0022-538X/00/$04.00⫹0
Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Suppression of Acute Viremia by Short-Term Postexposure Prophylaxis
of Simian/Human Immunodeficiency Virus SHIV-RT-Infected Monkeys
with a Novel Reverse Transcriptase Inhibitor (GW420867) Allows
for Development of Potent Antiviral Immune Responses
Resulting in Efficient Containment of Infection
KAZUYASU MORI,1,2* YASUHIRO YASUTOMI,3SHUZO SAWADA,4FRANCOIS VILLINGER,5 KAZUSHIGE SUGAMA,6BRIGITTE ROSENWITH,7JONATHAN L. HEENEY,7KLAUS U¨ BERLA,8
SHUDO YAMAZAKI,1AFTAB A. ANSARI,5ANDHELGA RU¨ BSAMEN-WAIGMANN9*
AIDS Research Center1and Tsukuba Primate Center for Medical Sciences,2National Institute of Infectious Diseases, Tokyo 162-8640, Department of Bioregulation, Mie University School of Medicine, Tsu 514-8507,3Bayer Yakuhin, Osaka
532-0003,4and Omtest Laboratory, Kyurin Corporation, Kitakyushu 806-0046,6Japan; Department of Pathology and Laboratory Medicine, Emory University School of Medicine, Atlanta, Georgia 303225; Department of
Virology, Biomedical Primate Research Center, 2288 GH Rijswijk, The Netherlands7;and Institut fu¨r Virologie, Universita¨t Leipzig, D-04103 Leipzig,8and Department of Virus Research,
Pharma Research Center, Bayer AG, D-42096 Wuppertal,9Germany Received 19 January 2000/Accepted 3 April 2000
A nonnucleoside reverse transcriptase (RT) inhibitor, GW420867, was tested for postexposure prophylaxis (PEP) in rhesus macaques experimentally infected with 100 50% tissue culture infective doses of a chimeric simian/human immunodeficiency virus (SHIV) containing the RT gene of HIV-1 (SHIV-RT). Animals were ei-ther mock treated, or treated for 4 weeks starting at 8 or 24 h postinfection (p.i.) with GW420867. While such therapy led to undetectable plasma viremia in three of six monkeys, a transient plasma viremia was noted in the other three treated animals at 2 to 4 weeks following cessation of therapy. Following this transient viremia all drug-treated animals showed low or undetectable levels of plasma viremia up to the last sample examined at 90 weeks p.i. Despite low and/or undetectable viremia, virus-specific cytotoxic T lymphocyte and viral Env-specific proliferative responses were seen in the peripheral blood mononuclear cells of both mock- and drug-treated animals as early as 3 weeks p.i. Such virus-specific cellular responses, however, were better maintained in the drug-treated animals than the mock-treated animals. In contrast to the virus-specific cellular response, the magnitude and kinetics of virus specific humoral responses appeared to correlate with the detection of viremia. These data support the view that a short-term PEP with GW420867 permits the generation and main-tenance of long-lasting virus-specific cell-mediated immune responses while markedly reducing viral loads to undetectable levels for a prolonged period of time (90 weeks) and leads to long-term disease protection. This model provides a unique means to define mechanisms and correlates of disease protection.
A dramatic decrease in viral load coincident with a recovery of CD4⫹T-cell counts has been noted in human
immunode-ficiency virus (HIV)-infected patients following highly active antiretroviral therapy (HAART) (2, 10, 11). However, it has been noted that while such therapy does lead to recovery of CD4⫹T-cell levels, it fails to establish effective anti-HIV
im-mune responses, as evidenced by a rebound of viremia which has been noted among HIV-infected individuals in whom HAART was discontinued (31). Of great concern is the finding that latent HIV infection may persist for more than several decades even in successful HAART cases (7, 27, 30). These findings indicate a need for defining therapeutic modalities that will not only reduce viral load but concomitantly allow for
the induction of a quality and quantity of immune responses that will continue to maintain low viral loads and gradually either eliminate latent virus-infected cells and/or reduce and/or eliminate the likelihood of disease. However, the precise na-ture of the host immune response(s) that is capable of con-taining viral load and/or preventing disease remains to be de-fined. Some insights have been gained on this issue by the study of the host antivirus-specific immune responses in HIV type 1 (HIV-1)-infected long-term nonprogressors and in nonhuman primates infected with attenuated simian immunodeficiency virus (SIV) viral constructs (13, 15). However, to date, there have been limited if any studies of the use of chemotherapeutic drugs that reduce viral loads yet allow for the development of the quality and quantity of antiviral immune responses out-lined above.
