0022-538X/97/$04.0010
Copyright © 1997, American Society for Microbiology
Sequence Variation within a Nonstructural Region of the
Hepatitis G Virus Genome
YURY E. KHUDYAKOV,1* MIAN-ER CONG,1,2MARIA-TERESA BONAFONTE,1 SULAIMAN ABDULMALEK,1,3BARBARA L. NICHOLS,1STEPHEN LAMBERT,1
MIRIAM J. ALTER,1ANDHOWARD A. FIELDS1
Hepatitis Branch, Division of Viral and Rickettsial Diseases, National Center for Infectious Diseases,
Centers for Disease Control and Prevention, Atlanta, Georgia1; Institute of Virology, Chinese
Academy for Preventive Medicine, Beijing, People’s Republic of China2; and
Specialty Laboratories, Inc., Santa Monica, California3
Received 6 December 1996/Accepted 10 June 1997
Nine sets of nested PCR primers from a 2.6-kb region of the hepatitis G virus (HGV) genome at nucleotide positions 5829 to 8421 were designed and used to analyze serum specimens obtained from patients with community-acquired non-A, non-B hepatitis who were HGV RNA positive. One set of primers was found to be most efficient in detecting HGV and was subsequently used to test 162 HCV-positive and 11 HCV-negative plasma units obtained from individual paid donors. HGV RNA was detected in 30 (17.3%) plasma units, 2 of which were found among the 11 HCV-negative specimens. A complete set of nine PCR fragments was obtained from two patients with community-acquired acute non-A, non-B hepatitis and from four paid donors. All PCR fragments were sequenced and were shown to have a nucleotide similarity of 85.9 to 92.3% and a derived amino acid similarity of 96.0 to 99.0%. The majority of nucleotide changes occurred in the third position of codons. The HGV nucleotide and protein sequences obtained in this study were compared with HCV sequences. Based on this analysis the 2.6-kb fragment was predicted to encode the C-terminal part of the putative NS4b, the entire NS5a, and almost the complete NS5b proteins. Putative protease cleavage sites separating these proteins were also predicted. In serial specimens obtained from the two HGV-infected patients, no significant variations were found in the HGV nucleotide and derived amino acid sequences over time. The HGV sequences obtained from one patient showed no changes over 6 months, whereas more than 99.0% homology was observed for sequences from the second patient over 2.5 years. Heterogeneity analysis performed on 10 sequences obtained in this study and corresponding regions from 6 known full-size sequences of the HGV genomes demonstrated notable discrete heterogeneity consistent with the existence of HGV genetic groups or types.
The hepatitis G virus (HGV) is an agent that was recently discovered in the plasma of a patient with community-acquired acute hepatitis (13). This patient was originally diagnosed with non-A, non-B, non-C hepatitis, based upon negative results for all known hepatitis serological markers. However, retesting of this plasma by a second-generation immunoassay for the de-tection of anti-HCV antibody and by PCR using primers from the 59end of the HCV genome demonstrated that the patient was also infected with HCV. Immunoscreening of the cDNA library for anti-HGV activity obtained from this plasma al-lowed the identification of HGV (13).
Recently, the discovery of three more hepatitis viruses was reported (23, 24). Two of these, hepatitis GB virus A (GBV-A) and GBV-B, were isolated from tamarins that were experimen-tally infected with the serum obtained on the third day of jaundice from a 34-year-old surgeon (7, 23). The third, GBV-C, was identified in human sera by using a set of degen-erate PCR primers designed for specific amplification of a small DNA fragment derived from the NS3 gene of HCV-, GBV-A-, and GBV-B-like viruses (24). Direct sequence com-parison demonstrated that HGV and GBV-C are different isolates of the same agent. HGV or GBV-C has sequence homology with HCV, GBV-A, and GBV-B (12, 14). All four
viruses (HGV, HCV, GBV-A, and GBV-B) are closely related and belong to the familyFlaviviridae(12, 14).
The HGV genome has been detected in different human populations in different parts of the world (1, 11, 13, 24). Early experiments suggested a possible association between HGV or GBV-C infection and hepatitis (1, 10, 12, 13, 23, 24, 26); however, current data have failed to confirm this (3).
The HCV genome shows substantial variability, suggesting the existence of several genetically distinct types (5, 16, 22), which may have clinical and diagnostic implications (5). Be-cause of a notable resemblance to HCV, HGV is expected to have similar discrete heterogeneity consistent with grouping into genotypes. The first reports on sequencing of small re-gions from the putative helicase gene (11, 24, 26), the ns5 gene (25), and the 59noncoding region (15) implied that HGV or GBV-C isolates from different geographic areas have such a genetic heterogeneity. Sequencing and comparison of the pri-mary structure of the entire HGV genomes from different parts of the world strongly confirm this original observation (8, 12, 13, 20, 21, 27). However, classification into genotypes or subtypes must await considerably more sequence information from different regions of the HGV genome and from many more different geographic regions.
