Copyright © 1998, American Society for Microbiology
Expression of the pRb-Binding Regions of E1A Enables
Efficient Transformation of Primary Epithelial Cells by v-src
ROBERT S. FISCHER†
ANDMARGARET P. QUINLAN*
Department of Microbiology and Immunology, University of Tennessee Health Science Center,
Memphis, Tennessee 38163
Received 29 September 1997/Accepted 23 December 1997
Primary cultures of rat embryo fibroblasts have been shown to be resistant to transformation by dominant
oncogenes such as v-src. We sought to determine if similar resistance is displayed by primary epithelial cells,
and, if so, whether an immortalizing oncogene such as E1A could enhance transformation of primary epithelial
cells by v-src. Transformation of primary rat epithelial cells by v-src was synergistically enhanced when E1A
expression plasmids were cotransfected with a v-src expression plasmid. Foci were more numerous and
observed earlier (9 to 14 days) with E1A plus v-src than with v-src alone (18 to 28 days). This cotransformation
ability was abrogated by deletions in CR1 or CR2 of E1A, which encode the binding regions for the pRb family
and are responsible for E1A-mediated cell cycle activation. Mutations in the p300 binding site or the second
exon, which abolish immortalization, did not affect v-src cooperation, in contrast to ras and adenovirus E1B.
While kinase activation was required for growth in soft agar, differential activation of Src kinase did not
correlate with transformation efficiency. Cell morphology and actin structures were not dramatically impacted
by E1A expression; thus, hypertransformation, as previously described for ras cotransformation, was not
observed with v-src and second-exon mutants of E1A. However, growth rates for cells expressing both E1A and
v-Src were higher than those for cells expressing only v-Src. These results suggest that functions involved in
cell cycle activation encoded by E1A first exon can enhance v-src transformation of primary epithelial cells.
Oncogenic transformation is a multistep process that
in-volves the loss of cellular growth controls maintained by tumor
suppressor genes, as well as the acquisition of enhanced
pro-liferative capacity and morphological alterations provided by
dominant oncogenes (for reviews, see references 4 and 85). In
support of this model, primary cells have been shown to be
resistant to transformation by dominant oncogenes acting
alone and seem to require the concerted action of at least two
oncogenes to become transformed (23, 46–48, 71; for a review,
see reference 37). Immortalized fibroblasts, on the other hand,
are much more sensitive than similar primary cells to
transfor-mation by a variety of dominant oncogenes, including v-ras and
v-src (21, 38, 55). The mechanism(s) behind this resistance to
transformation is beginning to be understood and may include
specific cell cycle blocks (30, 77, 82). In combination with the
fact that more than 80% of human solid tumors are of
epithe-lial origin, these observations suggest that primary epitheepithe-lial
cells would provide an excellent model system to study
molec-ular signals involved in carcinogenesis.
The adenovirus (Ad) E1A gene is one of several DNA
tu-mor virus oncogenes that have been used to study oncogene
cooperative transformation of primary cells (for a review, see
reference 2). E1A has been shown to immortalize primary
cells, such that they maintain many of their differentiated
char-acteristics (35, 64, 65). In addition, E1A can cooperate with
Ha-ras and Ad E1B, as well as polyomavirus mT (pmT), to
transform primary cells (14, 71, 81, 88). The pmT binds to and
activates the c-Src protein (5, 10). This c-Src stimulation is
required for cellular transformation by pmT (39). However,
pmT cannot transform primary cells without the assistance of
an immortalizing oncogene (11, 71). In the present study, we
have extended these observations by investigating the ability of
v-src to transform primary epithelial cells in the presence or
absence of E1A expression. When baby rat kidney (BRK) cells
were transfected with v-src, inefficient transformation was
ob-served. However, when v-src was cotransfected with Ad 5 E1A
12S, marked improvement in transformation efficiency was
noted. Synergistic enhancement of v-src transformation did not
require full immortalization functions of E1A but only
func-tions encoded by the conserved regions 1 and 2 (CR1 and
CR2), corresponding to the ability of E1A to activate quiescent
BRK cells into the cell cycle (65) and to the ability to bind the
pRb family of proteins (16, 89). Transformation by Ha-ras is
subject to dramatic modulation of transformation by E1A (13,
80). This was not observed with v-src cooperation.
v-src-trans-formed cells exhibited a loss of actin organization, in the
pres-ence or abspres-ence of E1A, and inhibition of v-Src with radicicol
(45) caused reestablishment of prominent actin stress fibers.
This suggests that the transformation by E1A and v-src may be
reversible and that the morphologic alterations observed were
due to the kinase activity of v-Src and not to functions encoded
by E1A. This is in contrast to transformation of primary
epi-thelial cells by Ha-ras and E1A. These data imply that
activa-tion of the cell cycle in primary epithelial cells by E1A is
sufficient to allow efficient oncogenic transformation of such
cells by v-src and that this transformation is not subject to
modulation by E1A.
MATERIALS AND METHODS
Cells, transfections, and plasmids. 293 cells are human kidney cells trans-formed by Ad E1A and E1B (26) and were maintained in Dulbecco modified Eagle medium (DMEM)–5% fetal calf serum (FCS). BRK cells were prepared from 5-day-old rats as previously described (71). Transfections on BRK cells were performed at 2 days postplating by a modification of the calcium phosphate precipitation technique (90), as previously described (71). pJH v-src (expressing the wild-type [WT] SH2 and SH3 domains fused to the COOH-terminal portion
* Corresponding author. Mailing address: Dept. of Microbiology
and Immunology, University of Tennessee Health Science Center,
Memphis, TN 38163. Phone: (901) 448-8219. Fax: (901) 448-8462.