Our laboratory has been studying a nonnucleosidic class of the quinoxalines as inhibitors of HIV-1 reverse transcriptase (RT), using HBY 097 as the first clinical candidate (16, 19, 20). A search for a compound with improved pharmacokinetics led to the discovery of HBY 1293A (now GW420867), which al-lows a single daily dosing to reach and maintain therapeutic levels in humans (data not shown). Toward the goal of deter-mining the efficacy of this drug, we utilized rhesus macaques * Corresponding author. Mailing address for Kazuyasu Mori:
Tsukuba Primate Center for Medical Sciences, National Institute for Infectious Diseases, 1 Hachimandai, Tsukuba 305-0843, Japan. Phone: 81-298-37-2121. Fax: 81-298-37-0218. E-mail: [email protected] .jp. Mailing address for Helga Ru¨bsamen-Waigmann: Department of Virus Research PH-R AI Virology Pharma Research Centre, Bayer AG, Aprather Weg, Postfach 101709, D-42096 Wuppertal, Germany. Phone: 49-0202-36-4143. Fax: 49-202-36-4162. E-mail: [email protected].
5747
on November 9, 2019 by guest
http://jvi.asm.org/
potentially identify disease-protective immune responses. Re-sults of the studies performed constitute the basis of this re-port.
MATERIALS AND METHODS
Virus.The chimeric SHIV-RT consists of a SIVmac239 virus backbone in which the SIV RT gene was replaced by the HIV-1 HxB2 RT gene as previously described (25). This SHIV-RT has been shown to induce AIDS in experimentally infected rhesus monkeys (23, 25). The parent viral stocks were prepared by DNA transfection of proviral DNA into COS-1 cells. The virus stock used in the studies was prepared by propagation of the COS-1 virus stock in phytohemagglutinin-activated rhesus monkey peripheral blood mononuclear cells (PBMC) (14). The p27 antigen concentration of the virus stock was determined by using a com-mercial SIV Gag antigen enzyme-linked immunosorbent assay (ELISA) kit (Coulter, Tokyo, Japan). The TCID50of the virus stock was determined utilizing herpesvirus saimiri-transformed cynomolgus CD4⫹T cells (1).
GW420867 (formerly HBY 1293A).A quinoxaline class of the nonnucleoside RT inhibitors [S -3-ethyl-6-fluoro-4-isoproxycarbonyl-3,4-dihydro-quinoxaline-2(1H)-one] was provided initially by Hoechst and Bayer AG (Wuppertal, Ger-many) and subsequently by Glaxo Wellcome (Greenford, Middlesex, United Kingdom). The compound was dissolved with dimethyl sulfoxide at 1 g per ml, and then aliquots were stored at⫺20°C. Prior to administration into animals, the compound was diluted with a solvent consisting of polyethylene glycol 400, glycerol, and water (960:60:100).
Animal experiment study plan.Nine rhesus macaques (male, 2 years old, 2 to 3 kg) were screened and found to be seronegative for SIV, simian T-cell leuke-mia virus, B virus, and type D retroviruses prior to initiation of the study. All animals were housed in individual cages and maintained according to the rules and regulations of the National Institute of Infectious Diseases and guidelines for experimental animal welfare. GW420867 was administered subcutaneously to the animals at a dose of 15 mg per kg of body weight, twice a day (12-h interval) for a total of 28 days. All animals were infected intravenously with 100 TCID50 of SHIV-RT. The first group was treated with GW420867 starting at 8 h p.i. and then received treatment twice daily at 12-h intervals for 28 days. The second group was treated with GW420867 starting at 24 h p.i. and then received treat-ment twice daily for 28 days. The third group was administered an equivalent dose of the solvent that was used to dissolve GW420867 starting at 8 h p.i. and then received treatment twice daily for 28 days and served as a control group.
Pharmacokinetics of GW420697.Initial in vitro and in vivo studies were performed in an effort to define the optimal dosage to be administered to the monkeys. The 50% inhibitory concentration of this drug for SHIV-RT is com-parable to the value previously defined for several subtype B field isolates of HIV, which ranged from 0.15 to 1.4 nM utilizing human PBMC cultures (data not shown). The 90% inhibitory concentration of the drug determined using a macaque CD4⫹T-cell line (1) was found to be 8 nM (2.24 ng per ml). We estimated that the effective minimal in vivo concentration in blood to achieve for GW420867 would be 50 ng per ml. During our experimental study, in which the monkeys were subcutaneously administered the compound at 15 mg per kg of body weight twice daily for 4 weeks, the drug level maintained in the blood was significantly higher than the calculated minimal effective concentration (data not shown).
Sampling of specimen.Blood samples were collected from each of the nine animals prior to the initiation of the study (baseline) and then at regular time intervals as specified for each of the assays described herein. Biopsies of inguinal lymph nodes from one animal in the control group and from one animal in each of the two drug treatment groups were performed at 8 weeks p.i.
Plasma viral load.Viral RNA was purified from 0.2 ml of plasma with a commercial viral RNA isolation kit (Boehringer Mannheim, Tokyo, Japan) and
separated on a 2% agarose gel by electrophoresis and stained with SYBR green (Takara). Measurement of amplified RNA was achieved utilizing a fluorescence imaging analyzer (FLA2000; Fuji Film, Tokyo, Japan), and the viral RNA load was calculated using an Excel spreadsheet (Microsoft, Tokyo, Japan) as previ-ously described (26). The sensitivity of plasma viral RNA detection by this technique was determined to be 1,000 copies per ml of plasma.
