB] per ml) for 30 min at 37°C. Following DNase treatment, the nucleic acid was again phenol-chloroform ex-tracted and EtOH precipitated. The RNA was then resuspended in H2O, and its concentration was determined by optical density. The RNA was stored at270°C until further analysis.
Isolation of virion-associated RNA and protein.Virion-associated protein and nucleic acid was isolated by first centrifuging media (30 ml) from three 100-mm-diameter plates of transfected cells at 1,8003g for 10 min. The supernatant was
then transferred to a fresh tube and centrifuged again. The virus was concen-trated from the clarified media by centrifugation through a 5-ml cushion con-sisting of 20% (wt/vol) sucrose-TSE (10 mM Tris [pH 7.5]–100 mM NaCl–1 mM EDTA) for 2 h at 25,000 rpm (83,0003g) in an SW28 rotor. The virus pellet was
then resuspended in 0.5 ml of PK buffer (50 mM Tris [pH 7.5]–100 mM NaCl–10 mM EDTA–1% SDS), and 50ml was removed and stored at220°C for subse-quent protein analysis. Proteinase K (500mg/ml) was added to the remaining viral lysate and incubated at 37°C for 30 min. Following this incubation, the nucleic acid was phenol-chloroform extracted and EtOH precipitated in the presence of 20mg of Escherichia coli tRNA. The nucleic acid pellet was resus-pended and incubated in DNase treatment buffer for 30 min at 37°C. The nucleic acid was again phenol-chloroform extracted and EtOH precipitated. The RNA pellet was then resuspended in H2O and stored at270°C until further analysis.
Western blot analysis.Cytoplasmic (1/10 plate equivalent) or virion-associated (1/3 plate equivalent) protein was suspended in protein loading buffer (10 mM Tris [pH 6.8]–2% SDS–10% glycerol–5 mg of dithiothreitol per ml–0.04% brom-phenol blue). Protein samples were subjected to SDS-polyacrylamide gel elec-trophoresis (PAGE) analysis (10% acrylamide; ratio of bisacrylamide to acryl-amide, 29:1) at a constant current of 40 mA for 4 to 6 h (47). Following electrophoresis, proteins were transferred to a nitrocellulose membrane in a Bio-Rad trans-blot cell with Tris–glycine buffer (25 mM Tris–180 mM glycine) at 250 mA for 15 to 18 h at 4°C. Following transfer, the nitrocellulose membrane was rinsed for 5 min in PBS and then incubated for 1 h at room temperature in blocking buffer (PBS containing 5% dry milk and 3% bovine serum albumin). Following blocking, the membrane was incubated for 1 h in antibody incubation buffer (PBS containing 5% dry milk and 1% bovine serum albumin) and a 1/200 dilution of a rabbit anti-Gag serum. This anti-Gag serum was generated in our laboratory by the injection of bacterium-expressed, histidine-tagged Gag protein into rabbits. Following incubation with this primary antibody, membranes were rinsed five times over a period of 30 min with PBS containing 0.05% Tween 20.
on November 9, 2019 by guest
http://jvi.asm.org/
The membrane was then incubated in antibody incubation buffer containing a 35S-conjugated donkey anti-rabbit antibody (catalog no. SJ434; Amersham) at a concentration of 0.2ml/ml (vol/vol) for 1 h. Membranes were again rinsed as described above, dried, and subjected to phosphorimage analysis (model 425S; Molecular Dynamics, Sunnyvale, Calif.).
RNase protection analysis.Antisense riboprobes for RNase protection anal-ysis were transcribed with T7 RNA polymerase (New England Biolabs). The template used for transcription was pGEM(600-900) that had been linearized with HindIII, proteinase K treated, phenol-chloroform extracted, and EtOH precipitated. Approximately 2.0mg of template DNA was incubated at 37°C for 30 min in a 20-ml reaction volume under the following buffer conditions: 40 mM Tris (pH 8.0), 10 mM dithiothreitol, 6 mM MgCl2, 2 mM spermidine, 2 U of RNasin ribonuclease inhibitor (Promega) per ml, 400mM (each) recombinant ATP (rATP), rGTP, and rUTP, 20mM rCTP, and 50mCi of [a-32P]CTP (800 Ci/mmol; 20 mCi/ml; catalog no. PB20382; Amersham). Following transcription, 10 U of RNase-free DNase I (BMB) was added and this mixture was incubated at 37°C for 15 min to degrade the DNA template. Unincorporated nucleotides were removed by passing the transcription reaction mixture over a Quick Spin Sephadex G-50 column (BMB) according to the manufacturer’s directions.
Ten micrograms of cytoplasmic RNA or 1 plate equivalent of virion-associated RNA was mixed with 106cpm (approximately 200 fmol) (determined by Ceren-kov counting) of riboprobe. These RNAs were then EtOH precipitated and resuspended in 30ml of hybridization buffer {50 mM PIPES [piperazine-N,N9 -bis(2-ethanesulfonic acid)] (pH 6.8), 80% formamide, 400 mM NaCl, 1 mM EDTA}, heated to 95°C for 2 min, and then incubated at 50°C for 15 to 20 h. Following hybridization, the mixture was incubated at room temperature for 1 h and then brought to a volume of 300ml with RNase digestion buffer (10 mM Tris [pH 7.5]–300 mM NaCl–5 mM EDTA–2mg of RNase T1per ml–10mg of RNase A per ml). This mixture was then incubated at 37°C for 30 min. The RNase digestion procedure was terminated by the addition of SDS and proteinase K to final concentrations of 1% and 500mg/ml, respectively. The RNA was then phenol-chloroform extracted and EtOH precipitated along with 20mg of E. coli tRNA.
Pellets were resuspended in 10ml of RNA loading buffer (80% formamide–10 mM EDTA–0.1% bromphenol blue–0.1% xylene cyanole) and heated to 95°C for 2 min. This mixture was then subjected to PAGE (5% acrylamide [ratio of bisacrylamide to acrylamide, 29:1]–8 M urea–10 mM Tris–10 mM boric acid–2 mM EDTA). DNA molecular weight markers were generated bya-32P[dCTP] end labeling with the Klenow fragment of the plasmid pGEM11Zf(2) (Pro-mega) that had been digested with HpaII. Following electrophoresis, gels were dried under vacuum at 80°C for 2 h. The dried gels were then subjected to phosphorimage analysis.
RESULTS
Analysis of RNA encapsidation by NC mutants.
To further
characterize the role of each of the Cys-His boxes of the HIV-1
NC protein in RNA encapsidation, a series of mutant virus
ex-pression plasmids derived from the WT plasmid pMSM
D
Env2
were constructed (previously described [56]) (Fig. 1A). pMSM
D
Env2 is a derivative of the replication competent proviral clone
pNL4-3 (1) that contains both a deletion within the env gene
and a point mutation that creates a premature stop codon
within the env gene. Since the env gene product is not required
for either virus particle production (36) or RNA encapsidation
(39, 61, 70), pMSM
D
Env2 can be considered WT for RNA
encapsidation and will be referred to as pWT throughout the
remainder of the text.