In this study, nine HGV PCR fragments covering a 2.6-kb nonstructural region were obtained from two patients with community-acquired hepatitis and from four paid plasma do-nors. Each PCR fragment was sequenced, and fragments were compared to each other and to HCV. Sequence analysis re-vealed a discrete heterogeneity of nucleotide sequences from * Corresponding author. Mailing address: Hepatitis Branch, MS
A-33, Division of Viral and Rickettsial Diseases, National Center for Infectious Diseases, Centers for Disease Control and Prevention, 1600 Clifton Rd., Atlanta, GA 30333. Phone: (404) 639-2335. Fax: (404) 639-1563. E-mail: [email protected].
6875
on November 9, 2019 by guest
http://jvi.asm.org/
different HGV isolates. This observation adds credence to the idea that distinct HGV genetic groups exist.
MATERIALS AND METHODS
Serum specimens.Ten serum specimens obtained from 10 patients that were HGV RNA positive by PCR (8a) were identified through the Centers for Disease Control and Prevention Sentinel Counties study of community-acquired viral hepatitis (2).
Serum samples (n5173) from paid blood donors were randomly selected from a collection of anti-HCV-positive units reposited at Boston Biomedica Inc. (West Bridgewater, Mass.). Initially, each plasma unit obtained for this study tested anti-HCV positive by a screening assay, which was performed at the donation site. However, upon repeat testing of these units with a second-gener-ation anti-HCV enzyme immunoassay (Abbott Laboratories, Chicago, Ill.) and supplemental testing with a semiautomated dot blot immunoassay (MATRIX; Abbott Laboratories) or recombinant immunoblot assay (RIBA-3; Ortho Diag-nostic Systems, Inc., Raritan, N.J.), the anti-HCV status could not be confirmed for 11 specimens, while the status of 3 specimens was identified as indeterminate because of immunoreactivity with a single HCV protein.
All plasma units were tested by nested reverse transcription-PCR for the presence of the HCV genome. The protocol for HCV RNA extraction from serum and cDNA preparation has been described (4). The protocol for nested PCR was 30 cycles of amplification with a pair of external primers (59-CTGTGAGGAACTACTGTCTTC and 59-GGTGCACGGTCTACGAGA CCT) and 30 cycles of amplification with internal primers (59-CAGAAAGCGT CTAGCCATGGCGTT and 59-CCCTATCAGGCAGTACCACAA). The am-plification cycle was as follows: 40 s at 94°C, 20 s at 60°C, and 45 s at 72°C. Usually, 1ml of the external-primer PCR mixture was used for the second round of amplification, which used internal primers.
HGV PCR.HGV RNA was extracted from 70ml of serum with the Tri-Reagent kit (Molecular Research Center, Cincinnati, Ohio) according to the manufacturer’s protocol. Extracted RNA was incubated for 90 min at 42°C with 500 pmol of random primers, 40 nmol of deoxynucleoside triphosphate(s), 25 U of RNase inhibitor, and 30 U of avian myeloblastosis virus reverse transcriptase (all reagents from Boehringer Mannheim, Indianapolis, Ind.). Three microliters of this reaction mixture was used for first-round PCR, which was done with external primers. One microliter of the first-round PCR product was used for second-round PCR, which was done with internal primers.
Nine sets of nested PCR primers from a 2.6-kb region of the HGV genome at nucleotide positions 5829 to 8421 were designed based on the prototype HGV PNF2161 sequence (13) kindly provided by GeneLabs, Inc. Set 1 amplified a PCR fragment of 270 bp and contained external primers YK853 (59-GCGAGC CTAGTCTTTGACTTC) and YK857 (59-TGATCCACTCCCACAGGTCC) and internal primers YK854 (59-ATGGCGGGGAAACTTTCATC) and YK856 (59-CCCACCTGTACCTCATCCACCTTA). Set 2 amplified a PCR fragment of 374 bp and contained external primers YK858 (59-ACAGTTACTTTCAGCA AGTTGAC) and YK861 (59-CCAGACGTCCCGAAGGCAACAACC) and in-ternal primers YK859 (59-TGCTCCGGCGCCTGAGCCTCAC) and YK860 (59-GGAGAGGCGACGTTTCACCGTAAC). Set 3 amplified a PCR fragment of 387 bp and contained external primers YK862 (59-TGGTGACGTTCTGAA TGGGCAAC) and YK865 (59-GTCTCCGTCCCAATGTCAATGGAC) and in-ternal primers YK863 (59-CAGTTTACTCTACCAAGCTGTGCC) and YK864 (59-GATGAAACCTCAGAGGGTGCCAC). Set 4 amplified a PCR fragment of 383 bp and contained external primers YK866 (59-GGTCTCATGGGACGCG GACGCTC) and YK869 (59-TCTTGGATGACCTCGAATGAGTC) and