0022-538X/93/041778-10$02.00/0
Copyright X 1993,American SocietyforMicrobiology
The Acidic
Amino-Terminal
Region
of
Herpes Simplex
Virus
Type
1
Alpha
Protein
ICP27
Is
Required
for
an
Essential
Lytic
Function
STEPHEN A. RICE,`* VIVIANLAM,' ANDDAVID M.KNIPE2
Departmentof Biochemistty, University ofAlberta, Edmonton, Alberta, Canada T6G
2H7,1
andDepartment of Microbiology and Molecular Genetics, Harvard Medical School, Boston, Massachusetts 021152Received 28 September1992/Accepted28 December 1992
Theherpes simplexvirus type 1(HSV-1)aprotein ICP27 regulatesthe transition between thedelayed-early
and late phasesof the viral infection. Previousgeneticanalyseshave suggestedthat theimportant functional domains of ICP27 maptoitscarboxyl-terminal half. Onestrikingfeature of theprimarysequenceofICP27, however,isanextremelyacidicregionnearits amino terminus. To determine whether thisregionisrequired forICP27function,wedeleted the sequencesin the ICP27genewhich encodeit(codons 12 through 63). In transient expression assays, the deletion mutant was unable to efficiently repress the expression of a
cotransfected reportergeneor toefficiently complementthegrowth ofd27-1,an HSV-1 ICP27null mutant. These results suggested that the acidicregionofICP27is involved inaregulatoryfunctionrequiredforlytic growth. Totest this possibilityfurther, weintroduced the mutant allele into the HSV-1 genomeby marker transfer. Twoindependentlyderived isolates of the mutantvirus, designateddl-2a anddl-2b,wererecovered and analyzed. Both isolateswere defective forgrowth in Vero cells, exhibitinga 100-fold reduction in virus
yield compared with the wild-type infection. Vero cells infected with the dl-2 isolates showed a three- to
eightfold reduction in viral DNAreplication, a moderate reduction in theexpressionof viral ygenes,and a
delayintherepressionof
13
genes.Thephenotypeof the dl-2 isolates differssubstantiallyfromthephenotypes ofpreviously isolatedICP27 mutants, which show much more severe defects in viralgene expression. Our results demonstrate that the amino-terminal half of ICP27 participates in its regulatory activities in both infected and transfected cells.Thelyticinfection ofasusceptiblecellby herpes simplex virus type 1 (HSV-1) consists of a temporally regulated programof viralgeneexpression,viralDNAreplication, and virion assembly (reviewed in reference 36). The HSV-1
genome, a linear molecule of double-stranded DNA, is approximately 152,000 bp in size and encodes over 70
proteins. Soon after the virus penetrates the cell, the viral
genome enters the nucleus,where the viralgenes are tran-scribedbythe host RNApolymeraseII. The firstgenestobe transcribed are called ot (or immediate-early) genes. As discussed inmoredetailbelow,theotgenesencodeproteins whichregulatethe infectiousprocess. One function of thea
proteinsisto induce the transcription of the (or delayed-early)genes.Theprotein productsof the genesarelargely enzymes involved in viral DNA synthesis. Soon after the expressionof proteins,viral DNAreplication begins.The
processof viral DNAreplicationis criticalfor theexpression of the last set of viral genes, the ry (or late) genes. The y
genesfor the mostpart encode proteins which are
compo-nents of the virus particle or are involved in its assembly.
Therygeneshave been subdividedaccordingtotheextentto
which their expression requiresviral DNAreplication; the expressionof y-1 (or leaky-late)genesis reduced butreadily
detectable in the absence of DNA replication, while the expression of -y-2 (or true-late) genes is more stringently
dependent on DNA replication. Following -y gene
expres-sion, DNA-containing virions are assembled in the cell
nucleus and then mature and exit the cell via a complex
interactionwith thehost secretory apparatus.
* Corresponding author. Electronic mail address: steve.rice
@darwin.biochem.ualberta.ca.
Genetic studies have demonstrated that twoof thefiveao
proteins haveregulatory roles which are essential for viral
growth (7, 25, 29, 37). One ofthese, infected cell protein 4 (ICP4), is the major transcription-activating protein of HSV-1; it isrequired throughout infection for the transcrip-tion of the L and y genes (14, 29, 47). The 175-kDa ICP4
polypeptidehas anintrinsicsequence-specific DNA-binding
activity (19), but it is not yet clear how this activity is involved in transcriptional activation (40, 41). The second essential a protein is the 63-kDa ICP27 polypeptide. As
discussed below, ICP27 appears to mediate the transition between the and y phases of viral infection. Like ICP4,
ICP27 is a phosphoprotein which accumulates in the cell nucleus (1, 20, 49), but its biochemical properties havenot
been characterized ingreatdetail.Recently, itwasreported
that purified ICP27 can bind to zinc ions and can interact
with single-stranded DNA (46).
The analysis of viral mutants containing temperature-sensitive mutations in theICP27genefirst demonstrated that
ICP27hasan essentialregulatoryrole during infection (37).
Subsequently, viral deletion mutantswhich do not express
ICP27 or any fragment of it were isolated (25, 33). Such mutantsaredefective forgrowth in normal cells butcan be
efficiently propagatedinICP27-expressing cell lines. These
mutants exhibit a 5- to 10-fold reduction in viral DNA replication, fail to repress gene expression at late times, and show dramatically reduced levels of y-1 and y-2 gene
expression. Several other ICP27 virus mutants, including five nonsense mutants and one in-frame deletion mutant,
havebeen isolated (26, 33). One nonsense mutant,n504R, has a phenotype which is of particular interest (33). The
n504R mutant encodes an ICP27 polypeptide in whichthe 1778
on November 9, 2019 by guest
http://jvi.asm.org/
carboxyl (C)-terminal eight
amino acid residuesarereplacedby
fournew residues(the wild-type [WT] protein
consist of 512residues).
Cells infected with then5O4Rmutant exhibit defects in theexpression
of-y-2
genes butshow normal levels of viral DNAreplication
and y-1 gene expression. On the basis of thephenotype
ofthe n5O4R mutant, we proposedthatICP27carriesout twofunctionswhich contributetothe
P--y
transition,
onewhichstimulates-y-1 geneexpressionand viral DNAreplication
andasecondwhich inducesy-2genes,independent
of DNAreplication (33).
Transient-expression experiments
have alsoprovided ev-idence for theregulatory
activities of ICP27. ICP27 can eitherpositively
ornegatively
affect the expression of cotransfected reporter genes(3, 8, 32,
39,44).
Positiveregulation
of reporter genes in manycaseshas been showntobea
synergistic
effectwhichrequires
asecondherpesviraltransactivator,
eitherICP4,
ICPO(another
oa-regulatory
pro-tein),
or thepseudorabies
virus major immediate-early pro-tein.However,
in some cases ICP27can actin the absence ofother transactivators to stimulateexpression of reporter genes(5, 32, 38).
Negative regulation
of reporter genes is somewhatmorecomplicated
in thatICP27usuallyhas little effectby
itself but appearstoinhibit activationbyherpesvi-ral transactivators. The
cis-acting
elements in the reporter genes which mediatepositive
andnegative
regulation have not beendefinitively
identified,
but recent studies have indicated that mRNAprocessing signals
are critical to the responseofareporter gene toregulation
by ICP27.Specif-ically,
3' sequences involved in pre-mRNA cleavage andpolyadenylation
appear to mediatepositive
regulation byICP27
(5, 38),
while intron sequences appear necessary fortransrepression (38).
These studies suggest that ICP27canregulate
geneexpression
at aposttranscriptional
level in transfectedcells, possibly by affecting
mRNAprocessing.
Several mutationalstudies have been carried outwith the aim of
mapping
the functional domains ofICP27which are involved inactivating
andrepressing
geneexpression
in transfected cells(17,
26,
35). Although
these studies havedemonstrated that the
positive
andnegative regulatory
ac-tivities ofICP27 canbe
distinguished
genetically,
therearestill several unresolved
questions
about where the functionalregions
ofICP27map. Forexample,
bothHardwicke et al.(17)
and McMahan and Schaffer(26)
concluded that theregions important
forrepression
and activationmapped
toC-terminal half of the ICP27 molecule.