Three HIV-1 expression plasmids that contain mutations
which disrupt the Cys-His boxes of the NC protein were
con-structed (Fig. 1B). pNC1 contains mutations that create
cys-teine-to-tyrosine substitution mutations at both the first and
second cysteine residues of Cys-His box 1 of the NC protein.
Similarly, pNC2 contains mutations that create
cysteine-to-tyrosine substitution mutations at both the first and second
cysteine residues of Cys-His box 2 of the NC protein. pNC2
also contains mutations that create a lysine-to-threonine
sub-stitution mutation at amino acid position 38 of NC. pNC12
combines the mutations of both pNC1 and pNC2.
The WT and mutant virus expression plasmids were
trans-fected into 293 cells, a transformed primary embryonic human
kidney cell line (29). Cytoplasmic and virion-associated
frac-tions were collected as described in Materials and Methods.
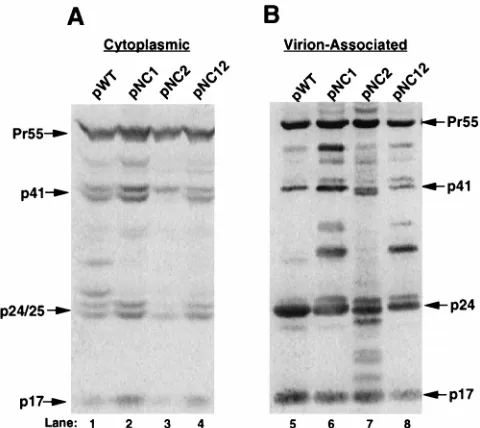
Western blot analysis of cytoplasmic protein with Gag
anti-serum indicated that cells transfected with mutant virus
expres-sion plasmids pNC1, pNC2, or pNC12 (Fig. 2A, lanes 2 to 4,
respectively) produced similar levels of cytoplasmic Gag
pro-tein compared to cells transfected with pWT (Fig. 2A, lane 1).
Moreover, cytoplasmic Gag protein expressed from pNC1,
pNC2, or pNC12 (Fig. 2A, lanes 2 to 4) exhibited a pattern of
proteolytic processing similar to that of pWT (Fig. 2A, lane 1).
Western blot analysis of virion-associated Gag protein
indi-cated that the amount of virus particles produced from pNC1,
pNC2, or pNC12 (Fig. 2B, lanes 6 to 8) was similar to the
number produced from pWT (Fig. 2B, lane 5). However,
dis-ruption of the first Cys-His box or both Cys-His boxes reduced
the efficiency of proteolytic processing in virus particles, as
evidenced by the increased accumulation of processing
inter-mediates (Fig. 2B, compare lanes 6 and 8 with lane 5).
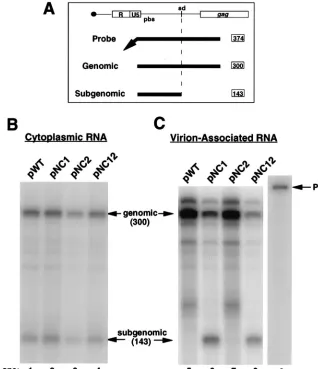
[image:3.612.312.544.73.288.2]To determine the efficiency of RNA encapsidation by the
NC Cys-His box mutant virus, quantitative RNase protection
was used to measure total cytoplasmic and virion-associated
RNA produced from 293 cells transfected with WT or mutant
virus expression plasmids. The antisense riboprobe used in the
RNase protection analysis spans the major subgenomic splice
donor, allowing for the distinction between genomic and
sub-genomic viral RNA (Fig. 3A). RNase protection analysis
indi-cated that the relative amount of genomic and subgenomic
viral RNA within the cytoplasm of transfected cells was similar
FIG. 1. Diagram of pMSMDEnv2, protein domains of the Gag polyprotein precursor, and the amino acid sequence of WT and mutant HIV-1 NC proteins. (A) Diagram of virus expression plasmid pMSMDEnv2 (previously described [56]), referred to as pWT in the text. This plasmid was derived from the repli-cation-competent proviral clone NL4-3 (1) and contains an in-frame premature stop codon at bp 6359 as well as a deletion from bp 6365 to 7252 within the env gene. (B) Protein domains of Pr55gag, the amino acid sequence of WT HIV-1
NC, and the substitution mutations present in the mutant NC protein generated by the mutant virus expression plasmids. The Gag polyprotein precursor contains the MA(p17), CA(p24), NC(p7), and p6 protein domains as well as the p2 and p1 spacer peptides. The NC protein of HIV-1 is 55 amino acids in length and contains two Cys-His box motifs located at amino acid positions 15 through 28 and 36 through 49. The cysteine and histidine residues involved in the coordi-nation of Zn21are in large type (pWT). pNC1 contains mutations that create
cysteine-to-tyrosine substitution mutations at amino acids 15 and 18 of the NC protein domain. pNC2 contains mutations that create cysteine-to-tyrosine sub-stitution mutations at amino acids 36 and 39 of the NC protein domain as well as lysine-to-threonine substitution mutations at amino acid 38 of NC. pNC12 contains the mutations that create the six substitution mutations from both pNC1 and pNC2.
on November 9, 2019 by guest
http://jvi.asm.org/
for WT (Fig. 3B, lane 1) and mutant viruses (Fig. 3B, lanes 2
to 4). Subgenomic RNA made up about 40% of the total
cytoplasmic viral RNA in each case (Table 1). As expected, the
mutations introduced into the NC protein domain did not
significantly affect the overall amount of cytoplasmic viral
RNA expressed or the extent of the splicing of viral RNA at
the major subgenomic splice donor.
The relative amount of viral RNA encapsidated by WT and
mutant viruses was determined by RNase protection analysis.
Since the amount of viral protein for each sample was similar
(Fig. 2B, lanes 5 to 8), the amount of genomic RNA, as
de-termined by quantitative RNase protection analysis, is
indica-tive of the efficiency of genomic RNA encapsidation. RNase
protection analysis of cytoplasmic and virion-associated RNA
is shown in Fig. 3C, and the results of several independent
experiments are summarized in Table 1. NC1 virus was
re-duced in RNA encapsidation to about 19% of the WT level
(Fig. 3C, compare lanes 5 and 6, and Table 1), suggesting that
the first Cys-His box of the NC protein is important for efficient
genomic RNA encapsidation. NC2 virus was also reduced in
RNA encapsidation, but only to about 73% of the WT level
(Fig. 3C, compare lanes 5 and 7, and Table 1). In the case of
NC12 virus, in which mutations were introduced into both
Cys-His boxes, the efficiency of genomic RNA encapsidation
was reduced to about 7% of the WT level (Fig. 3C, compare
lanes 5 and 8, and Table 1). Taken together, these data suggest
that both Cys-His boxes of HIV-1 contribute to efficient
genomic RNA encapsidation but that the first is more
impor-tant than the second for efficient encapsidation of genomic
RNA into assembling virus particles.