inter-nal primers YK867 (59-TATGGCCCTGGGCAAAGTGTTACC) and YK868 (59-TCCTCCTGCGAGGAGGACACCGAC). Set 5 amplified a PCR fragment of 399 bp and contained external primers YK870 (59-ATGCCTGTATG GGGAGAAGACATC) and YK873 (59-CCAAGGTTTCTTGCCTAGCCAC) and internal primers YK871 (59-ATCGCCAGCACTTATCTCGGTTAC) and YK872 (59-CTTGTCACATTCAAAGGTAAGTTC). Set 6 amplified a PCR fragment of 400 bp and contained external primers YK874 (59-CTGATGTTGC TAGCCTGTGTGAGA) and YK877 (59-ACCGACACCTTAGATCCCCAGC CC) and internal primers YK875 (59-AGAACCATACAGCCTATTGTGACC) and YK876 (59-CCTTACAGTCCTTATTGCTTCCTC). Set 7 amplified a PCR fragment of 435 bp and contained external primers YK878 (59-CAAGGTGAC CTTCTGGCGTGCTCC) and YK881 (59-TCCACACAGATGGCGCAAGGG GTC) and internal primers YK879 (59-TCATGATAAGTACCTCGTGG ACTC) and YK880 (59-GACTCCCATAGCTTGAGCATCTCC). Set 8 ampli-fied a PCR fragment of 417 bp and contained external primers YK882 (59-CCCCCCTGGACTTCCGGATA) and YK885 (59-ATATGATCAAGCAG TCATCGCC) and internal primers YK883 (59-CTGAAAAGCTCATCTTG GGAGACC) and YK884 (59-GAGAGACATTTTTCAGCCCCACTC). Set 9 amplified a PCR fragment of 212 bp and contained external primers YK886 (59-TATGCCTCAGGCACCATGGTCAC) and YK889 (59-AATGATGCATG ATATGAGGGCTC) and internal primers YK887 (59-CGGTGAGAGGTAT TGCAGATCCTC) and YK888 (59-CGTACCCATAGCTCGCTAGGGCTC). All PCR fragments were significantly overlapped in order to facilitate assembly of the entire 2.6-kb sequence.
Set 6 was found to be the most efficient set of primers for HGV RNA detection. Set 6 allowed amplification of a PCR fragment from all specimens that
were confirmed to be HGV RNA positive by PCR by another set of primers. For some specimens, set 6 was the only primer pair that produced a PCR fragment. However, it was noticed after sequencing of a few PCR fragments that primer YK875 often produced a mismatch at the very 39end, where an A residue was often substituted for a C residue. Surprisingly, this dramatic mismatch had no noticeable effect on PCR amplification, at least in those specimens that were found to be HGV positive. Nevertheless, YK875 was modified to 59-CAGAACCATACAGCCTATTGTGAC (YK1183) to avoid this mismatch. Although no detectable increase in sensitivity was observed, all further experi-ments used YK1183 in place of YK875. No single specimen tested with these nine sets of primers yielded all nine PCR fragments, and additional primers were designed to obtain missing PCR fragments. The design of these additional primers was based upon new sequence information obtained from sequencing PCR fragments for each individual specimen.
Sequencing.The primary structure of PCR fragments was determined with an automated sequencer (373 DNA sequencer; Applied Biosystems, Foster City, Calif.) according to the manufacturer’s protocol. In all cases, internal PCR primers were used as sequencing primers.
Computer-assisted sequence analysis.Nucleotide and amino acid sequence comparisons were performed with the MegAlign program from the Lasergene software package (DNASTAR Inc., Madison, Wis.) and the PILEUP program from the Wisconsin Sequence Analysis Package (Genetics Computer Group, Madison, Wis.). Evolutionary distances between pairs of sequences were deter-mined by the DNADIST program from the PHYLIP package (J. Felsenstein, University of Washington, Seattle). The computed distances were used to con-struct phylogenetic trees by the program NEIGHBOR. Additionally, the pro-gram MegAlign was used to construct phylogenetic trees.
Nucleotide sequence accession numbers.GenBank accession numbers for the HGV full-size genome sequences used in this study are as follows: GBV-C from West Africa, U36380; HGV PNF2161 from North America, U44402; HGV R10291 from North America, U45966; HGV D87255 from Japan, D87255; HGV U63715 from East Africa, U63715; and HGV U75356 from China, U75356. Sequences of the nonstructural region of HGV variants were deposited in Gen-Bank with accession no. U96121 for PRC5118, U96119 for JFA2113, U96123 for HGV-14, U96124 for HGV-16, U96126 for HGV-31, and U96125 for HGV-26. A follow-up specimen corresponding to PRC5118 was sequenced and given GenBank accession no. U96122. For JFA2113, three additional isolates were obtained and deposited under GenBank no. U96117, U96118, and U96120 (see below for details).