However,
wefound that the N-terminal half of the ICP27 molecule exhibited alowbut measurable transactivation
activity (35).
Inaddition,
wefound that atruncated ICP27
molecule, entirely lacking
the C-terminal
region
definedby
Hardwicke et al. as therepression domain,
still showedaWTability
totransrepress geneexpression
(35).
McMahan and Schaffer found that a very similar truncation mutant exhibitedpartial
repression
activity (26).
These apparentdiscrepancies
might
beex-plained
if the different types of mutations that have beenanalyzed
lead to differentregulatory effects,
or if ICP27contains
multiple
transactivationorrepression
domains.Onefeature ofICP27which is
striking
isthehigh
propor-tion of acidic amino acid residuesinits amino(N)-terminal
region. Twenty-five
of the first 64 residues inICP27
consist of eitherglutamic
oraspartic
acid. Inaddition, ICP27,
a knownphosphoprotein
(49),
contains nineserine residues in thisregion
which arepotential phosphate
acceptors and hencemight
contribute tothe overallnegative charge.
Sim-ilarhighly
acidicregions
are found in manytranscription
factors,
including
the HSV-1 VP16protein,
andarethought
tostimulate
transcription
viaaninteraction withone or morecomponents of the RNA polymerase II transcription com-plex (43). It was therefore of interest toexamine whetherthe acidic N terminus of ICP27 plays a role in its function. We specifically deleted the sequencesencoding thisregion from the ICP27 gene and assayed the mutant gene in transfection assays and in the context of the viral genome. Our results indicate that theN-terminal acidic region contributes to the regulatory activities of ICP27 in both transfected and in-fected cells.
MATERIALS AND METHODS
Cells, viruses, and infections. All infections and transfec-tions were carried out in Vero cells, obtained from the American Type Culture Collection, Rockville, Md., or in V27cells, aline derived from Vero cells after stable trans-fection with the ICP27 gene (33). The cells were propagated inDulbecco modified Eagle medium plus 10% supplemented newborncalfserum (Calf Supreme; GIBCO). HSV-1 strain
KOS1.1, originally provided by M. Levine (University of
Michigan,AnnArbor),wasthe WT strain of HSV-1 used in
allexperiments. Infections were carried out at a multiplicity
of 10PFU per cell.
Construction ofplasmids. Plasmid pBS27, containing the intact ICP27 gene, was constructed by cloning a 2.4-kb HSV-1 BamHI-SstI fragment derived from plasmid pBH27 (32) into the BamHI and SstI sites of pGEM3Zf-(Promega
Biotec, Madison,Wis.). Plasmid pSVSK4, which expresses
ICP4 under the control of the simian virus 40 (SV40)
early-regionpromoter,wasconstructed in two steps.First, a
342-bp HindIII-PvuII SV40 DNA fragment containing the
SV40 enhancer and early-region promoter was cloned into
pUC19. Toaccomplish this,thefragmentwasisolated from
plasmid pSV2-CAT (15), the 3' recessed HindIII end was
filled in with the Klenowfragment of Escherichia coli DNA
polymeraseI, and the fragmentwasligated into the XbaI site
of pUC19 by usingXbaI linkers. A clone, pSVE-cx, was
isolated which had the SV40fragment oriented such that the
SV40promoterwasdirectedtoward the SalI site of pUC19.
Next, the ICP4 gene, derivedfrom plasmid pSG1-EK1 (30),
wasclonedas aSallfragment into the SalI site of pSVE-a.
Aplasmid, pSVSK4,wasisolated which contained the Sall
fragmentoriented such that theSV40 promoterwasadjacent
tothe 5' end of the ICP4 gene. ThisplasmidexpressesICP4 after transfection into Verocells, asjudgedby immunofluo-rescenceandWesternimmunoblot analysis (31).
Construction of the deletion mutant plasmids pBSdl-2a
andpBSdl-2bwasdone intwosteps.First,
oligonucleotide-directedmutagenesiswasusedtoconstructplasmidmutants
pBSpml2andpBSpm64,which containedpointmutations in
the ICP27 coding region. The mutations altered codons 12
and 64, respectively, by creating XhoI restriction sites.
Mutagenesis was performed by the gapped heteroduplex
technique (22, 27), using the oligonucleotides AATTG
ACCTCGAGCTGGACCTCTC and
CGATACTCGA-iGC
CGCTCGCC
(underlined
nucleotides denote alterationsfromtheWTsequence), respectively,for thepBSpml2and
pBSpm64 mutants. Foreach plasmidmutant, two
indepen-dently derived isolateswere obtained anddesignated aand
b. To construct the desired
deletion,
pBSpml2a andpBSpm64aplasmidDNAsweredigestedwithEcoRI (which
cuts atthe 3' end of the HSV-1
insert)
andXhoI. Thelarge3,650-bp EcoRI-XhoIfragmentfrom
pBSpml2a
wasligatedto the smaller
1,800-bp
XhoI-EcoRIfragment
frompBSpm64a,resultingina
plasmid, pBSdl-2a,
which encodedan ICP27 gene in which codons 12
through
63 had beenon November 9, 2019 by guest
http://jvi.asm.org/
deleted. An identical procedure was carried out with plas-midspBSpml2b andpBSpm64b, generatingplasmid pBSdl-2b. The mutations introduced into pBSdl-2a and pBSdl-2b
were confirmed by DNA sequencing. As expected, the
manipulations resulted in a 156-bp deletion in the5' coding
region of the ICP27gene,removing codons 12through63. In
addition, the mutation altered codon 64 from aspartic to
glutamic acid.
Fortransferof themutant alleles into the HSV-1genome, thepBSdl-2a and pBSdl-2b mutationswereengineered into aplasmid, pPs27pdl (35), which contains the ICP27geneon a6.1-kb HSV-1 insert. Thiswas donebycloning the 2.4-kb BamHI-SstI fragments of pBSdl-2a and pBSdl-2b into pPs27pdl in place ofthe correspondingWTfragment. The resultingplasmidswere designated pPsdl-2a andpPsdl-2b.
This step was done to increase the extent of HSV-1 se-quences flanking the mutation, thereby increasing the
ex-pected frequency of in vivohomologous recombination.
Analysis ofmutant plasmids. Transfections were carried
out by the calcium phosphate technique as previously
de-scribed (16, 32). For chloramphenicol acetyltransferase (CAT) assays, cellextracts wereprepared after 2 days and
relative CAT activitieswere measured by enzyme assayof an appropriate dilution of the cell extract (32). The CAT activities werenormalized according tothe protein content of each extract. Western blotting was carried out as
de-scribed previously (35), using a 1:300 dilution of H1113, a
monoclonal antibody directed against ICP27 (1). The virus complementation assays werecarriedout asdescribed
pre-viously (35) exceptthatat2 h postinfection (hpi), the cells
weretreated for2min withaglycine-saline solution (pH 3.0) as described by Cai et al. (4). This treatment significantly lowered the level ofbackground unadsorbed virus without significantly affecting virus yields (34).
Isolationofmutantviruses. Theisolation of ICP27mutant
viruseswascarriedoutbyamarkertransferprotocol thatwe
have previously described (33). Briefly, plasmids pPsdl-2a and pPsdl-2b were cleaved with PstI and individually
cotransfectedintoV27cellswith infectiousd27-lacZ1 DNA. Theprogenyfrom thecotransfectionwereplated inV27cells
in the presence of
5-bromo-4-chloro-3-indolyl-3-D-galacto-pyranoside. Potential viral recombinants were isolated as
clear plaques against the background of parental blue plaques.Totestwhether the isolates encodedadeleted form
ofICP27,individualclearplaqueswereusedtoinfect 25-cm2 flasksofV27cells. Thecultureswereharvested whenall the
cells showed cytopathic effects. The resulting virus stocks
were diluted 1:10 and used to inoculate cultures of Vero cells. Proteins were harvested at 8.5 hpi and subjected to
Western blotanalysis using theH1113 antibody. Several of theplaquesgaverisetostockswhich produced an
immuno-reactive protein of the expected size (approximately 52 kDa). Two such plaques, arising from marker transfers carriedoutwithpPsdl-2a and pPsdl-2b DNAs, respectively,
wereplaque purified in V27 cells and designateddl-2a and dl-2b. Southern blotanalysis of the viral DNAs confirmed that the dl-2a and dl-2b alleles had been successfully
introduced into the recombinant viral genomes in place of
theWTICP27gene(34).