Mutations in the first Cys-His box of NC result in an
in-crease in encapsidation of subgenomic RNA.
In WT HIV-1,
subgenomic RNA makes up only about 2% of the
encapsi-dated viral RNA. However, NC1 and NC12 mutant viruses
encapsidated subgenomic RNA with a markedly increased
ef-ficiency (54 and 52% of the total encapsidated viral RNA,
respectively) (Fig. 3C and Table 1). Furthermore, this increase
in the percentage of subgenomic RNA in virus particles
rep-resented an actual increase in the encapsidation of subgenomic
RNA and not merely a decrease in the encapsidation of
geno-mic RNA. These data indicate that Cys-His box 1 of the HIV-1
NC protein is important not only for the efficient encapsidation
of genomic RNA but also for the specific recognition of
genomic RNA and the concomitant exclusion of subgenomic
RNA during the assembly of virus particles. Mutations in the
second Cys-His box only marginally increased the amount of
subgenomic RNA found to be associated with NC2 virus
ticles compared to the amount associated with WT virus
par-ticles (Table 1 and Fig. 3C, compare lanes 5 and 7).
Composite virus particles composed of both WT and mutant
NC proteins encapsidate RNA with a similar efficiency and
specificity as WT particles.
If multiple functional NC domains
on separate Gag molecules are required for encapsidation,
coexpression of NC1 or NC12 mutant Gag protein with WT
Gag protein might be expected to dominantly interfere with
RNA encapsidation mediated by WT Gag. Conversely, if only
a subset of Gag molecules in the viral capsid need to contain a
functional NC domain, then coexpression of NC1 or NC12
mutant Gag with WT Gag protein might not significantly affect
encapsidation. Thus, 293 cells were cotransfected with
equimo-lar amounts of WT and mutant virus expression plasmids.
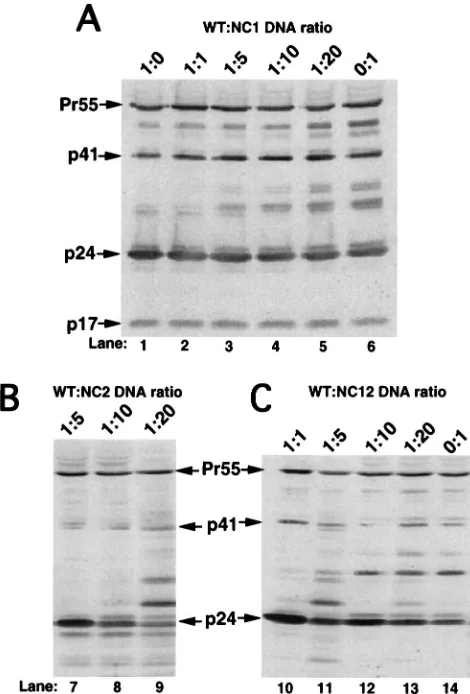
Western blot analysis indicated that similar amounts of virus
were released from cells cotransfected with WT and NC
mu-tant plasmids compared to the amount released from cells
transfected with WT plasmid alone (Fig. 4A, compare lane 1 to
lanes 2 to 4). Interestingly, RNase protection analysis indicated
that virus generated from cells coexpressing WT Gag and any
of the mutant NC Gag proteins retained the ability to
effi-ciently encapsidate genomic viral RNA (Fig. 5A, lanes 1 to 4).
A summary of the relative genomic RNA encapsidation
effi-ciency from several independent experiments is shown in the
bottom panel of Fig. 5A. Moreover, the composite virus
par-ticles excluded subgenomic viral RNA with an efficiency similar
to that of the WT virus (Fig. 5A).
[image:4.612.51.291.69.283.2]To further examine the encapsidation efficiency of
hetero-geneous virus particles, the encapsidation efficiency of virus
particles produced from the coexpression of various
combina-tions of the NC mutants was determined. As expected,
West-ern blot analysis indicated that similar amounts of virus were
released from cells cotransfected with the various
combina-tions of NC mutant expression plasmids (Fig. 4B, lanes 5 to 8).
RNase protection analysis indicated that virus produced from
cells cotransfected with pNC2 and either pNC1 or pNC12
encapsidated genomic RNA with a relatively high efficiency
(approximately 74 or 89% that of WT, respectively) (Fig. 5B,
compare lanes 7 and 9 with lane 6). A summary of the relative
genomic RNA encapsidation efficiencies from several
indepen-dent experiments is shown in the bottom panel of Fig. 5B. In
contrast, virus produced from cells cotransfected with pNC1
and pNC12 had a drastically reduced ability to encapsidate
genomic RNA, having a relative RNA encapsidation level of
approximately 10% compared to that of WT (Fig. 5B, compare
lanes 6 and 8). Thus, these data are in concordance with the
idea that RNA encapsidation requires that only a subset of the
FIG. 2. Western blot analysis of cytoplasmic and virion-associated Gag pro-tein isolated from 293 cells transfected with WT and mutant NC virus expression plasmids. Cytoplasmic protein lysate or virion-associated protein was subjected to SDS-PAGE through a 10% polyacrylamide gel, followed by Western blot analysis with an anti-Gag serum. A35S-conjugated antibody and phosphorimage analysis were used to visualize Gag protein. Viral protein products are identified by arrows and approximate molecular weight. The relative amount of Gag pro-tein was determined by comparison with a standard curve derived from serial dilutions of Gag protein (data not shown). Phosphorimage analysis of Western blots with serial dilutions of Gag protein indicated that this type of analysis is within the linear range of the assay. (A) Representative Western blot analysis of cytoplasmic protein isolated from 293 cells transfected with pWT, pNC1, pNC2, and pNC12 (lanes 1 to 4, respectively). Lysate from 1/10 plate equivalent was analyzed. (B) Representative Western blot analysis of virion-associated protein isolated from the media of 293 cells transfected with pWT, pNC1, pNC2, and pNC12 (lanes 5 to 8, respectively). Approximately one-third plate equivalent of virion-associated Gag protein was analyzed.
on November 9, 2019 by guest
http://jvi.asm.org/
Gag proteins in a virus particle contain a WT copy of Cys-His
box 1. The absence of a WT copy of Cys-His box 1 also results
in a higher amount of subgenomic RNA becoming associated
with virus particles (Fig. 5B, lane 8). These results, taken
to-gether, provide additional evidence that Cys-His box 1 is more
important than Cys-His box 2 in genomic RNA encapsidation
and in the specificity of genomic RNA encapsidation.