RESULTS AND DISCUSSION
Identification of HGV-positive specimens. Using the
PNF2161 HGV prototype sequence (13) we designed nine sets of nested PCR primers. All primers were designed to amplify overlapping PCR fragments of approximately 400 bp that col-lectively spanned the 2.6-kb nonstructural region at nucleotide positions 5829 to 8421. Based on a comparison of HGV and HCV prototype nucleotide sequences, the selected 2.6-kb HGV region for this study was predicted to encode the C-terminal NS4b region, the entire NS5a region, and a significant part of NS5b. All nine sets of PCR primers were applied to 10 serum specimens previously identified as HGV RNA positive by PCR. One set of primers, amplifying a 400-bp fragment derived from the NS5 region, was found to be the most efficient for the identification of the HGV genome (see Materials and Methods). This set (set 6) amplified a fragment from all HGV-positive serum specimens tested, whereas other sets often failed.
Set 6 was used to test the 173 plasma units obtained from individual paid donors for HGV RNA. A total of 30 units were found to be HGV PCR positive: 27 (16.9%) of the 159 HCV-positive units, 2 (18.2%) of the 11 HCV-negative units, and 1 of 2 units that yielded anti-HCV-indeterminate and HCV PCR-negative results.
Sequences of the nonstructural regions from six HGV
vari-ants.Two specimens (PRC5118 and JFA2113, drawn 6 and 4.5
years, respectively, after onset of illness) from patients with community-acquired acute non-A, non-B hepatitis and four specimens from plasma donors were used for sequencing. Specimen PRC5118 was positive for HCV RNA by PCR and for anti-HCV antibody by both the screening and the supple-mental assays, while specimen JFA2113 lacked any markers of HCV infection. Three specimens (HGV-14, HGV-16, and
on November 9, 2019 by guest
http://jvi.asm.org/
HGV-31) from paid plasma donors were positive for both HCV RNA and antibody, and one specimen (HGV-26) was negative for all markers of HCV infection. All six sequences were compared to the sequences of the corresponding region from the known HGV full-size genomes (PNF2161, R10291, D87255, U63715, and U75356) and to GBV-C sequences (8, 12, 13, 21, 27). The results of this analysis are presented in Fig. 1. The nucleotide sequences of these six new HGV variants were 86.4 to 91.5% similar to each other. When the prototype HGV strains (8, 13, 21, 27) and GBV-C (12) sequences were used in the homology analysis, the similarity varied from 85.9 to 92.3%, with the greatest divergence being observed between GBV-C and PNF2161 or R10291 HGV and between HGV D63715 from East Africa and U75356 from China. The highest similarity was observed between PNF2161 and HGV-26. Among the new variants identified in this study, HGV-14 and JFA2113 were the least homologous (86.2%). The deduced amino acid sequences were 94.5 to 99.5% similar. The highest protein similarity was observed for PRC5118 and Japanese HGV D87255 (2). The lowest amino acid sequence similarity was found for HGV-16 and Chinese HGV U75356 (27).
In general, all of the derived HGV sequences from within the 2.6-kb nonstructural region demonstrated relatively low variability. This observation may be explained by significant conservation of the primary structure because of important, or even essential, functions provided by these proteins in the HGV replication cycle. Alternatively, the low variability may
be due to the origin of the isolates, since all of them came from North America. Various genotypes of HCV predominate in different parts of the world. It is important to note that exper-imental conditions can also affect the interpretation of se-quence comparisons, and since HGV variants were identified by PCR, the design of the primers may have also introduced a selection bias. Because the complete nucleotide sequence for the entire 2.6-kb region was obtained only for those serum specimens that were amplified by several sets of primers, an additional selection bias may have been introduced.
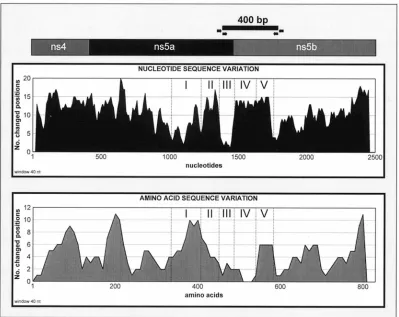
Nucleotide and amino acid sequence variations within
dif-ferent regions. Analysis of the sequence variations
[image:3.612.108.507.70.387.2]demon-strated that nucleotide changes are almost uniformly distrib-uted across the 2.6-kb region with only a few relatively conserved sites identified. The amino acid sequence showed relatively low variability with four regions demonstrating a high degree of variation (Fig. 1). It is interesting that one of the most variable protein regions is encoded by a relatively con-served RNA region identified as region I (Fig. 1). The opposite is true for variable regions II and IV, which demonstrate a high degree of conservation at the amino acid level. Region III, however, is very conserved at both the nucleotide and amino acid level. This region encodes a putative cleavage site between NS5a and NS5b (12), which may explain this observation. RNA regions IV and V are located at the 59 terminus of the pre-dicted NS5b gene. Both regions maintain almost identical de-grees of variability, with nucleotide changes affecting almost
FIG. 1. Hypothetical genetic organization of the HGV 2.6-kb DNA fragment and sequence variation within this fragment based upon alignment of 6 published sequences (8, 12, 13, 21, 27) and 10 sequences obtained in this study. The number of nucleotide or amino acid positions changed was based on a sliding window of 40 nucleotides or 40 aa calculated for each point. Regions I through V are described in Results and Discussion. The black horizontal bar shows the approximate location of a 400-bp PCR fragment amplified with primer set 6, shown by the arrows (see Materials and Methods).