Analysisofmutantviruses. To analyze viralDNA
replica-tion, 25-cm2 culture flasks of Veroor V27 cellswere mock
infected or wereinfected withWT HSV-1orvariousICP27 mutants. At 9 hpi, the medium was replacedwith 2 ml of
medium containing25 ,uCi of[3H]thymidine (NewEngland
Nuclear) per ml. At 12 hpi, the labeling medium was
re-placed with 3 ml of lysis buffer containing 10 mM Tris
hydrochloride (pH 8), 10 mM EDTA, 2% sodium dodecyl sulfate (SDS), and 100 ,ug of proteinase K per ml. After overnight incubation at 37°C, sodium acetate was added to 0.3 M, and the lysates were extracted once with phenol-chloroform-isoamyl alcohol (24:24:1) and once with chloro-form-isoamyl alcohol (24:1). Nucleic acids were precipitated after the addition of 2 volumes of ethanol and resuspended in 0.5 ml of 10 mM Tris hydrochloride (pH 8.0)-i mM EDTA (TE buffer). The samples were digested with 25,ugof RNase A per ml for 30 min at 37°C, phenol-chloroform-isoamyl alcohol extracted as described above, and made 0.8 M in sodium chloride. An equal volume of 13% polyethylene glycol (average molecular weight, 8,000) was added. The samples were incubated on ice for 1 h and then centrifuged for 10min at 13,000 x g. The DNA pellets were washed with 70% ethanol, dried, and resuspended in TE buffer. DNA concentration were determined by measuring UV absor-bance at260 nm. Equal amounts of each DNA preparation (4 ,ug) were digested withEcoRI andXbaI and electrophoresed on a 0.9% agarose gel. After electrophoresis, the gel was treating with a fluorography-enhancing agent (Entensify; DuPont Corp.), dried, and exposed to X-ray film at -70°C.
For analysis of protein synthesis, infected cells were pulse-labeled for 30 min with 15 ,uCi of [35S]methionine-cysteine (Tran35S-label; ICN Biomedicals) per ml.
Harvest-ing of infected-cell proteins and SDS-polyacrylamide gel electrophoresis (PAGE) in 9.25% polyacrylamide gels were performed as described previously (13, 21).
Cytoplasmic RNA for Northern (RNA) blot analysis was prepared at 12 hpi by extraction of the infected cells with 0.5% Nonidet P-40 (24), followed by phenol-chloroform extaction of the supernatant and ethanol precipitation. The RNA was then resuspended in TE buffer, digested with RNase-free DNase (Boehringer Mannheim), phenol-chloro-formextracted, and ethanol precipitated. Ten micrograms of each RNAsample was subjected to electrophoresis through denaturing formaldehyde-agarose gels and transferred to GeneScreen filters (DuPont). Hybridization of32P-labeled DNAprobes, radiolabeled by the random primer extension method (2), was carried outovernight at 68°C in 6 x SSC(1x SSC is 0.15 M sodium chloride plus 0.015 M sodium cit-rate)-5x Denhardt solution (lx Denhardt solution is 0.2% [wt/vol] bovine serum albumin, Ficoll [molecular weight, 400,000], and polyvinyl pyrrolidone)-0.5% SDS-250 ,ug of denatured salmon sperm DNA per ml. The filters were washed twice at 650C in 2x SSC-0.1% SDS and then three times at650C in 0.2x SSC-0.1% SDS. The dried filters were thenexposed to X-ray film. Plasmid DNAs were used for the hybridization probes. For the ICP8 (UL29) gene probe, plasmid pE/3583 (12) was used. Plasmid pBICP5 served as theICP5(UL19) gene probe. This plasmid was generated by cloning theBamHI insert from the bacteriophage M13 clone, ICP5a (14), into the BamHI site of pUC19. Plasmid pEcoRI-BamHI-I-I (11) was used as theglycoprotein C (UL44) gene probe.
RESULTS
Analysis ofplasmids encoding anICP27moleculelacking an acidic N-terminal region. To test the functional role of the acidic N-terminal region of ICP27, we used site-specific mutagenesis to alter anICP27-encoding plasmid so that the sequences encoding this region were deleted. The parental plasmid was pBS27, which contains a 2.4-kb HSV-1 DNA insertencoding the intactICP27gene,including the 5' and 3' sequences needed for its expression in uninfected Vero cells
on November 9, 2019 by guest
http://jvi.asm.org/
B
I
Sp Ss
WT
A
4.
dl-2
1V
3--d d27-1
x
9 n504R
200 bp
FIG. 1. Structure and coding potential of WT and mutantICP27 genes. The horizontal linesrepresentHSV-1 DNA sequences con-tained in the plasmids and viruses used in this study. The parenthe-ses indicate deletion ofDNA sequences, while the letters denote relevantrestrictionenzyme sites (B, BamHI; Sp, SspI; Ss, SstI; X,
XbaI).Thearrowsbelow the lines represent the ICP27 polypeptides encodedby each allele. The dl-2polypeptide possesses an N-ter-minal in-framedeletion, while then5O4Rpolypeptide (33) possesses ashortC-terminal truncation due to the insertion of anXbaI-stop codonlinker into theSspIsite. The pBS series of plasmids used in this studycarryinserts of the BamHI-SstI fragment.
(32, 37). The mutant derivative, pBSdl-2, contained a dele-tion in the N-terminalcoding region of the ICP27 gene which resulted in the removal of codons 12 through 63 (Fig. 1). Twenty of the 52 codons deleted encode negatively charged
asparticand glutamic residues, while 9 encode serine
resi-dues which are potential sites of phosphorylation. Two
independently derived isolates ofthe mutant plasmid were
recovered and designated pBSdl-2a and pBSdl-2b. These were analyzed separately in the experiments described be-low.
Wefirstinvestigatedwhether the mutant ICP27plasmids had alterations in the ability to transactivate gene expression in transfected cells. The reporter gene used was gBCAT (-175), aconstructin which the HSV-1 glycoprotein B gene promoter drivesexpression of the CAT gene (32). We have
previouslyshown that this gene is inducedby ICP27, either
alone or in concert with other herpesvirus transactivators
(32). WTor mutantICP27plasmidswerecotransfected into
Vero cells with pgBCAT(-175), and 2 days later the cells
were harvested and tested for CAT activity. As anegative
control, we also transfected the mutant ICP27 plasmid
pBH-59R(35), which encodesatruncatedICP27molecule of
59 amino acid residues. This mutant has previously been shown to be defective for both the transactivation and
transrepression functions ofICP27 (35). The results of the
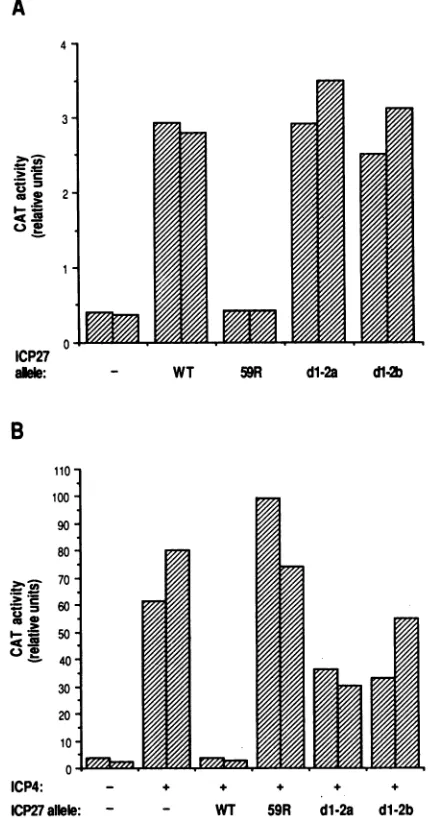
CAT assay are shown in Fig. 2A. As expected, the WT ICP27 plasmid showed a significant (seven- to eightfold)
induction of CAT activity, whereas plasmid pBH59R was
not able to transactivate expression of the reporter gene.