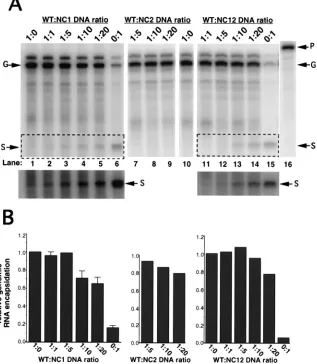
To test further the idea that successful RNA encapsidation
requires only a partial complement of WT NC protein, various
ratios of WT and NC mutant virus expression plasmids were
cotransfected into cells and the resulting virus particles were
analyzed for the ability to encapsidate viral RNA. Again,
West-ern blot analysis indicated that at all ratios of transfected DNA
tested, the amount of virus released was similar (Fig. 6). The
efficiency of genomic RNA encapsidation remained relatively
high, even at WT-to-NC1 plasmid ratios of up to 1:20 (Fig.
7A). A summary of the relative genomic RNA encapsidation
[image:5.612.141.459.70.439.2]FIG. 3. RNase protection analysis of cytoplasmic and virion-associated RNA isolated from 293 cells transfected with WT and mutant NC virus expression plasmids. (A) Region of complementarity between the antisense riboprobe and the 59region of HIV-1 RNA. The probe is 374 nt in length. The diagnostic fragment for full-length genomic RNA is 300 nt in length, and the diagnostic fragment for subgenomic RNA that is spliced at the major subgenomic splice donor is 143 nt in length. (B and C) Riboprobe was generated by in vitro transcription with T7 RNA polymerase of HindIII-linearized pGEM(600-900). Transcription reactions were performed in the presence of [32P]CTP. The probe was used in RNase protection analysis as described in Materials and Methods. The protected fragments resulting from RNase protection analysis were resolved on a 5% polyacrylamide-8 M urea gel. The gel was dried, and radiolabeled probe was visualized by phosphorimage analysis. The diagnostic bands for genomic and subgenomic RNA are indicated (arrows). Undigested probe (P) is shown in lane 9. (B) Ten-microgram amounts of total cytoplasmic RNA isolated from cells transfected with pWT, pNC1, pNC2, and pNC12 (lanes 1 to 4, respectively) were analyzed by RNase protection. (C) Virion-associated RNA isolated from the media of approximately one plate equivalent of cells transfected with pWT, pNC1, pNC2, and pNC12 (lanes 5 to 8, respectively) was analyzed by RNase protection.
TABLE 1. Summary of analysis of cytoplasmic and
virion-associated RNA from 293 cells transfected
with WT and NC mutant virus
expression plasmids
aVirus
Mean6SD of cytoplasmic
RNA
Mean6SD of virion-associated RNA
% Subgenomicb Relative genomic % Subgenomicb
WT
39
6
3
1.0
2
6
0.7
NC1
34
6
2
0.19
6
0.04
54
6
5
NC2
44
6
3
0.73
6
0.08
8
6
4
NC12
36
6
4
0.07
6
0.01
52
6
5
aCytoplasmic and virion-associated RNAs were isolated as described in
Ma-terials and Methods and in the legend for Fig. 3.
bCalculations of percentages of cytoplasmic subgenomic RNA and
virion-associated subgenomic RNA were corrected for the differences in protected probe lengths. Values reported were derived from at least three independent experiments.
on November 9, 2019 by guest
http://jvi.asm.org/
efficiencies from several independent experiments is shown in
Fig. 7B. Analysis of virus produced from cells cotransfected
with various ratios of pWT and pNC12 virus expression
plas-mids indicated that near-WT levels of genomic RNA
encapsi-dation can also occur at ratios of WT to NC12 plasmids as high
as 1:20 (Fig. 7A). Finally, virus produced from cells
cotrans-fected with various ratios of pWT and pNC2 virus expression
plasmids retained WT levels of genomic RNA encapsidation
up to WT-to-NC2 ratios of 1:20 (Fig. 7A, lanes 7 to 9). This last
observation was not surprising given that homogeneous NC2
particles retain a near-WT level of genomic RNA
encapsida-tion (Fig. 3C and Table 1).
The relative amount of subgenomic RNA associated with
virus particles is also shown in Fig. 7A. As the ratio of WT to
NC1 or WT to NC12 was increased, the amount of subgenomic
RNA that was associated with virus particles also increased
(Fig. 7B), indicating that as the amounts of NC1 and NC12
mutant proteins increased, the ability of these heterogeneous
virus particles to discriminate between genomic and
subgeno-mic RNA was reduced.
Since the primary Gag gene products expressed from pNC1,
pNC12, and pWT were indistinguishable by Western blot
anal-ysis, it was difficult to directly compare the levels of Gag
pro-tein expression in the cotransfection experiments. To directly
determine whether the various transfection conditions were
actually producing virus particles that contained the predicted
types and amounts of Gag proteins, a virus expression plasmid
containing a 168-bp, in-frame deletion within the gag gene was
used (Fig. 8A). This deletion results in the production of a Gag
precursor that lacks 56 amino acids of the CA protein domain,
and a similar mutant has been previously described (76).
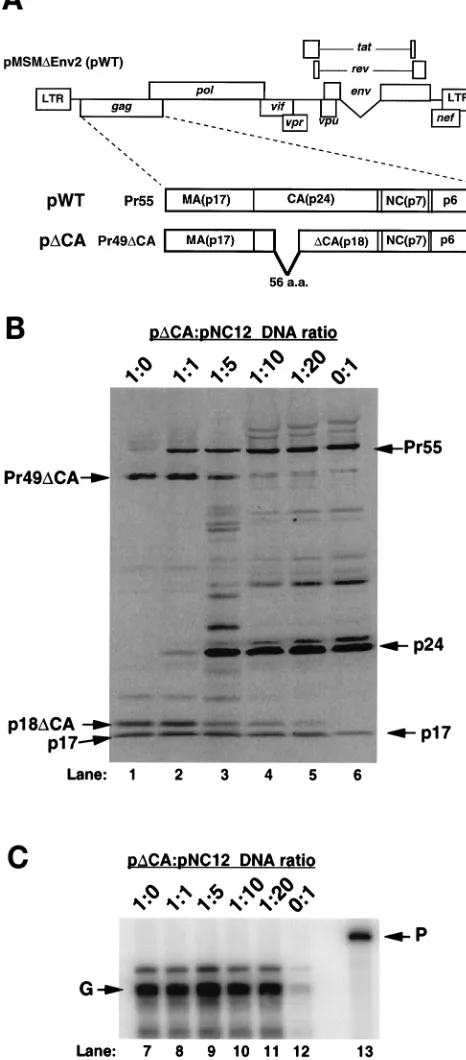
Pre-vious characterization of p
D
CA indicated that it was
compe-tent for virus particle production and, as shown below,
D
CA
virus particles are capable of efficient RNA encapsidation.
Western blot analysis of virion-associated Gag protein
iso-lated from cells transfected with various ratios of p
D
CA and
pNC12 is shown (Fig. 8B). As expected, transfection of p
D
CA
resulted in the production of virus that contained a smaller
Gag precursor (Pr49
D
CA) than did virus produced from
pNC12 (Pr55). The
D
CA virus also contained a smaller mature
CA protein (p18
D
CA) than the NC12 virus did (p24) (Fig. 8B).