on November 9, 2019 by guest
http://jvi.asm.org/
30% of all nucleotide positions among nine HGV isolates. However, protein regions IV and V demonstrate two very different patterns: region IV is highly conserved, whereas re-gion V is rather variable. Another important feature of this entire area is that RNA regions IV and V are flanked by highly conserved sequences that appear to be particularly suitable for designing PCR primers. This observation was confirmed by the finding that the most sensitive set of primers found in this study was derived from these conserved regions. Because of these properties, regions IV and V may represent an ideal location to perform genotype and/or phylogenetic analysis for HGV.
Synonymous mutations.A comparison of HGV nucleotide
sequences revealed a substantial number of silent mutations of codons, which may explain the high degree of homology be-tween the predicted protein sequences. Region IV (Fig. 1) provides the most notable examples of such mutations. As discussed above, the nucleotide sequence of this region and the encoded amino acid sequence are constrained differently: the nucleotide sequence demonstrates significant changes, while the amino acid sequence is highly conserved. Another inter-esting example of synonymous mutations was found within the JFA2113 sequence between nucleotide positions 8277 and 8306 (data not shown). This sequence contains a region in which 9 of 10 consecutive codons are affected by mutations, although none resulted in amino acid changes.
Predicted protein cleavage sites.The 2.6-kb region of the
HGV genome analyzed in this study encodes the C-terminal
part of NS4b, all of NS5a, and a significant part of NS5b, as predicted by comparison of HCV and HGV sequences. Align-ment of sequences revealed the identity of RNA polymerase domains within the NS5b protein encoded by both the PRC2161 (13) and GBV-C (12) strains. Alignment analysis also allowed the prediction of two protease cleavage sites. One site was predicted at amino acids 1893 to 1896, separating NS4b from NS5a, and another at aa 2307 to 2313, separating NS5a from NS5b (Fig. 2). Neither of these predicted sites contained cysteine residues, while both sites contained nega-tively charged amino acids common to HCV and HGV. The HGV cleavage site separating NS5a and NS5b had more sim-ilarity with the HCV site than did the site between NS4b and NS5a. This observation allowed the prediction of the boundary between NS5a and NS5b more accurately at positions 2309 and 2310, with alanine as the C-terminal residue for NS5a and serine as the N-terminal residue for NS5b. This observation strongly confirms previous prediction of the NS5a-NS5b cleav-age site made for the GBV-C polyprotein alone (12). It is interesting that when the entire 2.6-kb region was aligned with HCV sequences, a deletion of two codons, encoding both pre-dicted protease cleavage sites, within the HGV sequence was identified (Fig. 2b).
HGV sequences from serial serum specimens.In addition to
sequence variations between HGV isolates from different in-fected persons, another important characteristic of HGV ge-nome stability is sequence variation over time for HGV strains
FIG. 2. (a) Amino acid homology between the HCV NS4b-NS5a and NS5a-NS5b protein cleavage sites and the corresponding protein regions within the HGV polyprotein. (b) Nucleotide homology between the HCV genome regions encoding the NS4b/NS5a and NS5a/NS5b protein cleavage sites (9) and corresponding regions of the HGV genome. The HCV cleavage sites (a) and the HCV genome regions encoding these cleavage sites (b) are indicated by downward arrows. The hypothetical HGV protein cleavage sites are shown with brackets and upward arrows. HCV sequences used for the comparison are HCV1 (6), HCVJ6 (17), HCVJ8 (18), and HCVG9 (19).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.92.522.66.380.2]obtained from individual patients. To analyze this sequence variation, we selected additional specimens from patients from whom PRC5118 and JFA2113 were isolated. For the former, a 6-month follow-up specimen was subsequently sequenced over the entire 2.6-kb region (GenBank accession number is U96122). For the latter patient, three additional specimens were identified, two samples that were collected 1 and 2 years before the specimen (see above) already sequenced (GenBank accession no. U96117 and U96118, respectively) and a third specimen (GenBank accession no. U96120) that was collected 6 months after the sequenced specimen. Analysis of these sequences showed that only minor nucleotide changes oc-curred over 30 months for the patient from whom JFA2113 was obtained, and no changes were found over the 6-month period for the patient from whom PRC5118 was obtained (data not shown). Percent pairwise homology of the HGV nucleotide sequences varied from 99.3 to 99.7% for all four specimens tested from the former patient, with derived amino acid sequence homology varying between 99.2 and 99.8%. Nu-cleotide and amino acid substitutions were uniformly scattered across the entire region. Thus, for the analyzed 2.6-kb region of the HGV genome, the nucleotide sequence is very stable and does not undergo considerable change over the course of in-fection in individual patients.