Both isolates of plasmid pBSdl-2 showed transactivation levels that were comparable to that of the WT plasmid.
These results demonstrate that theacidic N-terminalregion
is not requiredforthe abilityofICP27 to transactivate the
gBCAT(-175) reporter gene.
Next, we tested the ability of the mutant
plasmids
totransrepress geneexpressionintransfected cells. We
exam-ined whether themutant
plasmids
could mediate the repres-sion of an ICP4-transactivated reporter gene,8-CAT,
in which the HSV-1 ICP8 gene promoter drives CATexpres-z, c
._
o X
2
1-ICP27 Ode
0-- WT 59R dl2a dl.b
B
~.p
:~0
a
r-C)00
110- 100-90 80- 70- 60-50' 40 30 20 10
0
ICP4: - + + + + +
ICP27allele: - - WT 59R d12a d1-2b FIG. 2. Evidence that thedl-2plasmidmutantsaredefective for transrepression but not transactivation. WT or mutant plasmids
weretested incotransfection assaysfor thetransactivation (A)or transrepression(B) functions of ICP27. (A) Transactivation assay. Vero cell cultures were transfected in duplicate with 6 ,ug of pgBCAT(-175) plus,whereindicated,4 ,ugofWTor mutantICP27 plasmid. The cellswere harvested after 2days,andrelative CAT activitieswere determined. Duplicatesamples are shown as side-by-side bars. (B) Transrepression assay. Vero cell cultureswere
transfected induplicate with6 ,ugofp8CAT and,whereindicated,2 ,ugofpSVSK4, encoding ICP4. Where indicated, 4 ,ugofWTor mutantICP27plasmidwasalso added.Analysisandrepresentation of CAT activitiesisasinpanelA.
sion(44).Thep8CATplasmidwastransfectedalone,witha
plasmidencoding ICP4,orwith
plasmids
encodingICP4plusWTor mutantICP27plasmids.As
expected,
cotransfection of the ICP4 plasmidgreatly
transactivated the ICP8-CAT reporter gene (Fig. 2B). Consistent withprevious
observa-tions,theaddition ofa
plasmid encoding
WTICP27dramat-ically inhibited ICP4-mediated transactivation
(18-
to31-fold), while the control
plasmid pBH-59R
had little if anyeffect.
Compared
with the WT construct, bothpBSdl-2
isolates were defective in the
ability
to repress 8-CAT Ion November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.61.300.79.225.2] [image:4.612.334.548.82.491.2]TABLE 1. Complementationof d27-1growth by ICP27-encoding plasmids
Plasmidtransfecteda Virusyield(PFU/ml)' Complementation
index(%)C
pUC19 <1.0 x 101
pBS27 1.3 x 105 100
pBSdl-2a 2.5 x 103 1.9
pBSdl-2b 1.0 x 104 7.7
a Approximately 3 x 106Verocellswere transfected with theplasmids
indicated. Onedaylater, the cellswereinfectedwith 2 PFU ofd27-1percell. Theinfected cellswereharvested after24h.
b Virus titers in the culturelysates (10 ml)weredeterminedbyplaqueassay onV27 cells.
c Expressedasyield relativetothat for the transfectionwithpBS27DNA.
transactivation, showing onlytwo- to threefoldrepression. These results suggest that the acidic N-terminal region of ICP27hasanimportant role in thetransrepression function ofICP27.
To determine whether the acidic region of ICP27 is in-volvedinanactivitynecessaryforlyticviralreplication,we
examined whether the mutant plasmids could complement the growth of a viral mutant, d27-1 (33), which does not
expressICP27because ofalargedeletion(Fig. 1).Vero cells
were transfected with pUC19, pBS27, pBSdl-2a, or
pBSdl-2b DNA. Thefollowing day, the cellswereinfected with d27-1 and allowed to undergo one infectious cycle. Virusyieldwas determined by plaqueassay on a cell line, V27 (33),which ispermissivefor d27-1growth asthe result of the expression of a stably transfected ICP27 gene. No viral growth was detected in the culture transfected with
pUC19,whereas transfectionwithpBS27increased theyield of d27-1 by more than 4 orders of magnitude (Table 1). Transfection with the pBSdl-2 plasmids resulted in virus
yields that were 2 to 8% of the WT value. These results suggestthat the N-terminal acidicregionofICP27isrequired forafunctionneeded for the efficientreplicationof the virus
inVero cells.
However,it ispossiblethat deletion of the acidicregionof ICP27causes themutantpolypeptideto be lessstable than the WT protein. If so, we could not conclude that the deletion directlyaffected aregulatory activityofICP27. To testthestabilityof themutantpolypeptides,wetransfected
Vero cells with the WTor mutantplasmids. Theexpressed proteins were analyzed by SDS-PAGE and Western blot
analysis, using a monoclonal antibody specific for ICP27
(Fig. 3A). The pBSdl-2 plasmids encoded smaller ICP27-related proteins with apparent molecular sizes of 52 kDa, consistent with thepredicted size. Furthermore,themutant
proteins accumulatedtolevelsthatwerecomparable tothat ofthe WTprotein expressed from thepBS27 plasmid. We also used immunofluorescence microscopy to examine the cellular localization of the mutant proteins in transfected cells. Likethe WTprotein,themutantproteinswerealmost
exclusively nuclear (34). Therefore, the defects in
transre-pression andvirus complementation exhibited by the dl-2 plasmid mutants appear to result directly from an altered
regulatory function of ICP27.
The acidicamino-terminal region of ICP27is required for lytic viral growth. To study the function of the acidic N-terminalportionofICP27inthe viralinfection,weuseda
marker transferprotocol(33)tointroduce the dl-2 allele into theHSV-1genomein placeof the WTICP27gene. Marker transfer andgrowthof themutantvirus stockswerecarried
A
B
es cn C
m m In
C2
kd
-92.5
_VSW a- -69
-46
WT d1-2a
kd -97.4 -69
-46
FIG. 3. Western blotanalysisofICP27polypeptides expressed in transfected and infected cells.(A)Expression in transfected cells. Vero cellswere transfected with the plasmids indicated, and the total cell lysateswereprepared after 2 days.The proteins in the lysates were separated by SDS-PAGE, electrophoretically
trans-ferred to nitrocellulose, and probed with H1113, a monoclonal antibodyspecificfor ICP27(1).Asacontrol, proteins from HSV-1-infected Vero cells wereelectrophoresedin theright-handlane. The molecularsizesof'4C-labeled standardsareindicatedatthe left.(B) Expressionin infected cells.Vero cellswereinfectedwiththe WT virus ordl-2a, and the total proteins were isolated at the times indicated at the top. Western blotanalysiswas carriedout asfor panel A. The sizes ofprotein standards areindicated attheright.
outinICP27-expressingV27cells,sothatpotential
deficien-cies in viralgrowthwould becomplementedby the expres-sion of WT ICP27. Twoindependentisolates of the desired mutant wereobtainedanddesignated dl-2aanddl-2b. These mutants weregenerated usingthedl-2a and dl-2b plasmid
DNAs, respectively, as gene donors. Southern blotting
experimentsconfirmed that both viralisolatescontained the
dl-2mutant alleles atthe ICP27 locus(34).
The dl-2 stocks generally failed to form plaques when titrated on Vero cells, indicating that the deletion had
inactivated an essential function of ICP27. However, in
some experiments the mutant stocks formed very minute
plaqueson Vero cells. Therefore, tostudy theeffect ofthe
dl-2 mutation on viral growth in more detail, single-cycle
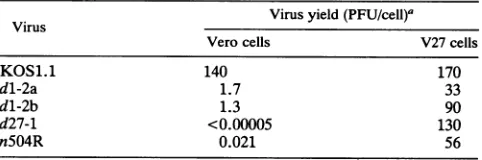
virusgrowthexperimentswereperformedat amultiplicityof infection of 10(Table 2). Thedl-2a anddl-2b stocks were both defective for growth in Vero cells in this assay,
repli-catingapproximately100-fold lessefficientlythan the
[image:5.612.60.297.102.166.2]paren-tal WTstrain,KOS1.1. Additionalexperiments showed that the dl-2 mutants gave a similar low yield if the infections
TABLE 2. Growth properties of HSV-1 ICP27 mutants
Virus Virusyield
(PFU/cell)a
Verocells V27cells
KOS1.1 140 170
dl-2a 1.7 33
dl-2b 1.3 90
d27-1 <0.00005 130
n504R 0.021 56
aVero cells(2x106)wereinfectedatamultiplicityofinfection of10PFU per cell. The same inocula were used toinfectparallelculturesof 3 x106V27 cells. Thecultureswereharvestedat 24hpi,andvirus yieldwasdetermined by plaque assayonV27cells.