As the ratio of p
D
CA:pNC12 was decreased (Fig. 8B, lanes 2
to 5), the levels of Pr55 increased, while the levels of Pr49
D
CA
decreased. At the same time, the levels of p24 increased, while
the levels of p18
D
CA decreased. Quantitative phosphorimage
analysis indicated that the population of virus particles
pro-duced in this experiment mirrors the relative amount and types
of virus expression plasmids used in these cotransfections.
RNase protection analysis was performed to measure the
RNA encapsidated by composite virus particles composed of
D
CA and NC12 Gag protein. The amount of virion-associated
[image:6.612.315.545.291.581.2]RNA paralleled that of particles made up of WT and NC12
Gag protein (Fig. 7 and 8C). Moreover, even though the level
FIG. 4. Western blot analysis of virion-associated protein isolated from the media of 293 cells cotransfected with equimolar amounts of WT and NC mutant plasmids or combinations of the NC mutant plasmids. Western blot analysis was performed as described in the legend for Fig. 2. (A) Representative Western blot analysis of virion-associated protein isolated from the media of approximately one-third plate equivalent of cells transfected with pWT (lane 1) or cotransfected with pWT and each of the NC mutant plasmids (lanes 2 to 4). (B) Representative Western blot analysis of virion-associated protein isolated from the media of approximately one-third plate equivalent of cells transfected with pWT (lane 8) or cotransfected with combinations of each of the NC mutant plasmids (pNC1/ pNC2, pNC1/pNC12, and pNC2/pNC12) (lanes 5 to 7, respectively).
FIG. 5. RNase protection analysis of virion-associated RNA isolated from the media of 293 cells cotransfected with WT and mutant NC plasmids or combinations of NC mutant plasmids. RNase protection analysis was performed as described in the legend for Fig. 3. The riboprobe is shown in lane 5 (P). The diagnostic fragments for genomic (G) and subgenomic (S) RNAs are indicated (arrows). An average genomic RNA encapsidation efficiency determined from the results of several independent experiments is represented by bar graphs. The standard deviation (SD) from the mean is represented by error bars. The relative genomic encapsidation efficiency for pWT was normalized to 1.0 for each exper-iment. (A) Representative RNase protection analysis of virion-associated RNA isolated from the media of cells transfected with pWT (lane 1) or cotransfections of pWT with pNC1, pNC2, and pNC12 (lanes 2 to 4, respectively). The total amount of DNA transfected was kept constant. (B) Representative RNase pro-tection analysis of virion-associated RNA isolated from the media of cells trans-fected with pWT (lane 6) or cotransfections of pNC1/pNC2, pNC1/pNC12, and pNC2/pNC12 (lanes 7 to 9, respectively). The total amount of DNA transfected was kept constant.
on November 9, 2019 by guest
http://jvi.asm.org/
of WT
D
CA protein was significantly reduced as the ratio of
p
D
CA to pNC12 was decreased, the level of RNA
encapsida-tion remained relatively high (Fig. 8C). This result
demon-strated that WT Gag protein, expressed from p
D
CA, can
trans-complement mutant Gag protein, expressed from pNC12,
resulting in efficient RNA encapsidation.
Composite virus particles composed of both WT and mutant
NC proteins can replicate.
Since virus particles produced from
cotransfection of cells with pWT and any of the NC mutant
expression plasmids retained the ability to efficiently
encapsi-date genomic RNA, it was of interest to determine whether
these particles could replicate. To test for replication, virus
particles were produced by cotransfection of 293 cells with the
various HIV-1 expression plasmids and a plasmid that
ex-presses an amphotropic murine leukemia virus envelope
gly-coprotein (pSV-A-MLV) (63). Virus particles were then
as-sayed for infectivity with CD4-LTR/
b
-Gal indicator cells as
previously described (23, 43). Virus produced from
transfec-tion of cells with pWT produced approximately 8
3
10
3blue
cells/ml, while neither pNC1, pNC2, nor pNC12 produced
de-tectable numbers of blue cells (Table 2). Interestingly,
cotrans-fection of cells with pWT and either pNC1, pNC2, or pNC12
yielded significant amounts of infectious virus (Table 2).
Fi-nally, cotransfection of various combinations of the NC
mu-tants produced significantly lower numbers of infectious
vi-ruses than did WT (Table 2). These results indicated that a full
complement of WT NC is not required for infectivity of virus
particles.
DISCUSSION
We have further characterized the role of the NC protein in
the efficient and specific encapsidation of HIV-1 genomic
RNA. Our data suggest that Cys-His box 1 is more important
for RNA encapsidation than Cys-His box 2, consistent with
previous reports (25, 27). Our finding that composite particles
must contain at least some copies of WT Cys-His box 1 further
suggests that Cys-His box 1 is more important than Cys-His box
2 in HIV-1 RNA encapsidation. The observation that HIV-1
particles containing mutations in Cys-His box 1 have a reduced
ability to specifically encapsidate genomic RNA over
sub-genomic RNA is consistent with the work of others (8, 79). In
contrast, Poon et al. demonstrated that mutations affecting
some of the charged amino acids of HIV-1 NC did not lead to
an increase in the encapsidation efficiency of subgenomic viral
RNA (66).
A subset of the cis-acting encapsidation elements present on
HIV-1 genomic RNA is also found on subgenomic RNA (56,
58). Also, encapsidation elements described for the avian
leu-kosis viruses are present on both genomic and subgenomic
RNAs (38, 44). It is probable that exonic RNA encapsidation
elements are capable of promoting suboptimal encapsidation
of subgenomic RNA into assembling virus particles, even
though they lack intronic encapsidation signals. Along the
same lines, reticuloendotheliosis virus vectors containing
ex-onic RNA elements are encapsidated more efficiently than
vectors lacking these elements (22). Similarly, HIV-1 and Rous
sarcoma virus subgenomic RNAs are encapsidated with high
efficiency compared to nonviral RNA (38, 54), and HIV-1
subgenomic RNA is encapsidated more efficiently than
nonvi-ral RNA (8). Finally, experiments were performed to measure
the encapsidation of highly expressed nonviral RNAs, and it
was found that there was not an increase in the encapsidation
of these nonviral RNAs by NC1 or NC12 mutant virus (data
not shown). These results reinforce the idea that the
encapsi-dation signal is multipartite and that maximal encapsiencapsi-dation
requires both exonic and intronic sequences (56).