Proviral DNA load.PBMC samples were purified from heparinized blood utilizing standard Ficoll-Hypaque gradient centrifugation. Cellular DNA was purified from 106cells with a commercial DNA purification kit (Qiagen, Tokyo, Japan). ThegagDNA was amplified with a commercial DNA PCR kit with Ex
TaqDNA polymerase (Takara). Viral DNA standards were prepared by dilution of the plasmid containing the 5⬘half of SIVmac239 (17). Thenefsequence of viral DNA was amplified to determine the proviral DNA load in samples which had fewer than 10 copies by nested PCRs using two sets of primer pairs as previously described (5). The first primer pair included F34C (nucleotides [nt] 9065 to 9082) (5⬘-CCTACCTACAATATGGGT-3⬘) and F35C (nt 9800 to 9778) (5⬘-CCTCTGACAGGCCTGACTTGCTT-3⬘), and the second primer pair in-cluded N3 (nt 9182 to 9201) (5⬘-GAAGATGGATACTCGCAATC-3⬘) and N4 (nt 9552 to 9533) (5⬘-TAATCCTGCCAATCTGGTAT-3⬘). The sensitivity of this technique was determined to be one copy per 100,000 cells. The relative numbers of PBMC and lymph node mononuclear cells (LNMC) harboring pro-viral DNA was determined by the analysis of DNA from fourfold dilutions of the initial 105cells run in parallel with the viral DNA standards described above. DNAs from the samples were separated by agarose gel electrophoresis and stained with SYBR green, and quantification was achieved using a fluorescence imaging analyzer (FLA2000, Fuji Film).
Quantitative virus isolation (QVI).Fourfold dilutions of PBMC (starting with 106in 0.5 ml) were cocultured with 2.5⫻105C8166 cells (in 0.5 ml) in duplicate wells of 24-well plates. The cocultures were incubated for 3 to 4 weeks, and individual wells were scored for the presence of syncytia. Culture supernatants were subjected to RNA PCR for the detection of viral RNA to confirm viral replication as previously described (23).
Anti-SIV ELISA.A 1:100 dilution of each plasma sample in phosphate-buff-ered saline (pH 7.4) containing a blocking reagent (Dainippon Seiyaku, Osaka, Japan) was assayed for the presence of SIV-specific antibodies using stan-dard ELISA techniques. The 96-well microtiter plates were precoated with a SIVmac239 virion lysate as previously described (14). The OD492was recorded and utilized as a relative measure of antibody titer.
CTL assay.The cytotoxic T-lymphocyte (CTL) assay method used has been previously described (30). In brief, PBMC samples stored at ⫺150°C were thawed and cultured in RPMI 1640 medium with concanavalin A (5g per ml) at 106PBMC per ml for 3 days, washed and then maintained for another 3 days in medium supplemented with human interleukin 2 (2 U per ml). These effector cells were then cocultured for 7 days with autologous herpesvirus papio-trans-formed B-lymphoblastoid cell lines (B-LCL), which were previously either in-fected with recombinant vaccinia virus (rVV) expressing the SIVmac239gag-pol, SIVmac239env, or the parental VV (NYCBH strain) for 16 h at 37°C. The VV constructs were obtained via the courtesy of D. Panicali (Therion Corp., Cam-bridge, Mass.). For the CTL assay, similarly infected autologous (B-LCL) target cells were labeled with51Cr and then incubated at 104cells/well with various concentrations of effector cells for 5 h. Supernatant fluids from each of the target cells (104cells per well) incubated alone with medium was used to calculate spontaneous release, and supernatant fluid from each of the target cells (104cells per well) incubated with Triton X-100 was used to calculate 100% release. Each effector-to-target cell combination was performed in triplicate. Specific net lysis was calculated as the percentage of SIV Env- or Gag-Pol-specific lysis minus the percentage of lysis obtained using the control VV (NYCBH)-infected target cells. The control lysis was always⬍10%.
SIV Env-specific cell proliferation.The method utilized has been previously described (13). In brief, PBMC samples were thawed and triplicate cultures of
on November 9, 2019 by guest
PBMC (4⫻105per well) were incubated with irradiated autologous B-LCL (1⫻ 105 per well) that had been previously infected (for 16 h) with either VV containing SIVmac239envor control VV. Cultures were performed in triplicate, and [3H]thymidine was added to each well 8 h prior to harvest on day 5. The incorporation of [3H]thymidine was measured by standard liquid scintillation counting. The standard deviation of the triplicate samples was⬍10%. The stimulation index was calculated by dividing the mean uptake of [3H]thymidine values of the PBMC cocultured with the VV SIVenv-infected autologous cells by the mean uptake of [3H]thymidine values obtained by coculture of an aliquot of the same PBMC with the VV (NYCBH)-infected autologous cells. Because depletion of CD4⫹cells from PBMC at 8 weeks p.i. with monoclonal antibody 19thy5D7 (17) in the presence of complement resulted in the loss of theenv -specific proliferative response in the positive PBMC specimen, the majority of the cells stimulated with Env proteins were assumed to be CD4⫹T cells.