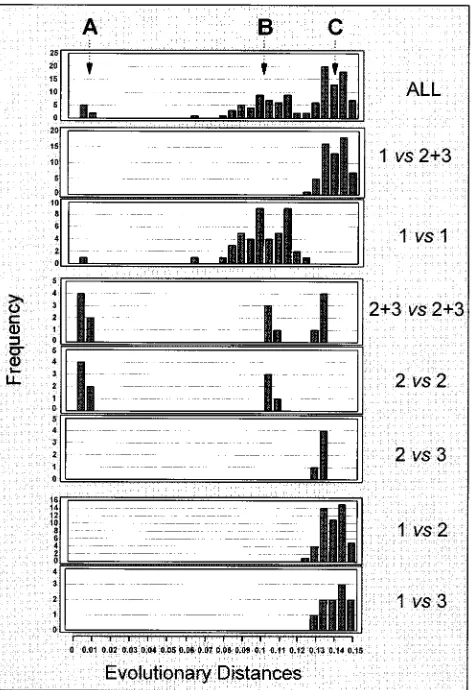
Heterogeneity of HGV. All 10 sequences from the HGV
2.6-kb nonstructural region obtained in this study were com-pared with the corresponding regions of five HGV sequences (8, 13, 21, 27) and one GBV-C sequence (12). Phylogenetic analysis of all 16 sequences was performed, and the results are presented in Fig. 3 and 4. The phylogenetic tree presented in Fig. 3 contains three major branches, 1, 2 and 3, which may represent three different genetic groups. The evolutionary dis-tances were not normally distributed when all 16 sequences were analyzed (Fig. 4). Three distinct peaks, A, B, and C, were identified. The deviation from a normal distribution may be attributed to the small number of observations included in this analysis and/or may reflect discrete HGV heterogeneity sug-gesting the existence of distinct genetic groups. Peak A corre-sponds to sequence comparisons between HGV isolates from serial specimens obtained from individual patients and, thus, presents variations within a single HGV strain. Peak B corre-sponds to sequence homologies between related HGV strains obtained from different patients, while peak C, indicating se-quences with highest evolutionary distances, implies the
[image:5.612.62.298.71.434.2]pres-FIG. 3. Proposed phylogenetic tree. PRC5118-3 and -4 and JFA2113-11, -13, -17 and -18 are sequences obtained from follow-up specimens (see Results).
FIG. 4. Frequency distribution of evolutionary distances between pairs of nucleotide sequences of the entire 2.6-kb nonstructural region from 10 HGV isolates and the corresponding regions from 5 HGV isolates (8, 13, 21, 27) and 1 GBV-C isolate (12). A, B, and C show three major trends in variability of the HGV nucleotide sequence. Each panel shows a frequency distribution of evolu-tionary distances, as follows: ALL, all 16 sequences; 1vs213, sequences of group 1 and groups 2 and 3 together (Fig. 3); 1vs1, sequences from group 1; 213vs
213, sequences from groups 2 and 3 together; 2vs2, sequences from group 2; 2
vs3, sequences from group 2 and group 3; 1vs2, sequences from group 1 and group 2; and 1vs3, sequences from group 1 and group 3.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.320.556.294.639.2]ence of distinct HGV groups. However, when sequences from only group 1, which were obtained from different individuals, were compared a normal distribution of evolutionary distances was observed (Fig. 4). Groups 2 and 3 do not have enough members for such an analysis. Also, a normal distribution was observed when sequences from different groups were com-pared to each another (Fig. 4). Thus, groups 1, 2, and 3 (Fig. 3) may represent three different HGV genotypes. However, this phylogenetic analysis failed to show full separation between genetic groups identified by peak C and subgroups identified by peak B in Fig. 4. This may pose a significant problem for establishing a firm genetic classification of different HGV vari-ants into genotypes and subtypes.