Since mutations within Cys-His box 1 reduce the ability of
the assembling virus particle to discriminate between genomic
and subgenomic RNAs it is likely that Cys-His box 1 is required
for recognition of a sequence or element derived, at least in
part, from the viral intron. Such an RNA element either could
be fully contained on the intron of viral RNA or could
con-tribute to the formation of a higher-order structure composed
of exonic and intronic sequences. It is interesting that a similar
phenotype, a loss in discrimination between the encapsidation
of genomic and subgenomic RNAs, has been observed
follow-ing disruption of stem-loop 3 in the 5
9
region of the HIV-1
[image:7.612.52.287.70.417.2]genome (56) or deletion of HIV-1 intronic sequences (54). We
suggest that the recognition of genomic RNA involves
inter-action between Cys-His box 1 and such a cis-acting sequence.
An extension of this idea is that other domains of Gag
recog-nize cis-acting elements that are located upstream of the major
FIG. 6. Western blot analysis of virion-associated Gag protein isolated from the media of 293 cells cotransfected with varying ratios of WT to NC mutant plasmid. Western blot analysis was performed as described in the legend for Fig. 2. For each ratio, the total amount of virus expression plasmid transfected was kept constant. (A) Representative Western blot analysis of virion-associated Gag protein isolated from the media of cells cotransfected with WT to NC1 plasmid ratios of 1:0, 1:1, 1:5, 1:10, 1:20, and 0:1 (lanes 1 to 6, respectively). (B) Repre-sentative Western blot analysis of virion-associated Gag protein isolated from the media of cells cotransfected with WT to NC2 plasmid ratios of 1:5, 1:10, and 1:20 (lanes 7 to 9, respectively). (C) Representative Western blot analysis of virion-associated Gag protein isolated from the media of cells cotransfected with WT to NC12 plasmid ratios of 1:1, 1:5, 1:10, 1:20, and 0:1 (lanes 10 to 14, respectively).
on November 9, 2019 by guest
http://jvi.asm.org/
subgenomic splice donor and present on both genomic and
subgenomic RNAs (58).
The exact status of the Gag precursor proteins during RNA
recognition and encapsidation is unclear. The recognition of
genomic RNA may be mediated through a single Gag
mono-mer or by multiple Gag monomono-mers. Virus particles produced
by transfection of WT-to-NC mutant DNA ratios as high as
1:20 had encapsidation efficiencies near WT levels. One
inter-pretation of these observations is that a limited number of Gag
molecules need to interact with RNA for efficient
encapsida-tion to occur. Since only two RNA molecules are encapsidated
into each virus particle, we favor the possibility that very few
Gag molecules are required for initial recognition of RNA.
However, another possibility is that the WT Gag molecules
present in heterogeneous virus particles can act in trans to
coordinate the formation of multi-Gag complexes composed of
both WT and mutant molecules, thus allowing mutant
mole-cules to contribute to RNA recognition and encapsidation. The
NC protein of HIV-1 is found associated with viral RNA in
mature particles mostly through nonspecific interactions (16).
It would be interesting to determine whether NC is still found
to be associated with viral RNA in heterogeneous virus
parti-cles containing both WT and mutant NC proteins.
Mutations in either Cys-His box 1 or 2 result in a reduction
in replication of at least 4 orders of magnitude, while the
reduction in RNA encapsidation is only 1 order in magnitude,
suggesting that these NC mutations likely affect other roles in
viral replication. Other roles that have been attributed to
Cys-His boxes include nonspecific coating of RNA to protect the
RNA from degradation (49), promotion of DNA strand
trans-fer during reverse transcription, promotion of dimerization of
viral RNA, and the annealing of replication primer tRNA to
the primer binding site (18, 37, 41, 48, 67, 75). In contrast,
heterogeneous particles produced from the cotransfection of
WT and any of the NC mutant virus expression plasmids
re-sulted in virus particles capable of highly efficient replication.
This result indicates that no step in nucleic acid replication
requires a full complement of WT NC.
[image:8.612.141.458.76.440.2]Mutations in the first Cys-His box resulted in a reduction in
proteolytic processing of Gag. Perhaps mutations of Cys-His
FIG. 7. RNase protection analysis of virion-associated RNA isolated from the media of 293 cells cotransfected with varying ratios of WT to NC mutant plasmids. RNase protection analysis was performed as described in the legend for Fig. 3. The riboprobe is shown in lane 16 (P). Fragments representing genomic (G) and subgenomic (S) RNA are indicated (arrows). (A) Representative RNase protection analysis of virion-associated RNA isolated from the media of cells cotransfected with various ratios of WT to NC mutant plasmids. The plasmids transfected are indicated above the gels, and the ratios of WT to NC mutant plasmids are indicated above each lane. A longer exposure of the boxed regions of the gels is shown below. (B) An average genomic RNA encapsidation efficiency was determined from several independent experiments, and the SD from the mean is represented by error bars. The relative genomic encapsidation efficiency for pWT was normalized to 1.0 for each experiment.
on November 9, 2019 by guest
http://jvi.asm.org/
box 1 lead to a defect in processing due to a cis effect. In other
words, when mutations are present in Cys-His box 1, the
cleav-age sites in the Gag precursor are altered in a way that does not
allow efficient processing by the viral protease. However, this
defect in processing was not observed in experiments in which
NC1 or NC12 mutant Gag molecules were coexpressed with
WT Gag (Fig. 4A). This processing defect was not observed in
the NC1/NC2 or NC2/NC12 cotransfection experiments either
(Fig. 4B). On the surface, these results suggest that a cis-acting
processing defect can be rescued in trans by Gag molecules
that can be processed. However, we propose that efficient
proteolytic processing requires the encapsidation of viral or
cellular RNA. In cases in which a processing defect is
ob-served, the virus particles contain less viral RNA and perhaps
less total RNA than do WT virus particles (Fig. 3 and 5). Other
reports have also suggested a potential role for RNA during
virus particle assembly and maturation. Sheng and
Erickson-Viitanen demonstrated that in vitro proteolytic cleavage of
p15, the NC-p6
gagprocessing intermediate, by HIV-1 protease
is RNA dependent (73). Campbell and Vogt found that
effi-ciency of in vitro assembly of HIV-1-like particles was
dramat-ically increased when RNA was present (12).
If RNA is required for Gag processing, viral RNA per se is
probably not required. Western blot analysis from the
cyto-megalovirus mutant CMV
259D21 (56), which is deficient in
HIV-1 RNA encapsidation due to a cis-acting defect, does not
exhibit a similar defect in the efficiency of proteolytic
process-ing (data not shown). It is likely that CMV
259D21 virus
parti-cles contain quantities of nonviral RNA sufficient for
process-ing, whereas the trans-acting encapsidation mutants, NC1 and
[image:9.612.50.283.77.607.2]FIG. 8. Diagram of pDCA and Western blot and RNase protection analysis of 293 cells cotransfected with varying ratios of pDCA and pNC12. (A) pDCA contains a 168-nt deletion from NsiI (bp 1251) to PstI (bp 1419). This deletion results in the production of a truncated Gag precursor containing a 56-amino-acid (a.a.) deletion within the CA domain. The Gag precursor produced from pDCA has an approximate molecular mass of 49 kDa (Pr49DCA), and the mature capsid protein produced from pDCA has an approximate molecular mass of 18 kDa (p18DCA). (B) Representative Western blot analysis of virion-asso-ciated Gag protein isolated from the media of cells cotransfected with pDCA to NC12 plasmid ratios of 1:0, 1:1, 1:5, 1:10, 1:20, and 0:1 (lanes 1 to 6, respectively). Western blot analysis was performed as shown in Fig. 2. The predicted protein products from pWT (Pr55, p24, and p17) and from pDCA (Pr49DCA, p18DCA, and p17) are indicated (arrows). Phosphorimage analysis indicated that the population of virus particles produced in this experiment mirrored the relative
TABLE 2. Infectivity of NC mutant virus particles and virus
particles produced from cotransfection of WT and
NC mutations assayed by CD4-LTR/
b
-Gal indicator cells
aPlasmid(s)b Relative no. of blue
cell-forming U/ml
pWT ...