I
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.314.555.617.697.2]were incubated for 2 days instead of 1 (34), indicating that
the defect in dl-2growthwas notsimplydueto adelay in the
kinetics of viral growth. Both isolates grew efficiently in V27 cells, demonstrating that the mutation could be comple-mented in trans by the WTICP27. Although the dl-2 isolates weredefective for growth in Vero cells, they were not nearly
as impaired as the ICP27 null mutant d27-1 and the ICP27
C-terminal mutant n5O4R (Fig. 1), which were reduced in
yield in Vero cells by more than 6 and 3 log units,
respec-tively, compared with the WT virus.
To testthepossibilitythatdl-2 polypeptidewasunstable
ininfectedcells,weanalyzedthesteady-state level of ICP27
proteins produced in the WT and dl-2amutant infections.
Totalproteins frominfected cellswere isolated at 4, 9, and
14hpiandsubjectedtoWestern blotanalysis (Fig. 3B).The
mutantICP27 protein synthesizedindl-2a-infectedcells had an apparent molecular size of 51 kDa, similar to the size
observed intransfected cells. At all time points tested, the mutant protein accumulated to levels that were equal or greater than the WT protein level. Similar results were observedforthemutantpolypeptide expressedby thed1-2b
virus (34). Furthermore, immunofluorescence experiments
indicated that themutantICP27proteins expressedin
dl-2a-anddl-2b-infectedVerocells accumulatedpredominatelyin
the cell nucleus(34). Therefore,thegrowthdefect of thedl-2
mutants in Vero cells does not appear to be due to either instability of the mutant protein or a defect in its nuclear localization.
The dl-2 mutants are deficientinviral DNAreplication.The
dl-2 isolatesarethe firstICP27virusmutants tobe isolated
which encode proteins with alterations specifically in the
N-terminal half of ICP27. It was therefore of interest to
characterizetheirphenotypesin detail.First,westudied the
ability ofthedl-2isolatestoreplicateviral DNA. Verocells
weremock infectedor wereinfected with either WTHSV-1,
dl-2a, ordl-2b, the ICP27 null mutantd27-1, or the ICP27
C-terminal mutantn5O4R. As a control, the same inocula
were used to infect parallel cultures of V27 cells. The cultureswere labeled with
[3H]thymidine
from 9to 12 hpi.Afterlabeling, thetotal DNA from the cultureswasisolated
and equal amounts were analyzed by restriction enzyme
digestion (EcoRI plus
XbaI),
gel electrophoresis, andfluo-rography.Theresultsfrom the Vero cellinfections indicated
the ICP27 mutants differed
significantly
in their abilitiestoreplicate viral DNA (Fig. 4A). Both isolates of the dl-2
mutant showed defects in viralDNA synthesis. Consistent
withourprevious results(33), the ICP27 nullmutantd27-1
also exhibited a
deficiency
inviralDNAreplication.
Densi-tometricanalysisof
autoradiograms
indicatedthat thedl-2aand dl-2bmutantsreplicated3- and8-fold lessDNA,
respec-tively, during the labeling
period
than did the WTvirus,
while the d27-1 mutant replicated 12-fold less DNA. The
n5O4R mutant, on the other hand, replicated viral DNAas
well asdid the WT virus. This result is consistent withour
previous phenotypic analysisof thismutant
(33).
All of thevirusesreplicated viralDNAtosimilarextentsin V27cells
(Fig. 4B), demonstrating that the differences seen in Vero
cellswere notdue tovariations in the titers of the inocula. These results indicate that the acidic
region
of ICP27 isrequiredfornormallevels of viral DNA
synthesis
in infectedcells.
The dl-2 mutants exhibit modest defects in viral gene
expression. We next studied viral gene
expression
indl-2-infected cells.
First,
we examinedthe ratesof viralprotein
synthesis atvarious times after infection. Vero cells either
were mock infectedorwere infected with the WT virus or
A
B
-W et cu C^4
r_ C C C t
E o ue Z Z >
Ii
X3fDCM-la Cz C n
-_ = _
,-__ _ _ - logo
Flu. 4. viral DNA replication in ICP27 mutant-infected cells. Vero(A)orV27(B) cellsweremock infectedor wereinfectedwith the viruses indicated at the top. The cells were labeled with [3H]thymidinefrom9 to12hpi.TotalDNAwasthenpurified,and equal amounts were digestedwith EcoRI plusXbaI and electro-phoresedon a0.9% agarosegel.Thegelwasprocessed for fluorog-raphy, dried, and exposed to X-ray film. The n504R genome containsanXbaI site at the 3' endof the ICP27 gene(Fig. 1)and thereforegives riseto anovel EcoRI-XbaIrestrictionfragment. with ICP27 mutants. At 4, 9, or 14 hpi, the cultureswere
pulse-labeled with
[35S]methionine-cysteine,
and thepro-teins were analyzed by SDS-PAGE and
autoradiography
(Fig. 5). Both isolates of dl-2 showed a pattern of viral
protein synthesisthatdifferedmodestly fromtheWTpattern
4h 9h 14h
x co 8 (> >yc.c ,_
Z~
gR o 7 CM
-1-2
-4
-5 -6
-8
- pgB - 15
[image:6.612.318.554.380.631.2]-27
FIG. 5. Protein
synthesis
in ICP27mutant-infected cells.Vero cellswere mock infected or were infected with the WT virusorICP27mutants. At4, 9,and 14
hpi,
thecellswerepulse-labeled
for 30 min with[35S]methionine-cysteine
and then harvested.Equal
fractions of each celllysate
wereanalyzed
by
SDS-PAGE andautoradiography.
Shownattheright
arethepositions
ofmigration
ofseveralHSV-1
proteins.
The WTICP27polypeptide
andthen504R
mutant ICP27
polypeptide
comigrate
on thisgel,
while the dl-2mutantICP27
polypeptides
arefoundatalowerposition (indicated
byan
asterisk).
on November 9, 2019 by guest
http://jvi.asm.org/
0 r-_ V C
E o C -a -a
A
-ICP8
B
S.I
Om_
ICP51..
C
;..*W .M-gC
FIG. 6. Accumulationof viralmRNAsinICP27 mutant-infected cells. Vero cells were mock infected or were infected with the viruses indicated at the top. At 12 hpi, cytoplasmic RNA was
prepared. Equalamountsof eachRNApreparationweresubjected
toNorthern blotanalysis using 32P-labeledprobesspecificfor ICP8 (A),ICP5(B),orgC(C)mRNA.
with the WT infection. This resultwas consistent with the protein synthesis analysis, which showed that ,B protein
repressionwassomewhat delayedin thedl-2 infections(Fig.
5, 9hpi). The d27-1 and n5O4R infections,however,showed
amore dramaticoverexpression ofICP8 mRNAat 12 hpi,
consistent with thehighlevels ofproteinsynthesisthatwere
observedatlate times. The expressionofa-y-1mRNA,that
encodingthemajorcapsid protein ICP5,was nextexamined
(Fig. 6B). Expression of ICP5 mRNA was reduced in the
dl-2 infections compared with the WT infection. A similar
reduction was seen in thed27-1 infection. Then5O4R
infec-tion, onthe otherhand, showedWT levels of ICP5 mRNA.
Thelevels of ICP5 mRNA in the various infections corre-lated qualitatively with the levels of ICP5protein synthesis
that were observed at late times (Fig. 5, 14 h). Finally,we
examinedtheexpressionofawell-characterized-y-2 mRNA,
thatencoding theglycoprotein C (gC; Fig. 6C).