RESULTS
Containment of SHIV infection by short-term post-exposure
prophylaxis with GW420867. We used GW420867 to treat
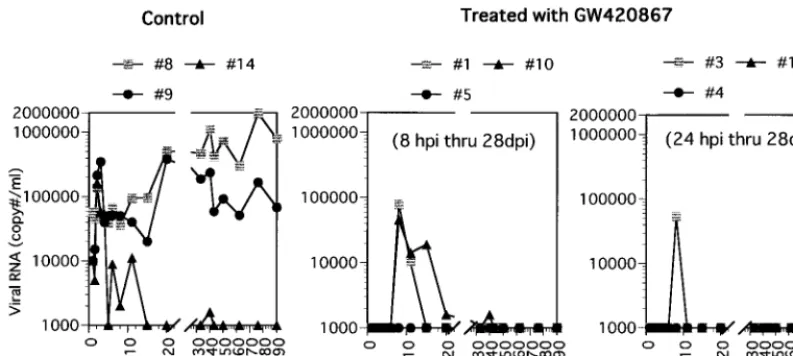
monkeys that were infected with a high dose (100 TCID50) of SHIV-RT. Treatment was initiated at 8 or 24 h p.i., with the drug or vehicle (in control animals) being administered twice a day for a total of 4 weeks. The effect of GW420867 therapy on plasma and cellular viral load was examined in samples ob-tained from each of the nine animals at various times p.i. As seen in Fig. 1, all three mock-treated animals showed signifi-cant plasma viremia, which reached set points in two of three animals shortly following the acute viremia phase. The third mock-treated animal showed transient plasma viremia follow-ing the acute phase but thereafter showed low levels of plasma viremia. In contrast, all six drug-treated animals showed unde-tectable levels (below the limits of RT-PCR detection, which was determined to be 1,000 copies/ml) of plasma viremia up to 3 weeks postcessation of therapy, regardless of the difference in the time of drug therapy initiation. Thereafter, three of six animals showed transient plasma viremia at about 4 weeks following cessation of drug therapy, which subsequently re-turned to undetectable levels. Studies of the cellular viral loads as determined by QVI showed a significant number of virus-infected cells in the mock-treated animals at various time points following infection. The frequency of virus replication-competent infected cells decreased with time in the control animals, presumably due to trapping of such cells in regional lymph nodes. In comparison, low but detectable levels of in-fected cells were noted in three of six drug-treated animals between 6 and 11 weeks p.i., a low level was detected at a single
time point in two of six drug-treated animals, and undetectable levels were noted in all the other remaining samples tested (Table 1).
Subsequent analyses of samples obtained at 50, 62, 76, and 90 weeks p.i. indicated that plasma viral RNA levels were maintained below 1,000 copies/ml in each of the six drug-treated animals. Results of the proviral DNA analysis (Table 2) correlated with the plasma viral loads. No proviral DNA was detected in PBMC samples at any time during antiviral treat-ment, while it was readily detectable in samples from the con-trol group early p.i. Also at later time points, a far lower level of infection was noted in the drug-treated group relative to the control group. These data nevertheless demonstrate that de-spite drug therapy, all the animals in both groups became infected, as evidenced by the detection of SHIV-RT DNA sequences at least at one time point in the PBMC and/or LNMC of each of these animals.
Humoral response in SHIV-infected monkeys treated with
GW420867. We studied the humoral response against
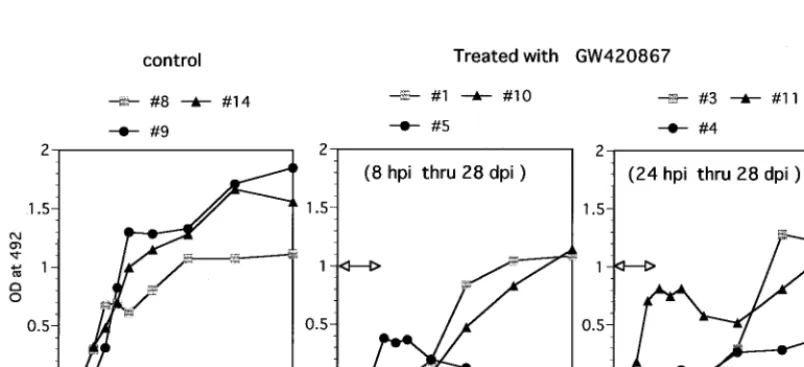
[image:3.612.104.501.73.251.2]SHIV-RT by using ELISA against SIV virion proteins. Infected but mock-treated (control) animals became seropositive at 4 weeks p.i.; thereafter, high antibody titers were sustained (Fig. 2). In contrast, a varied pattern of anti-SIV humoral response was noted in the drug-treated animals. One drug-treated animal (animal 5) showed a weak but positive transient antibody re-sponse (6 to 8 weeks p.i.) which was lost by 10 weeks p.i.
TABLE 1. QVI
Wk p.i.