1.0
pNC1...
0
pNC2...
0
pNC12...
0
pWT/pNC1... 0.8
6
0.50
pWT/pNC2... 0.3
6
0.1
pWT/pNC12... 0.5
6
0.3
pNC1/pNC2...
0
pNC1/pNC12...
0
pNC2/pNC12...
0
aThe CD4-LTR/b-Gal infectivity assay was performed as described in
Mate-rials and Methods.
bA total of 15mg of DNA was transfected per plate (7.5mg of virus expression
plasmid[s] and 7.5mg of the amphotropic envelope expression plasmid, pSV-A-MLV). In the case of cotransfection of virus expression plasmids, 3.75mg of each plasmid was transfected. Values for relative numbers of blue cell-forming units were normalized to pWT (1.0). The average number of blue cell-forming units for WT was 75,000. The SD from the mean is reported.
amount and types of virus expression plasmids used in these cotransfections. (C) Representative RNase protection analysis of virion-associated RNA isolated from the media of cells cotransfected with pDCA to NC12 plasmid ratios of 1:0, 1:1, 1:5, 1:10, 1:20, and 0:1 (lanes 7 to 12, respectively). RNase protection analysis was performed as shown in Fig. 3. Predicted fragments for genomic RNA (G) and undigested probe (P) are indicated.
on November 9, 2019 by guest
http://jvi.asm.org/
NC12, do not contain enough viral or nonviral RNA for proper
maturation.
ACKNOWLEDGMENTS
We thank Michael Havert and Mark Handley for critical reviews of
the manuscript, Kate Gerten for technical assistance, and Dan Loeb
for helpful discussion.
This work was supported by NIH grant ROI AI 34733. M.D.S. is a
Cremer scholar.
REFERENCES
1. Adachi, A., H. E. Gendelman, S. Koenig, T. Folks, R. Wiley, A. Rabson, and
M. A. Martin.1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J. Virol. 59:284–291.
2. Aldovini, A., and M. B. Feinberg. 1990. Transfection of molecularly cloned HIV-1 genomes, p. 147–175. In A. Aldovini and B. D. Walker (ed.), Tech-niques in HIV research. Stockton Press, New York, N.Y.
3. Aldovini, A., and R. A. Young. 1990. Mutations of RNA and protein se-quences involved in human immunodeficiency virus type 1 packaging result in production of noninfectious virus. J. Virol. 64:1920–1926.
4. Baudin, F., R. Marquet, C. Isel, J. L. Darlix, B. Ehresmann, and C.
Ehres-mann.1993. Functional sites in the 59region of human immunodeficiency virus type 1 RNA form defined structural domains. J. Mol. Biol. 229:382– 397.
5. Berg, J. 1986. Potential metal-binding domains in nucleic acid binding pro-teins. Science 232:485–487.
6. Berkowitz, R. D., and S. P. Goff. 1994. Analysis of binding elements in the human immunodeficiency virus type 1 genomic RNA and nucleocapsid pro-tein. Virology 202:233–246.
7. Berkowitz, R. D., J. Luban, and S. P. Goff. 1993. Specific binding of human immunodeficiency virus type 1 gag polyprotein and nucleocapsid protein to viral RNAs detected by RNA mobility shift assays. J. Virol. 67:7190–7200. 8. Berkowitz, R. D., Å. Ohagen, S. Ho¨glund, and S. P. Goff. 1995. Retroviral
nucleocapsid domains mediate the specific recognition of genomic viral RNAs by chimeric Gag polyproteins during RNA packaging in vivo. J. Virol.
69:6445–6456.
9. Bess, J. W. J., P. J. Powell, H. J. Issaq, L. J. Schumack, M. K. Grimes, L. E.
Henderson, and L. O. Arthur.1992. Tightly bound zinc in human immuno-deficiency virus type 1, human T-cell leukemia virus type 1, and other ret-roviruses. J. Virol. 66:840–847.
10. Bowles, N. E., P. Damay, and P. F. Spahr. 1993. Effect of rearrangements and duplications of the Cys-His motifs of Rous sarcoma virus nucleocapsid protein. J. Virol. 67:623–631.
11. Bryant, M., and L. Ratner. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. USA
87:523–527.
12. Campbell, S., and V. M. Vogt. 1995. Self-assembly in vitro of purified CA-NC proteins from Rous sarcoma virus and human immunodeficiency virus type 1. J. Virol. 69:6487–6497.
13. Clavel, F., and J. M. Orenstein. 1990. A mutant of human immunodeficiency virus with reduced RNA packaging and abnormal particle morphology. J. Vi-rol. 64:5230–5234.
14. Clever, J., C. Sassetti, and T. G. Parslow. 1995. RNA secondary structure and binding sites for gag gene products in the 59packaging signal of human immunodeficiency virus type 1. J. Virol. 69:2101–2109.
15. Dannull, J., A. Surovoy, G. Jung, and K. Moelling. 1994. Specific binding of HIV-1 nucleocapsid protein to PSI RNA in vitro requires N-terminal zinc finger and flanking basic amino acid residues. EMBO J. 13:1525–1533. 16. Darlix, J. L., C. Gabus, M. T. Nugeyre, F. Clavel, and F. Barre-Sinoussi.
1990. Cis elements and trans-acting factors involved in the RNA dimerization of the human immunodeficiency virus HIV-1. J. Mol. Biol. 216:689–699. 17. Davis, J., M. Scherer, W. P. Tsai, and C. Long. 1976. Low-molecular-weight
Rauscher leukemia virus protein with a preferential binding for single-stranded RNA and DNA. J. Virol. 18:709–718.
18. De Rocquigny, H., C. Gabus, A. Vincent, M. C. Fournie-Zaluski, B. Roques,
and J. L. Darlix.1992. Viral RNA annealing activities of human immuno-deficiency virus type 1 nucleocapsid protein require only peptide domains outside the zinc fingers. Proc. Natl. Acad. Sci. USA 89:6472–6476. 19. Dorfman, T., J. Luban, S. P. Goff, W. A. Haseltine, and H. G. Gottlinger.