Accumula-tionofgC mRNAwas only modestly reduced in thed1-2a
anddl-2b infectionscomparedwith the WTinfection, show-ing three- and twofold reduction, respectively, as deter-mined by densitometricanalysis. In contrast, accumulation
of gC mRNA was quite deficient in the d27-1 and n5O4R
infections. In thecaseof thed27-1 infection, gCmRNAwas
undetectable, while the levels in the n5O4R infectionwere
reduced 13-foldcomparedwith the WTinfection.
Theresultspresentedabovedemonstratethat thetwodl-2
isolates exhibit similar defects in viral gene expression.
Specifically, the dl-2 mutantsshow both areduction in the
expression of -y mRNAs and proteins and a delay in the
down-regulation of 1 mRNAs and proteins. These modest
deficiencies are in contrast to the more striking defects in
gene expression which are exhibited by the ICP27 null
mutantd27-1 and the ICP27C-terminalmutantn5O4R.
intworespects.First, thesynthesisof several-yproteinswas
somewhat deficient at the latertime points. This was most
easily seen for the high-molecular-weight ICP1-2 polypep-tide. Thesynthesisofseveral otheryproteins,suchasICP5,
wasalsoreduced buttoalesserextentthan that ofICP1-2. Second, the repression ofsynthesis of several ,B proteins, includingICP6 andICP8,wasdelayedinthedl-2 infections comparedwith the WT infection.
The patternof viralproteinsynthesisexhibitedbythedl-2 mutantswasquite different from the patterns shownbythe d27-1 and n504R mutants, both of which showed patterns thatwereconsistent with ourprevious analysis (33). First, the dl-2 mutants, like the WTvirus,wereabletoefficiently
repress the expression of the aprotein ICP4 at late times
afterinfection. Incontrast, neitherthe d27-1northe n504R mutant was able to efficiently down-regulate ICP4
expres-sion.Second,thedl-2mutantsappearedtohaveamuch less
severe defect in the repression of protein synthesis than
did thed27-1 and n504Rmutants. Third, thedl-2mutants
wereabletoinducethe synthesis of certain-yproteinssuch
asICP15,whichwerenotinducedto appreciable extentsin thed27-1 andn504R infections.
Tostudydl-2aanddl-2bgeneexpression inmoredetail, we examined the accumulation of specific mRNAs from
well-characterized 1, -y-1, and -y-2 genes. Vero cells were
mockinfectedor wereinfectedwith theWTvirusorICP27
mutants. Cytoplasmic RNA was isolated at 12 hpi and analyzed by Northern blot analysis. We first studied the expression of a mRNA, that encoding the major DNA-binding protein, ICP8 (Fig. 6A). The dl-2 infections both showedamodestoverexpressionof ICP8 mRNAcompared
DISCUSSION
Astrikingfeature of theprimarysequenceofICP27 is the
highproportionofacidicaminoacidresiduesinits
N-termi-nal region. Nearly 40% of the 64 N-terminal residues of
ICP27consist ofasparticor glutamic acid.The presenceof
an acidic region in this viral gene-regulatory protein is of interest for two reasons. First, acidic regions are found in manyviral and cellulartranscription factors and have been
implicatedintranscriptionalactivation. Theyarethoughtto
function viaaninteraction withone ormore componentsof the RNApolymeraseIItranscription complex(43). Second,
several other herpesviruses have been shown to possess openreading frames(ORFs)with some degree of amino acid homology to ICP27. These ORFs include varicella-zoster virus ORF 4 (6), equine herpesvirus 1 ORF 5 (45, 51),
Epstein-Barrvirus BMLF1 (6), andthe herpesvirus saimiri
52-kDa ORF (28). Although the homology between ICP27
and these ORFs ismostlylimitedtotheirC-terminal halves, allcontainacluster of acidic residues at their N termini. This observation raises the possibility that the acidic region of ICP27 is involved in afunction common to diverse herpes-viruses.
The acidic N-terminal region of ICP27 is required for efficient transrepression in transfected cells. To study the function of the acidic region of ICP27, we constructed an ICP27 gene inwhich the sequences encoding it (codons 12
through 63)were deleted. The mutant gene wastested in a
number of transfection assays designed to measure the
propertiesand functions ofICP27 in uninfected cells. These
experiments indicated that the N-terminal deletion did not
affect the stability or nuclear localization of ICP27 or its
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.113.246.80.340.2]ability to transactivate the expression of a reporter gene. However, the deletion did significantly reduce the ability of ICP27 to transrepress gene expression in transfected cells. This result is noteworthy because previous studies have concluded that the transrepressing activity of ICP27 maps to its C-terminal half (17, 26). Although it is clear that regions in theC-terminal half of the molecule are required for transre-pression, the only published data which might argue against an important contribution of the N-terminal half are the experiments of Hardwicke et al. (17). These investigators constructed plasmids having in-frame oligonucleotide inser-tions at many sites in the ICP27 coding region. It was found that insertions in the N-terminal half of the ICP27 gene, including insertions between residues 28 and 29 and residues 58 and 59, had no effect on transrepression. The combined results thus suggest that the specific amino acid sequence of theN-terminal region of ICP27 may not be as important as a general biochemical feature of this region, such as its high negative charge. If so, the N-terminal region ofICP27 may be similar to the negatively charged domains of transcription
factors,manyofwhich do not appear to depend on specific
aminoacid sequences (23). It will be of interest to determine
whether other acidic sequences can substitute for the acidic
regionof ICP27.
The acidic N-terminal region ofICP27 is required for an essentiallytic function. Our plasmidcomplementation assay suggested that the acidic region of ICP27 is required for efficient viral growth. To test this possibility directly, we introduced the mutant ICP27 allele into the HSV-1 genome
inplaceof the WT gene. Twoindependently derived isolates
of therecombinant virus, dl-2a and dl-2b, were recovered and separately characterized. This was done to exclude the
possibility that an unintended, secondary mutation might
contribute to the observed phenotype of the dl-2 mutant. However, the two isolates exhibited very similar, if not
identical, phenotypic properties. In addition, the growth
defectsof both mutants could be complemented by the WT
tCP27 expressed from V27 cells. These results together
show that the phenotype displayed by the dl-2 isolates
resulted directlyfrom the engineered mutation.
Single-cycle growth experiments indicated that the virus
yield from dl-2-infected Vero cells was reduced
approxi-mately100-fold compared with the WT infection. Therefore, we conclude that the acidic N-terminal region of ICP27 is
required foran essential lytic function. To gain insight into
thatfunction,weexamined viral DNA replication and gene
expression indl-2-infected cells. We found that the mutant
infections differed from the WT infection in two respects.
First,thedl-2isolates were deficient in viral DNA
replica-tion, showing athree- to eightfold reduction in the level of
DNAsynthesis. Second, the dl-2 mutants exhibited modest
deficiencies in viral gene expression. Specifically,
dl-2-infectedcells showed both a reduction in the level ofygene
expression and adelay in the repression of ,B gene
expres-sion.
Inhibition ofHSV-1 DNAsynthesisby avariety of means
results in both a reduction in -y gene expression and a
potentiation of 1 gene expression (36). Therefore, the
sim-plesthypothesisthat wouldexplain the phenotype of dl-2 is
that the primary effect of the mutation is to reduce the amount of viral DNAreplication. Thereduced amounts of
DNAreplicationindl-2-infected cellsmight result indirectly
in the observed alterations in viral gene expression. How-ever, at presentwe cannot exclude the possibility that the dl-2 mutation has a direct effect on gene expression in infected cells. Indeed, our finding that the dl-2 plasmid
mutant is defective for transrepression in transfected cells is consistent with this latter possibility.