Recently, evidence for the existence of three different HGV genotypes based on analysis of the sequence heterogeneity of the 59-terminal region of the HGV genome was reported (15). In a more recent publication, this observation was confirmed when full-length HGV genomic sequences were compared (20). Despite concordance in the number of genotypes found, it is difficult to determine whether the three genotypes found in this study and in the above-mentioned publications are iden-tical (15, 20). It is interesting that phylogenetic analysis per-formed on a small NS5 region derived from HGV isolates obtained from many parts of the world failed to identify HGV genetic types (25). This discrepancy suggests that not every region of the HGV genome contains a sufficient amount of genetic information to discriminate between different HGV genotypes. Thus, larger regions or carefully selected regions should be used for the HGV phylogenetic analysis.
In conclusion, although only a small number of sequences were available to perform a complex heterogeneity analysis of the HGV genome, making definitive conclusions on the extent of HGV heterogeneity difficult, the data obtained in this study indicate that the HGV genome is discretely heterogeneous, consistent with the grouping of HGV into subtypes and puta-tive genotypes.
REFERENCES
1.Alter, H. J.1996. The cloning and clinical implications of HGV and HGBV-C. N. Engl. J. Med.334:1536–1537.
2.Alter, M. J., H. S. Margolis, M. D. Krawczynski, F. N. Judson, A. Mares, W. J. Alexander, P. Y. Hu, J. K. Miller, M. A. Gerber, R. E. Sampliner, E. L. Meeks, and M. J. Beach.1992. The natural history of community-acquired hepatitis C in the United States. N. Engl. J. Med.327:1899–1905. 3.Alter, M. J., M. Gallagher, T. T. Morris, L. A. Moyer, E. L. Meeks, K.
Krawczynski, J. P. Kim, and H. S. Margolis.1997. Acute non-A-E hepatitis in the United States and the role of hepatitis G virus infection. N. Engl. J. Med.336:741–746.
4.Beach, M. J., E. L. Meeks, L. Mimms, D. Vallari, L. DuChatme, J. Spelbring, S. Taskar, J. B. Schleicher, K. Krawczynski, and D. W. Bradley.1992. Temporal relationships of hepatitis C virus RNA and antibody response following experimental infection of chimpanzees. J. Med. Virol.36:226–237. 5.Bukh, J., R. H. Miller, and R. H. Purcell.1995. Genetic heterogeneity of hepatitis C virus: quasispecies and genotypes. Semin. Liver Dis.15:41–63. 6.Choo, Q.-L., K. Richman, J. H. Han, K. Berger, C. Lee, C. Dong, C. Gallegos,
D. Coit, A. Medina-Selby, P. J. Barr, A. Weiner, D. W. Bradley, G. Kuo, and M. Houghton.1991. Genetic organization and diversity of the hepatitis C virus. Proc. Natl. Acad. Sci. USA88:2451–2455.
7.Denhardt, F., A. Holmes, R. Capps, and H. Popper.1967. Studies on the transmission of disease of human viral hepatitis to marmoset monkeys. I. Transmission of disease, serial passage and description of liver lesions. J. Exp. Med.125:673–687.
8.Erker, J. C., J. N. Simons, A. S. Muerhoff, T. P. Leary, M. L. Chalmers, S. M. Desai, and I. K. Mushahwar.1996. Molecular cloning and characterization of a GB virus C isolate from a patient with non-A-E hepatitis. J. Gen. Virol. 77:2713–2720.
8a.Gallagher, M.Personal communication.
9.Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice.
1993. Characterization of the hepatitis C virus-encoded serine protease: determination of proteinase-dependent polyprotein cleavage sites. J. Virol. 67:2832–2843.
10. Kao, J. H., P. J. Chen, and D. S. Chen.1996. GBV-C in the aetiology of fulminant hepatitis. Lancet347:120.
11. Kao, J. H., P.-J. Chen, S.-C. Hsiang, W. Chen, and D.-S. Chen.1996. Phylogenetic analysis of GB virus C: comparison of isolates from Africa, North America, and Taiwan. J. Infect. Dis.174:410–413.
12. Leary, T. P., A. S. Muerhoff, J. N. Simons, T. L. Pilot-Matias, J. C. Erker, M. L. Chalmers, G. G. Schlauder, G. J. Dawson, S. M. Desai, and I. K. Mushahwar.1996. Sequence and genomic organization of GBV-C: a novel member of the flaviviridae associated with human non-A-E hepatitis. J. Med. Virol.48:60–67.
13. Linnen, J., J. Wages, Z.-Y. Zhang-Keck, K. E. Fry, K. Z. Krawczynski, H. Alter, E. Koonin, M. Gallagher, M. Alter, S. Hadziyznnis, P. Karayiannis, K. Fung, Y. Nakatsuji, J. W.-K. Shih, L. Young, M. Piatak, C. Hoover, J. Fernandez, S. Chen, J.-C. Zou, T. Morris, K. C. Hyams, S. Ismay, J. D. Lison, G. Hess, S. K. H. Foung, H. Thomas, D. Bradley, H. Margolis, and J. P. Kim.1996. Molecular cloning and disease association of hepatitis G virus: a transfusion-transmissible agent. Science271:505–508.