Cellular viral load determined by quantitative virus isolation for animal no.a:
8b 9b 14b 1c 5c 10c 3d 4d 11d
2 16 (⫹) 16 (⫹) 16 (⫹) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺) 6 16 (⫹) 16 (⫹) 4 (⫹) 4 (⫹) 0 (⫺) 1 (⫹) 16 (⫹) 0 (⫺) 0 (⫺) 8 64 (⫹) 16 (⫹) 1 (⫹) 16 (⫹) 0 (⫺) 16 (⫹) 16 (⫹) 1 (⫹) 0 (⫺) 11 16 (⫹) 1 (⫹) 0 (⫺) 4 (⫹) 0 (⫺) 4 (⫹) 16 (⫹) 0 (⫺) 4 (⫹) 15 4 (⫹) 1 (⫹) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺) 0 (⫺)
aResults are given as the number of infected cells/106PBMC followed by the results of viral RNA PCR performed with supernatants of the cultures (⫹, positive;⫺, negative).
bControl.
[image:3.612.311.553.593.678.2]cGW420867 treatment 8 h p.i. through 28 days p.i. dGW420867 treatment 24 h p.i. through 28 days p.i.
FIG. 1. Plasma viral RNA in SHIV-RT-infected animals treated with GW420867 or left untreated. Viral RNA levels in the plasma are expressed as copy number per milliliter.
on November 9, 2019 by guest
http://jvi.asm.org/
despite undetectable viral loads in this animal (Fig. 1 and Table 1). A lymph node biopsy done on this animal showed that this animal was indeed infected but had low levels of proviral DNA (Table 2). Another drug-treated animal (animal 11) showed an initial antiviral antibody response similar to those seen in the control animals but then showed a transient decrease, with a return to high levels by 18 to 20 weeks p.i. This presumably anamnestic response followed the detection of in-fectious virus in PBMC samples from this animal at 11 weeks p.i. (Table 1). The relatively early seroconversion of drug-treated animals 5 and 11 suggests that the limited viral repli-cation that occurred in these two animals may have led to sufficient availability of antigen to induce antibody responses. Yet another drug-treated animal (animal 4) seroconverted by 11 weeks p.i. but maintained low but significant SIV-specific antibody titers relative to the other remaining three animals. The other three drug-treated animals showed a delay in the kinetics of the humoral antiviral response, with seroconversion at 11 weeks p.i., corresponding to 7 weeks postcessation of drug therapy. In general, therefore, the kinetics of antiviral antibody generation appeared to follow the levels of viremia
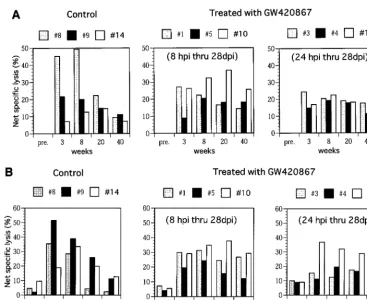
[image:4.612.54.293.82.178.2]in one of three control animals (animal 14), there was a delay in the kinetics of CTL activity despite similar levels of acute viremia in this animal (Fig. 1). In contrast to the control ani-mals, all of the drug-treated animals showed significant SIV Gag-Pol- and Env-specific CTL activity in samples from as early as 3 weeks p.i., despite the absence of detectable viral replication during the drug treatment period. More impor-tantly, again in contrast to the control animals, the CTL levels were maintained up to 40 weeks p.i. with minor variations (Fig. 3). It is important to note that viral load by itself does not account for such virus-specific CTL responses, but viral control may be due to other host responses in addition to competent helper T cells. These findings are reminiscent of the data that have documented the maintenance of HIV antigen-specific CTL responses seen in the PBMC of long-term nonprogressors in contrast to those individuals who progress to AIDS (13, 18). In addition to the virus-specific CTL response, the SIV env-specific proliferative responses in these animals were also stud-ied. Results showed that the PBMC from the control animals demonstrated a transient but weak proliferative response at 3 weeks p.i. This response remained low throughout the period of assay in two of the control animals (animals 8 and 9) (Fig. 4) and increased in the remaining animal (animal 14) that showed naturally developed good long-term control of viremia (Fig. 1).
FIG. 2. Humoral response against SHIV-RT. Shown are results of antibody ELISA analysis of plasma samples from the control animals and GW420867-treated animals. A 1:100 dilution of each plasma sample in phosphate-buffered saline (pH 7.4) containing a blocking reagent was assayed for SIV-specific antibody by using standard ELISA techniques with 96-well plates precoated with SIVmac239 virion lysate (14). The OD492was recorded and used as a relative measure of antibody titer.
The duration of drug treatment is indicated by the bar with arrowheads.
on November 9, 2019 by guest
[image:4.612.101.503.509.692.2]In marked contrast, PBMC samples from the drug-treated ani-mals showed significantly higher SIVenv-specific proliferative responses as early as 3 weeks p.i., and these responses were maintained throughout the period of assay. In select experi-ments, depletion of CD4⫹ T cells prior to the proliferation
assay led to marked diminution of the proliferative response, indicating indeed that the proliferative response involved CD4⫹T cells in these assays.