Stimulation of viral DNA replication byICP27. Our results demonstrate that ICP27 has a specific role in stimulating viral DNA replication in infected cells. However, ICP27 is not one of the seven HSV-1 replication proteins which are thought to participate directly in the DNA replication pro-cess (reviewed in reference 48). Indeed, these seven viral proteins are sufficient in transfected cells to direct replica-tion from an HSV-1 origin of replication contained on a plasmid molecule (ori-plasmid) (50). How then does ICP27
stimulate the level of viral DNA replication in infected cells? One possibility is that ICP27 stimulates the expression of one or more of the replication proteins, which are of the ,B kinetic class. This explanation appears unlikely, however, because the major 13 genes that we examined, ICP6 and ICP8, were not underexpressed in either dl-2 or d27-1 infections. In fact, these genes were overexpressed at late times. This latter observation raises a second possibility, which is that the overexpression of 13 proteins inhibits viral DNAreplication to some extent. However, this explanation also appears unlikely because the n504R mutant shows elevated levels of 13 gene expression but is not defective for DNA replication. A third possibility, and the one that we favor, is that ICP27 stimulates DNA replication not by an effect on viral geneexpression but in a more direct capacity, perhaps by altering theposttranslational modification of host or viral proteins (26, 32, 44). Further experiments will be required to distinguish between these and otherpossibilities. It is ofinterest thatICP27 was identified as a stimulatory factor in the original ori-plasmid replication assays used to map the HSV-1 replication genes (50). At the time, it was suggested that ICP27 stimulated plasmid replication by transactivating one or more of the seven HSV-1 replication genes. However, we have shown that the dl-2 allele is not defective fortransactivation in a transfection assay but does lead to a viral DNAreplication defect when introduced into a recombinant virus. This finding suggests thatICP27 may stimulate DNA replication in the ori-plasmid replication assays by a mechanism other thantransactivation. It will be ofinterest to determine whether the dl-2 protein can stimu-late thereplication of an ori-plasmid in transfected cells.
ICP27 mediates multiple activities in infected cells. We previously suggested that ICP27 carries out two distinct regulatory functions during the HSV-1 infection (33). This hypothesis was based on the phenotypic analysis of several viralICP27 mutants, including the null mutant d27-1 and the C-terminal mutantn504R.Thed27-1 mutant showeddefects inviral DNAreplication, -y-1 gene expression, and -y-2 gene expression, whereas the n504R mutant exhibited only de-fects in -y-2 gene expression. Therefore, we proposed that ICP27 mediates two genetically separable activities in in-fected cells: (i) an activity that stimulates viral DNA repli-cation and fy-1 gene expression and (ii) an activity that transactivates -y-2 genes, independent of viral DNA replica-tion. Thephenotype of the dl-2 mutant can be considered in the context of this model if we hypothesize that it is defective in the first but not the second activity. This mutant exhibits adeficiency in -y-1 gene expression and viral DNA replication and shows a modest reduction in
y-2
gene ex-pression. However, the reduction in y-2geneexpression in dl-2-infected cells is consistent with the three- toeightfold defect in viral DNA replication that is observed and con-trasts quitedramatically with the more severe reductions in -y-2 gene expression which are observed in d27-1-infectedon November 9, 2019 by guest
http://jvi.asm.org/
cells
(in
which DNA replication is defective) and n5O4R-infected cells(in
which DNAreplication isnormal).However, someofourresults are notentirelyconsistent
with these
interpretation.
Forexample, d27-1-infected cells showastriking
defect in the induction ofICP5 and atleast twootherproteins
atearly
times(Fig. 5,4hpi).Thiseffect is notobserved in cellsinfectedby
dl-2orn504R. Therefore,d27-1- and dl-2-infected cells induce the -y-1
protein
ICP5 andsome otherproteins
withquite
different kinetics, eventhough they
appeartobesimilarly
defective for viral DNAreplication. Thus,
ICP27 may induce-y-l genesby
multiplemechanisms. In
addition,
ifthe defective functions ofdl-2 and n504R arecompletely independent,
then twomutants should becapable
ofintragenic complementation.
We have failedtoobserve suchcomplementation
(34), suggesting
thatthey
are notcompletely independent.
The currentevidence suggests that ICP27is a
multifunc-tional
protein.
It appearstoregulate
viral geneexpression
atboththe
transcriptional (18, 25)
andposttranscriptional
(38,42)
levelsand isrequired
for efficient viral DNAreplication(25, 33;
thisstudy).
Thefunctionalcomplexity
ofICP27 is underscoredbyourgenetic analyses. Ofthe six recombinant viruses that we haveisolated,
fivedisplay
demonstrablydifferent
phenotypes
with respect to viral gene expressionand DNA
replication (33;
thisstudy).
Theseresults suggest thatICP27,
like other viralregulatory
proteinssuch as the adenovirus ElAproteins (10)
and SV40 T antigen (9), iscomposed
ofmultiple
functional domains which mediate distinct activities.Although previous
studies havesuggestedthat the
important regulatory
activities ofICP27 aremedi-ated
by
itsC-terminalhalf,
ouranalysis
of thedl-2mutantindicates thattheN-terminalhalf also hasanessential role in
regulation.
The definitivemapping
of the functional domainsofICP27will
require
theisolation andanalysis
of additionalICP27gene mutations.
ACKNOWLEDGMENTS
WeareextremelygratefultoMin Gao forcarryingout someof the
preliminary characterization of the virus mutants. We are also indebted to Leslie Schiff, who read the manuscript and provided
experteditorialassistance.
This research was supported by an operating grant from the National Cancer Institute of Canada(to S.A.R.),anestablishment grant from the AlbertaHeritageFoundation for MedicalResearch
(toS.A.R.), andbyPublic Health ServicegrantAI20530 from the National Institute ofAllergyand Infectious Diseases(to D.M.K.). S.A.R.wassupported by aTerryFoxTeam DevelopmentAward from the NationalCancerInstitute of Canada.
REFERENCES
1. Ackermann, M.,D. K.Braun,L.Pereira,andB.Roizman. 1984. Characterization ofherpes simplexvirustype 1aproteins 0, 4, and27 with monoclonal antibodies.J.Virol. 52:108-118. 2. Ausubel, F.,R. Brent,R.Kingston, D.Moore, J. Seidman, J.
Smith,and K.Struhi (ed.).1987. Currentprotocolsin molecular
biology,p.3.5.9-3.5.10. John Wiley&Sons, Inc.,NewYork. 3. Block, T.,and R.Jordan. 1988.Herpes simplex virus type 1 a
genecontaining plasmidcan inhibitexpression regulatedfrom
an alpha promoter in CV-1 but not HeLa cells. Virus Res. 11:269-279.
4. Cai, W., S. Person, C. DebRoy, and B. Gu. 1988. Functional regionsand structuralfeatures of thegB glycoproteinofherpes
simplexvirustype 1: ananalysisoflinker insertionmutants.J.
Mol. Biol. 201:575-588.
5. Chapman,C.J.,J.D.Harris,M. A.Hardwicke,R.M.
Sandri-Goldin,M.K. L.Collins,and D. S.Latchman.1992.
Promoter-independentactivation ofheterologousvirusgeneexpression by
theherpes simplexvirusimmediate-early protein ICP27. Virol-ogy186:573-578.
6. Davison,A.J.,andP.Taylor. 1987. Genetic relations between varicella-zoster virus and Epstein-Barr virus. J. Gen. Virol. 68:1067-1079.
7. DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate early regulatory proteinICP4.J. Virol. 56:558-570.
8. Everett,R.D.1986. Theproductsofherpes simplex virustype 1(HSV-1) immediate-earlygenes1,2, and3canactivategene expression intrans.J. Gen. Virol. 68:2507-2513.
9. Fanning,E. 1992. Simian virus 40large T antigen: the puzzle, thepicture, andtheemerging picture. J. Virol. 66:1289-1293. 10. Flint, J.,and T.Shenk. 1989. AdenovirusElAprotein paradigm
viral transactivator.Annu. Rev. Genet. 23:141-161.
11. Frink,R.J.,R.Eisenberg,G.Cohen,andE. K.Wagner. 1983. Detailedanalysisof theportionof theherpes simplex virustype 1genomeencoding glycoprotein C. J. Virol. 45:634-647. 12. Gao, M.,and D. M.Knipe.1993.Intragenic complementation of
herpes simplex virus ICP8 DNA-binding protein mutants. J. Virol.67:876-885.