1993. Mapping of functionally important residues of a cysteine-histidine box in the human immunodeficiency virus type 1 nucleocapsid protein. J. Virol.
67:6159–6169.
20. Dupraz, P., S. Oertle, C. Meric, P. Damay, and P.-F. Spahr. 1990. Point mutations in the proximal Cys-His box of Rous sarcoma virus nucleocapsid protein. J. Virol. 64:4978–4987.
21. Dupraz, P., and P.-F. Spahr. 1992. Specificity of Rous sarcoma virus nucleo-capsid protein in genomic RNA packaging. J. Virol. 66:4662–4670. 22. Embretson, J. E., and H. M. Temin. 1987. Lack of competition results in
efficient packaging of heterologous murine retroviral RNAs and reticuloen-dotheliosis virus encapsidation-minus RNAs by the reticuloenreticuloen-dotheliosis virus helper cell line. J. Virol. 61:2675–2683.
23. Geraghty, R. J., and A. T. Panganiban. 1993. Human immunodeficiency virus type 1 has a CD4- and envelope glycoprotein-independent function. J. Virol.
67:4190–4194.
24. Gheysen, D., E. Jacobs, F. de Foresta, C. Thiriart, M. Francotte, D. Thines,
and M. De Wilde.1989. Assembly and release of HIV-1 precursor Pr55gag
virus-like particles from recombinant baculovirus-infected insect cells. Cell
59:103–112.
25. Gorelick, R. J., D. J. Chabot, A. Rein, L. E. Henderson, and L. O. Arthur. 1993. The two zinc fingers in the human immunodeficiency virus type 1 nucleocapsid protein are not functionally equivalent. J. Virol. 67:4027–4036. 26. Gorelick, R. J., L. E. Henderson, J. P. Hanser, and A. Rein. 1988. Point mutants of Moloney murine leukemia virus that fail to package viral RNA: evidence for specific RNA recognition by a “zinc finger-like” protein se-quence. Proc. Natl. Acad. Sci. USA 85:8420–8424.
27. Gorelick, R. J., S. M. Nigida, J. R. Bess, L. O. Arthur, L. E. Henderson, and
A. Rein.1990. Noninfectious human immunodeficiency virus type 1 mutants deficient in genomic RNA. J. Virol. 64:3207–3211.
28. Gottlinger, H. G., J. G. Sodrowski, and W. A. Haseltine. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA 86:5781– 5785.
29. Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J. Gen. Virol. 36:59–72.
30. Haffer, O., J. Garrigues, B. Travis, P. Moran, J. Zarling, and S. L. Hu. 1990. Human immunodeficiency virus-like, nonreplicating, gag-env particles as-semble in a recombinant vaccinia virus expression system. J. Virol. 64:2653– 2659.
31. Harrison, G. P., and A. M. Lever. 1992. The human immunodeficiency virus type 1 packaging signal and major splice donor region have a conserved stable secondary structure. J. Virol. 66:4144–4153.
32. Hayashi, T., T. Shioda, Y. Iwakura, and H. Shibuta. 1992. RNA packaging signals of human immunodeficiency virus type 1. Virology 188:590–599. 33. Jentoft, J. E., L. M. Smith, X. Fu, M. Johnson, and J. Leis. 1988. Conserved
cysteine and histidine residues of the avian myeloblastosis virus nucleocapsid protein are essential for viral replication but are not “zinc-binding fingers.” Proc. Natl. Acad. Sci. USA 85:7094–7098.
34. Jowett, J. M. B., D. J. Hockley, M. V. Nermut, and I. M. Jones. 1992. Distinct signals in human immunodeficiency virus type 1 Pr55 necessary for RNA binding and particle formation. J. Gen. Virol. 73:3079–3086.
35. Kaplan, A. H., J. A. Zack, M. Knigge, D. A. Paul, D. J. Kempf, D. W.
Norbeck, and R. Swanstrom.1993. Partial inhibition of the human immu-nodeficiency virus type 1 protease results in aberrant virus assembly and the formation of noninfectious particles. J. Virol. 67:4050–4055.
36. Karacostas, V., K. Nagashima, M. A. Gonda, and B. Moss. 1989. Human immunodeficiency virus-like particles produced by a vaccinia virus expres-sion vector. Proc. Natl. Acad. Sci. USA 86:8964–8967.
37. Karpel, R. L., L. E. Henderson, and S. Oroszlan. 1987. Interactions of retroviral structural proteins with single-stranded nucleic acids. J. Biol. Chem. 262:4961–4967.
38. Katz, R. A., R. W. Terry, and A. M. Skalka. 1986. A conserved cis-acting sequence in the 59 leader of avian sarcoma virus RNA is required for packaging. J. Virol. 59:163–167.
39. Kaye, J. F., and A. M. L. Lever. 1996. trans-Acting proteins involved in RNA encapsidation and viral assembly in human immunodeficiency virus type 1. J. Virol. 70:880–886.
40. Khan, R., and D. P. Giedroc. 1994. Nucleic acid binding properties of recombinant Zn21HIV-1 nucleocapsid protein are modulated by
COOH-terminal processing. J. Biol. Chem. 269:22538–22546.
41. Khan, R., and P. Giedroc. 1992. Recombinant human immunodeficiency virus type 1 nucleocapsid (NCp7) protein unwinds tRNA. J. Biol. Chem.
267:6689–6695.
42. Kim, H. J., K. Lee, and J. J. O’Rear. 1994. A short sequence upstream of the 59major splice site is important for encapsidation of HIV-1 genomic RNA. Virology 198:336–340.
43. Kimpton, J., and M. Emerman. 1992. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integratedb-galactosidase gene. J. Virol. 66: 2232–2239.
44. Knight, J. B., Z. H. Si, and C. M. Stoltzfus. 1994. A base-paired structure in the avian sarcoma virus 59leader is required for efficient encapsidation of RNA. J. Virol. 68:4493–4502.
45. Kohl, N. E., E. A. Emini, W. A. Schleif, L. J. Davis, J. C. Heimbach, R. A. F.
Dixon, E. M. Scolnick, and I. S. Sigal.1988. Active human immunodeficiency virus protease is required for viral infectivity. Proc. Natl. Acad. Sci. USA
85:4686–4690.
46. Konings, D. A. M., M. A. Nash, J. V. Maizel, and R. B. Arlinghaus. 1992. Novel GACG-hairpin pair motif in the 59untranslated region of type C retroviruses related to murine leukemia virus. J. Virol. 66:632–640.
![FIG. 1. Diagram of pMSM�gene. (B) Protein domains of Pr55precursor, and the amino acid sequence of WT and mutant HIV-1 NC proteins.(A) Diagram of virus expression plasmid pMSM[56]), referred to as pWT in the text](https://thumb-us.123doks.com/thumbv2/123dok_us/1251542.77488/3.612.312.544.73.288/diagram-protein-precursor-sequence-proteins-diagram-expression-referred.webp)