DISCUSSION
One of the goals of this study was to investigate whether treatment of monkeys as early as 8 to 24 h p.i. would be able to prevent the establishment of SHIV infection in vivo, as shown in previous studies using (R)-9-(2-phosphonylmethoxypropyl) adenine (PMPA) (24) and zidovudine, respectively (25). Clearly, GW420867 postexposure prophylaxis monotherapy
[image:5.612.120.487.69.369.2]FIG. 3. Virus-specific cellular immunity CTL activity in the PBMC samples of control and GW420867-treated animals was determined by using autologous B-LCL target cells previously infected with a VV construct containing SIVmacgag-pol(A) and a VV construct containing SIVmacenv(B). In addition, controls used B-LCL infected with VV wild type (NYCBH strain). The percentage of net specific lysis was calculated for each effector-to-target cell combination, and this value was subtracted from the value obtained with the wild-type VV target cell control. Only results of effector/target ratio of 80:1 are shown. pre., preinfection.
FIG. 4. In vitro proliferative response of PBMC against SIVmacenv-rVV-infected autologous B-LCL. PBMC used for this assay were aliquots of the same specimen used for the CTL assay. The stimulation index was calculated as the mean counts per minute of PBMC cultured withenv-rVV divided by the mean counts per minute of PBMC cultured with control VV. pre., preinfection.
on November 9, 2019 by guest
http://jvi.asm.org/
quired between very limited viral replication (below detectable levels) and sufficient pathways of antigen presentation to evoke long-lasting disease-protective host antiviral immune functions in the drug-treated animals. This balance appeared to have been preserved by early short-term treatment with this novel RT inhibitor. In addition, very low continuous or intermittent viral replication presumably results in a quality (viral epitope specificity) of virus-specific humoral response that may also play a role in disease protection. Thus, induction of such hu-moral response during the treatment period correlated with a more-effective restriction of virus replication in the treated animals. The third finding is that antiviral treatment during acute infection appeared to preserve mechanisms responsible for the development and maintenance of an antiviral host defense that included CTL effector function, which was most likely preserved by the continuous support of CD4⫹T helper
cell function in addition to other as yet undefined mechanisms. Because there was a more significant difference inenv-specific proliferative response than in CTL response between the treated groups and the control group, virus-specific CD4⫹T
cells may play a more important role than CTLs. The fourth finding is that it is possible that there are as yet to be defined additional effector mechanisms other than CTLs that play im-portant roles for the containment of the virus infection. This view is based on the results seen in one of the untreated control animals that suppressed acute viremia despite a relatively low CTL response during primary infection and another control animal that suppressed viremia weakly despite a relatively po-tent virus-specific CTL response. Chemokines and other cell-free factors have been identified as candidates for such unde-fined effector mechanisms (4, 8, 22). It is thus possible that virus infection-induced activation of CD4⫹and CD8⫹T cells
may function to secrete high levels of cell-free factors that play a dominant role in these select animals. Thus, orchestrated antiviral host responses consisting of mechanisms that permit low levels of viral replication sufficient to induce and maintain appropriate levels of virus-specific CD4⫹T helper responses,
which in turn facilitate the generation and maintenance of virus-specific CTL responses, in addition to undefined mecha-nisms may be the basis for the protective effect observed in the drug-treated animals. Support for this view has been recently documented with the use of a live attenuated SIV⌬nef immu-nization protocol (9). A more detailed characterization of the precise epitopes of the virus that permit the generation of the helper CD4 and cytotoxic T-cell responses coupled with iden-tification of the contributory role, if any, of the other effector immune mechanism(s) in such models, including the one de-scribed herein, may provide insights on the nature of the
im-influence of virus infection on host immune responses. Thus, the chemotherapy-assisted containment of the pathogenic vi-rus infection may differ quantitatively and qualitatively from the attenuated SIV infection in the context of protection from pathogenic infection. Recent data on a few HIV-1 infected patients for whom HAART was initiated early p.i. support the data reported herein, since discontinuation of HAART in these individuals did not result in viral rebound, suggesting that HAART may be able to induce immune responses capa-ble of containing viral loads (3). In contrast, as previously noted, discontinuation of HAART therapy initiated at later stages of HIV-1 infection did not lead to the induction of immune responses sufficient to contain viral rebound (31).
In the current controversy of early versus late therapy fol-lowing exposure to HIV, our results strongly suggest that ini-tiation of therapy past the acute viral infection stage may be too late and prevent the optimal development of antiviral re-sponse. If human studies come to the same conclusion, early treatment of a (presumed) HIV infection by selected well-tolerated and convenient drugs must become the rule, when-ever possible.
ACKNOWLEDGMENTS
We appreciate the generosity of and thank D. L. Panicali (Therion Biologics) for providing the recombinant vaccinia viruses expressing SIVmac239env, SIVmac251gag-pol, and the parental virus (NYBCH strain).
This work was supported by AIDS research grants from the Health Sciences Foundation and the Organization for Pharmaceutical Safety and Research in Japan.