14. Muerhoff, A. S., T. P. Leary, J. N. Simons, T. J. Pilot-Matias, G. J. Dawson, J. C. Erker, M. L. Chalmers, G. G. Schlauder, S. M. Desai, and I. K. Mushahwar.1995. Genomic organization of GB viruses A and B: two new members of theFlaviviridaeassociated with GB agent hepatitis. J. Virol. 69:5621–5630.
15. Muerhoff, A. S., J. N. Simons, T. P. Leary, J. C. Erker, M. L. Chalmers, T. J. Pilot-Matias, G. J. Dawson, S. M. Desai, and I. K. Mushahwar.1996. Sequence heterogeneity within the 59-terminal region of the hepatitis GB virus C genome and evidence for genotypes. J. Hepatol.25:379–384. 16. Ohba, K.-I., M. Mizokami, T. Ohno, K. Suzuki, E. Orito, Y. Ina, J. Y. N. Lau,
and T. Gojobori.1995. Classification of hepatitis C virus into major types and subtypes based on molecular evolutionary analysis. Virus Res.36:201–214. 17. Okamoto, H., S. Okada, Y. Sugiyama, K. Kurai, H. Iizuka, A. Machida, Y.
Miyakawa, and M. Mayumi.1991. Nucleotide sequence of the genomic RNA of hepatitis C virus isolated from a human carrier: comparison with reported isolates for conserved and divergent regions. J. Gen. Virol.72: 2697–2704.
18. Okamoto, H., K. Kurai, S. Okada, K. Yamamoto, H. Lizuka, T. Tanaka, S. Fukuda, F. Tsuda, and S. Mishiro.1992. Full-length sequence of a hepatitis C virus genome having poor homology to reported isolates: comparative study of four distinct genotypes. Virology188:331–341.
19. Okamoto, H., M. Kojima, M. Sakamoto, H. Iizuka, S. Hadiwandowo, S. Suwignyo, Y. Miyakawa, and M. Mayumi.1994. The entire nucleotide se-quence and classification of a hepatitis C virus isolate of a novel genotype from an Indonesian patient with chronic liver disease. J. Gen. Virol.75:629– 635.
20. Okamoto, H., H. Nakao, T. Inoue, M. Fukuda, J. Kishimoto, H. Iisuka, F. Tsuda, Y. Miyakawa, and M. Mayumi.1997. The entire nucleotide sequence of two GB virus C/hepatitis G virus isolates of distinct genotypes from Japan. J. Gen. Virol.78:737–745.
21. Shao, L., H. Shinzawa, K. Ishikawa, X. Zhang, M. Ishibashi, H. Misawa, N. Yamada, H. Togashi, and T. Takahashi.1996. Sequence of hepatitis G virus genome isolated from a Japanese patient with non-A-E-hepatitis: amplifica-tion and cloning by long reverse transcripamplifica-tion-PCR. Biochem. Biophys. Res. Commun.228:785–791.
22. Simmonds, P., E. C. Holmes, T. A. Cha, F. McOmish, B. Irvine, E. Beall, P. L. Yap, J. Kolberg, and M. S. Urdea.1993. Classification of hepatitis C virus into six major genotypes and a series of subtypes by phylogenetic analysis of the NS5 region. J. Gen. Virol.74:2391–2399.
23. Simons, J. N., T. J. Pilot-Matias, T. P. Leary, G. J. Dawson, S. M. Desai, G. G. Schlauder, A. S. Muerhoff, J. C. Erker, S. L. Buijk, M. L. Chalmers, C. L. Van Sant, and I. K. Mushahwar.1995. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA92:3401– 3405.
24. Simons, J. N., T. P. Leary, G. J. Dawson, T. J. Pilot-Matias, A. S. Muerhoff, G. G. Schlauder, S. M. Desai, and I. K. Mushahwar.1995. Isolation of novel virus-like sequences associated with human hepatitis. Nat. Med.1:564–569. 25. Viazov, S., M. Riffelmann, Y. Khudyakov, H. Fields, C. Varenholz, and M. Roggendorf.1997. Genetic heterogeneity of hepatitis G virus isolates from different parts of the world. J. Gen. Virol.78:577–581.
26. Yoshiba, M., H. Okamoto, and S. Mishiro.1995. Detection of the GBV-C hepatitis virus genome in serum from patients with fulminant hepatitis of unknown aetiology. Lancet346:1131–1132.
27. Zhou, Y.-S., H.-T. Wang, Y.-X. He, H.-L. Zhao, H.-C. Wang, W. Chen, and J.-J. Xu.Genomic organization of HGV from Chinese and distribution of HGV infection in China. Chin. J. Microbiol., in press.