13. Godowski,P.J.,and D. M.Knipe. 1983. Mutations in themajor
DNA-binding proteinofherpes simplexvirus type 1 result in increased levels of viralgeneexpression. J. Virol. 47:478-486. 14. Godowski,P.J.,and D. M.Knipe. 1986.Transcriptional control of herpesvirus gene expression: gene functions required for positive and negative regulation. Proc.Natl. Acad. Sci. USA 83:256-260.
15. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombinant genomes which expresschloramphenicol acetyl-transferase in mammalian cells. Mol. Cell. Biol. 2:1044-1051. 16. Graham,F.L.,and A.J.vanderEb.1973.Anewtechniquefor
theassayof human adenovirus 5 DNA.Virology52:456467. 17. Hardwicke, M. A., P. J. Vaughn, R. E. Sekulovich, R. E.
O'Connor,and R.M.Sandri-Goldin. 1989.Theregions
impor-tantfor theactivatorand repressorfunctionsofherpessimplex virustype 1aprotein ICP27maptotheC-terminal half of the molecule.J.Virol.63:4590-4602.
18. Jean,S.,and D. M.Knipe.Unpublisheddata.
19. Kattar-Cooley,P.,and K. W. Wilcox. 1989.Characterization of theDNA-binding propertiesofherpes simplexvirusregulatory protein ICP4. J. Virol. 63:696-704.
20. Knipe,D.M.,D.Senechek,S.A.Rice,andJ.L. Smith.1987. Stages in the nuclear association of the herpes simplex virus transcriptionalactivatorproteinICP4. J. Virol. 61:276-284. 21. Knipe,D.M.,andA. E.Spang. 1982. Definitionofa series of
stages in the association oftwo herpesviralproteins with the hostcellnucleus.J. Virol. 43:314-324.
22. Kramer, W., V. Drutsa, H.-W. Jansen, B. Kramer, M.
Pflugfelder,andH.-J.Fritz.1984. Thegapped duplex approach
to oligonucleotide mutation construction. Nucleic Acids Res. 12:9441-9456.
23. Ma, J.,and M. Ptashne.1987. Anewclass ofyeast transcrip-tional activators.Cell51:113-119.
24. Maniatis, T.,E. F.Fritsch,andJ.Sambrook. 1982. Molecular
cloning: alaboratorymanual. ColdSpring Harbor Laboratory, ColdSpring Harbor,N.Y.
25. McCarthy, A. M., L. McMahan, and P. A. Schaffer. 1989.
Herpes simplex virus type 1 ICP27 deletion mutants exhibit alteredpatternsoftranscriptionandareDNAdeficient.J.Virol. 63:18-27.
26. McMahan, L., and P. A. Schaffer. 1990. The repressing and
enhancing functions of the herpes simplex virus regulatory
proteinICP27maptothe C-terminalregionsand are required to modulate viralgeneexpressionveryearly in infection.J. Virol. 64:3471-3485.
27. Nabel, G.,and D. Baltimore. 1987. Aninducible transcription factor activatesexpressionof humanimmunodeficiency virusin Tcells.Nature(London)326:711-713.
28. Nicholas,J.,U. A.Gompels,M.A.Craxton,R.W.Honess. 1988. Conservationof sequence and function between theproductof the 52-kilodalton immediate-earlygene ofherpesvirus saimiri
on November 9, 2019 by guest
http://jvi.asm.org/
and theBMLF1-encoded transcriptional effector (EB2) of Ep-stein-Barrvirus. J. Virol. 62:3250-3257.
29. Preston, C. M. 1979. Control of herpes simplex virus type 1 mRNAsynthesis in cells infected with wild-type virus or the
temperature-sensitive mutanttsK. J. Virol.29:275-284. 30. Quinlan, M. P., and D. M. Knipe. 1985.Stimulation of
expres-sion ofaherpessimplex virus DNA-bindingprotein bytwoviral functions. Mol. Cell. Biol.5:957-963.
31. Rice, S. A. Unpublished data.
32. Rice, S. A., and D. M. Knipe. 1988.Gene-specific transactiva-tion by herpes simplex virustype 1aprotein ICP27. J. Virol.
62:3814-3823.
33. Rice, S. A., and D. M. Knipe. 1990. Geneticevidence fortwo
distincttransactivation functionsof the herpes simplex virusa
protein ICP27. J. Virol. 64:1704-1715. 34. Rice, S. A., and V. Lam.Unpublished data.
35. Rice, S. A., L. Su, and D. M. Knipe. 1989. Herpes simplex virus
a protein ICP27 possesses separable positive and negative regulatory activities. J. Virol. 63:3399-3407.
36. Roizman, B., and A. E. Sears. 1990. Herpessimplex viruses and theirreplication,p.1795-1842. In B. N. Fieldsand D. M. Knipe (ed.), Fundamental virology. Raven Press, New York.
37. Sacks, W.R., C. C. Greene, D. P. Aschman, and P. A. Schaffer. 1985. Herpessimplex virustypeIICP27 isanessential
regula-toryprotein. J. Virol. 55:796-805.
38. Sandri-Goldin, R. M., and G. E. Mendoza. 1992. A herpesvirus regulatory protein appears to act post-transcriptionally by af-fecting mRNA processing. Genes Dev.6:848-863.
39. Sekulovich,R.E.,K.Leary,and R. M.Sandri-Goldin. 1988.The
herpes simplex virus type 1 a protein can act as a
trans-repressor or a trans-activator in combination with ICP4 and
ICPO. J.Virol. 62:4510-4522.
40. Shepard, A. A., and N. A. DeLuca. 1991. Asecond-siterevertant ofadefectiveherpes simplex virus ICP4 protein with restored regulatory activities and impaired DNA binding properties. J.
Virol.65:787-795.
41. Smiley, J. R., D. C. Johnson, L.I.Pizer, and R. D. Everett. 1992. The ICP4 binding sites in the herpes simplex virus type 1
glycoprotein D (gD)promoter are notessential for efficient gD transcription during virus infection. J. Virol. 66:623-631. 42. Smith,I.L.,M. A. Hardwicke, and R. M.Sandri-Goldin. 1992.
Evidencethat the herpessimplex virus immediate early protein ICP27 acts post-transcriptionally during infection to regulate geneexpression. Virology 186:74-86.
43. Struhl,K. 1991.Acidconnections. Curr.Biol. 1:188-191. 44. Su,L., and D. M.Knipe. 1989.Herpessimplexvirusaprotein
ICP27caninhibitoraugment viralgenetransactivation. Virol-ogy170:496-504.
45. Telford, E.A., M.S.Watson, K.McBride, and A. J. Davison. 1992. The DNA sequence of equine herpesvirus-1. Virology 189:304-316.
46. Vaughn, P. J., K. J.Thibault, M. A. Hardwicke, and R. M. Sandri-Goldin. 1992. Theherpes simplex virus immediate early protein ICP27 encodes a potential metal binding domain and binds zincin vitro.Virology189:377-384.
47. Watson, R. J., and J. B.Clements.1980. Aherpes simplex virus type 1 function continuously required for early and late virus RNAsynthesis. Nature(London) 285:329-330.
48. Weller, S. K.1991. Genetic analysis of HSV-1 genesrequired for genome replication, p. 105-136. In E. K. Wagner (ed.), Herpesvirus transcription anditsregulation. CRCPress, Boca Raton, Fla.
49. Wilcox,K.W., A. Kohn, E.Sklyanskaya,and B.Roizman. 1980. Herpessimplex virusphosphoproteins. I.Phosphate cycleson
andoffsomeviralpolypeptides andcan alter theiraffinity for DNA.J. Virol. 33:167-182.
50. Wu,C.A., N. J. Nelson, D. McGeoch, and M. D. Challberg. 1988. Identification of herpes simplexvirus type 1 genes
re-quired for origin-dependent DNA synthesis. J. Virol. 62:435-443.
51. Zhao, Y., V. R. Holden, R. N. Harty, and D.J. O'Callaghan. 1992.Identification andtranscriptional analyses of the UL3 and UL4genesofequine herpesvirus 1,homologsof theICP27 and glycoproteinK genesofherpessimplex virus.J. Virol.
66:5363-5372.