REFERENCES
1.Akari, H., K. Mori, K. Terao, I. Otani, M. Fukasawa, R. Mukai, and Y. Yoshikawa.1996. In vitro immortalization of Old World monkey T lympho-cytes withHerpesvirus Saimiri. Virology218:382–388.
2.Autran, B., G. Carcelain, T. S. Li, C. Blanc, D. Mathez, R. Tubiana, C. Katlama, P. Debre, and J. Leibowitch.1997. Positive effects of combined antiretroviral therapy on CD4⫹T cell homeostasis and function in advanced HIV disease. Science277:112–116.
3.Balter, M.1999. Can immune systems be trained to fight HIV? Science 286:1470–1471.
4.Cocchi, F., A. L. Devico, A. Garzino-Demo, S. K. Arya, R. C. Gallo, and P. Lusso.1995. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8⫹T cells. Science270: 1811–1815.
5.Daniel, M. D., K. Kirchhoff, S. C. Czajak, P. K. Shegal, and R. C. Desrosiers. 1992. Protective effects of a live attenuated SIV vaccine with a deletion in the
nefgene. Science258:1938–1941.
6.Desrosiers, R. C., J. D. Lifson, J. S. Gibbs, S. C. Czajak, A. Y. Howe, L. O. Arthur, and R. P. Johnson.1998. Identification of highly attenuated mutants of simian immunodeficiency virus. J. Virol.72:1431–1437.
7.Finzi, D., J. Blankson, J. D. Siliciano, J. B. Margolick, K. Chadwick, T.
on November 9, 2019 by guest
Pierson, K. Smith, J. Lisziewick, F. Lori, C. Flexner, T. C. Quinn, R. E. Chaisson, E. Rosenberg, B. Walker, S. Gange, J. Gallant, and R. F. Siliciano. 1999. Latent infection of CD4⫹T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med.5:512–517.
8.Garzino-Demo, A., R. B. Moss, J. B. Margolick, F. Cleghorn, A. Sill, W. A. Blattner, F. Cocchi, D. J. Carlo, A. L. Devico, and R. C. Gallo.1999. Spon-taneous and antigen-induced production of HIV-inhibitory beta-chemokines are associated with AIDS-free status. Proc. Natl. Acad. Sci. USA96:11986– 11991.
9.Gauduin, M.-C., R. L. Glickman, S. Ahmad, T. Yilma, and R. P. Johnson. 1999. Immunization with live attenuated simian immunodeficiency virus in-duces strong type 1 T helper responses and beta-chemokine production. Proc. Natl. Acad. Sci. USA96:14031–14036.
10. Gulick, R. M., J. W. Mellors, D. Havlir, J. J. Eron, C. Gonzalez, D. McMa-hon, D. D. Richman, F. T. Valentine, L. Jonas, A. Meibohm, E. A. Emini, and J. A. Chodakewitz.1997. Treatment with indinavir, zidovudine, and lamivu-dine in adults with human immunodeficiency virus infection and prior anti-retroviral therapy. N. Engl. J. Med.337:734–739.
11. Hammer, S. M., K. E. Squires, M. D. Hughes, J. M. Grimes, L. M. Demeter, J. S. Currier, J. J. Eron, Jr., J. E. Feinberg, H. H. Balfour, L. R. Deyton, J. A. Chodakewitz, and M. A. Fischl.1997. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. N. Engl. J. Med.337:725–733.
12. Hiranuma, K., S. Tamaki, Y. Nishimura, S. Kusuki, M. Isogawa, G. Kim, M. Kaito, K. Kuribayashi, Y. Adachi, and Y. Yasutomi.1999. Helper T cell determinant peptide contributes to induction of cellular immune responses by peptide vaccines against hepatitis C virus. J. Gen. Virol.80:187–193. 13. Johnson, R. P., R. L. Glickman, J. Q. Yang, A. Kaur, J. T. Dion, M. J.
Mulligan, and R. C. Desrosiers.1997. Induction of vigorous cytotoxic T-lymphocyte responses by live attenuated simian immunodeficiency virus. J. Virol.71:7711–7718.
14. Kestler, H. W., III, D. J. Ringler, K. Mori, D. L. Panicali, P. K. Sehgal, M. D. Daniel, and R. C. Desrosiers.1991. Importance of thenefgene for mainte-nance of high virus loads and for development of AIDS. Cell65:651–662. 15. Klein, M. R., C. A. van Baalen, A. M. Holwerda, S. R. Kerkhof Garde, R. J.
Bende, I. P. M. Keet, J. M. Eeftinck-Schatenkerk, A. D. Osterhaus, H. Schuitemaker, and F. Miedema.1995. Kinetics of Gag-specific cytotoxic T lymphocyte responses during the clinical course of HIV-1 infection: a lon-gitudinal analysis of rapid progressors and long-term asymptomatics. J. Exp. Med.181:1365–1372.
16. Kleim, J. P., R. Bender, R. Kirsch, C. Meichsner, A. Paessens, M. Rosner, H. Rubsamen-Waigmann, R. Kaiser, M. Wichers, K. E. Schneweis, I. Winkler, and G. Riess.1995. Preclinical evaluation of HBY 097, a new nonnucleoside reverse transcriptase inhibitor of human immunodeficiency virus type 1 rep-lication. Antimicrob. Agents. Chemother.39:2253–2257.
17. Mori, K., D. J. Ringler, and R. C. Desrosiers.1993. Restricted replication of simian immunodeficiency virus strain 239 in macrophages is determined by
envbut is not due to restricted entry. J. Virol.67:2807–2814.
18. Rosenberg, E. S., J. M. Billingsley, A. M. Caliendo, S. L. Boswell, P. E. Sax,
S. A. Kalams, and B. D. Walker.1997. Vigorous HIV-1-specific CD4⫹T cell responses associated with control of viremia. Science278:1447–1450. 19. Rubsamen-Waigmann, H., E. Huguenel, A. Paessens, J. P. Kleim, M. A.
Wainberg, and A. Shah.1997. Second-generation non-nucleosidic reverse transcriptase inhibitor HBY 097 and HIV-1 viral load. Lancet349:1517. 20. Rubsamen-Waigmann, H., E. Huguenel, A. Shah, A. Paessens, H. J. Ruoff,
H. von Briesen, A. Immelmann, U. Dietrich, and M. A. Wainberg.1999. Resistance mutations selected in vivo under therapy with anti-HIV drug HBY 097 may differ from pattern selected in vivo. Antivir. Res.42:15–24. 21. Ruprecht, R., M. Gama-Sosa, and H. D. Rosas.1988. Combination therapy
after retroviral inoculation. Lancetii:239–240.
22. Tomaras, G. D., S. F. Lacey, C. B. McDanal, G. Ferrari, K. J. Weinhold, and M. L. Greenberg.2000. CD8⫹T cell-mediated suppressive activity inhibits HIV-1 after virus entry with kinetics indicating effects on virus gene expres-sion. Proc. Natl. Acad. Sci. USA97:3503–3508.
23. Ten Haaft, P., B. Verstrepen, K. Ueberla, B. Rosenwirth, and J. Heeney. 1998. A pathogenic threshold of virus load defined in simian immunodefi-ciency virus- or simian-human immunodefiimmunodefi-ciency virus-infected macaques. J. Virol.72:10281–10285.
24. Tsai, C. C., K. E. Follis, A. T. Sabo, W. Beck, R. F. Grant, N. Bischofberger, R. E. Benveniste, and R. Black.1995. Prevention of SIV infection in ma-caques in (R)-9-(2-phosphonylmethoxypropyl) adenine. Science270:1197– 1199.
25. Uberla, K., C. Stahl-Hennig, D. Bottiger, K. Matz-Rensing, F. J. Kaup, J. Li, W. A. Haseltine, B. Fleckenstein, G. Hunsmann, B. Oeberg, and J. Sodroski. 1995. Animal model for the therapy of acquired immunodeficiency syndrome with reverse transcriptase inhibitors. Proc. Natl. Acad. Sci. USA92:8210– 8214.
26. Watson, W., J. Ranchalis, B. Travis, J. McClure, W. Sutton, P. R. Johnson, S. L. Hu, and N. L. Haigwood.1997. Plasma viremia in macaques infected with simian immunodeficiency virus: plasma viral load early in infection predicts survival. J. Virol.71:284–290.
27. Wong, J. K., M. Hezareh, H. F. Gu¨nthard, D. V. Havlir, C. C. Ignacio, C. A. Spina, and D. D. Richman.1997. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science278:1291–1294. 28. Wyand, M. S., K. H. Manson, M. Garcia-Moll, D. Montefiori, and R. C.
Desrosiers.1996. Vaccine protection by a triple deletion mutant of simian immunodeficiency virus. J. Virol.70:3724–3733.
29. Yasutomi, Y., K. A. Reimann, C. I. Lord, M. D. Miller, and N. L. Letvin. 1993. Simian immunodeficiency virus-specific CD8⫹lymphocyte response in acutely infected rhesus monkeys. J. Virol.67:1707–1711.
30. Zhang, L., B. Ramratnam, K. Tenner-Racz, Y. He, M. Vesanen, S. Lewin, A. Talal, P. Racz, A. S. Perelson, B. T. Korber, M. Markowitz, and D. D. Ho. 1999. Quantifying residual HIV-1 replication in patients receiving combina-tion antiretroviral therapy. N. Engl. J. Med.340:1605–1613.
31. Zhang, L., S. R. Lewin, M. Markowitz, H. H. Lin, E. Skulsky, R. Karanicolas, Y. He, X. Jin, S. Tuttleton, M. Vesanen, H. Spiegel, R. Kost, J. van Lunzen, H. J. Stellbrink, S. Wolinsky, W. Borkowsky, P. Palumbo, L. G. Kostrikis, and D. D. Ho.1999. Measuring recent thymic emigrants in blood of normal and HIV-1-infected individuals before and after effective therapy. J. Exp. Med.190:725–732.