E-mail: [email protected].
† Present address: Department of Cell Biology, The Scripps
Re-search Institute, La Jolla, CA 92037.
2815
on November 9, 2019 by guest
http://jvi.asm.org/
of the Prague A v-src, which contains the kinase domain but lacks the regulatory tyrosine 527) was used at 5mg/60-mm-diameter dish, as were the E1A expression plasmids which have been described previously (13). To visualize transformed foci, cells were fixed with methanol at 14 days posttransfection and stained with Giemsa (Sigma, St. Louis, Mo.). For cell growth curves and Western analysis, whole transfected plates (.20 foci/plate) were trypsinized to establish a poly-clonal cell line and were used at low passage numbers (,10) in order to minimize the effects of clonal variation, except for v-src alone, which was derived from three separate clones that were pooled prior to assays. In addition, individual foci were cloned for additional growth curves and soft agar assays (Table 1). For cell growth curves, 105cells were seeded onto 60-mm-diameter dishes and grown in DMEM–5% FCS for the indicated number of days. Each time point represents at least four cell counts with a hemacytometer. The curves represent two inde-pendent experiments. Radicicol was used as previously described (58, 93), except that incubation in radicicol was carried out for 12 h (for Western blots) or 24 h (for immunofluorescence).
For fluorescence-activated cell sorting (FACS) analysis, cells were maintained in normal growth medium for 2 days prior to analyses. A total of 106cells were trypsinized, fixed with ethanol, and stained with propidium iodide for processing on a Coulter Profiler II analyzer (Coulter, Miami, Fla.). Phoenix Flow Multicycle analysis software was used to calculate the percentages of cells in G1, G2/M, and S phases in each population, as well as coefficients of variation (CV). The results represent at least two independent samples.
To test the ability of cell lines to grow independently of solid substrate, 60-mm-diameter dishes were coated with 2 ml of medium containing 0.5% agar (in DMEM-penicillin-streptomycin plus 5% FCS). The cells were then plated onto these dishes in the same medium at plating densities of 104, 105, and 106 cells per plate. The plates were incubated as described for cell line maintenance, with fresh medium (containing 0.5% agar) added every 3 to 4 days. Colony formation was assayed between 10 and 18 days after plating by overlaying the agar medium with 0.5 ml of p-iodonitrotetrazolium violet solution (0.5 g/liter in sterile water). Colonies were observed 24 h later. For samples exposed to radi-cicol, 0.1mg of radicicol per ml in agar medium was kept on cells, with fresh medium every other day.
Western blot analysis.Western blot analysis was performed essentially as described previously (58, 93). Briefly, approximately equal numbers of cells were labeled with (approximately 1mCi/23106cells) Trans-35S-Label (ICN, Costa Mesa, Calif.) overnight in DMEM with 5% FCS. The cells were washed twice with phosphate-buffered saline (PBS), lysed in boiling 23Laemmli sample buffer, scraped into tubes, and boiled for an additional 5 min. Lysates were centrifuged to remove debris, normalized to each other by scintillation counting, and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred to Immobilon SP in a Bio-Rad Western blot apparatus. The filters were blocked overnight in blocking buffer (0.7% fish gelatin in 10 mM Tris [pH 7.5], 150 mM NaCl, 0.1% Tween 20 [TBST]). The blots were probed with either anti-c-Src antibody (SC-019; Santa Cruz Biotechnologies, Inc., Santa Cruz, Calif.) or an antiphosphotyrosine antibody (Transduction Laboratories, Lexington, Ky.) in blocking buffer, followed by three washes with TBST. Anti-bodies were detected with anti-mouse-horseradish peroxidase conjugate (Sig-ma), followed by a chemiluminescence detection method, essentially as previ-ously described (58).
Immunofluorescence and photomicroscopy.Cells were seeded onto glass cov-erslips and maintained as described above. The cells were washed with cytoskel-eton buffer (32) and then fixed with 3.7% paraformaldehyde for 10 min at room
temperature, washed twice in cytoskeleton buffer, and permeabilized in 0.5% Triton X-100 in PBS. Nonspecific binding was blocked with 0.5% FCS–0.5% bovine serum albumin in PBS, and then cells were incubated in 1mg of phalloi-din-fluorescein isothiocyanate (FITC) per ml for 30 min, washed three times with PBS, and mounted in Airvol (Air Products, Allentown, Pa.). Fluorescence pho-tomicrographs were taken with a Zeiss Axiophot epifluorescence microscope fitted with an Optronics DEI-750 video camera system. For phase photomicros-copy, an Olympus CK2 microscope fitted with Minolta X-700 was used with Kodak Tri-X Pan ASA 400 film.
RESULTS
E1A first-exon functions enhance v-src transformation of
BRK cells.
To investigate possible cooperative transformation
of primary epithelial cells by E1A and v-src, a plasmid
express-ing v-Src was transfected alone or cotransfected with a plasmid
expressing either WT or mutated E1A 12S genes onto neonatal
BRK cells. Five functional regions of E1A have been
de-scribed, and mutations in all of these are represented by the
E1A mutants shown in Fig. 1 and have been previously
de-scribed (64, 88). As shown in Fig. 2, transformation of BRK
cells by v-src alone was inefficient; the visible foci that were
obtained were not observed until 18 to 28 days posttransfection
(data not shown). This is in contrast to Ha-ras transfections,
which never yield foci in the absence of E1A (71) (Fig. 2).
Transfections of v-src onto NIH 3T3 cells resulted in efficient
transformation (data not shown). When BRK cells were
co-transfected with v-src and WT 12S, efficient transformation was
observed, yielding numerous foci within 14 days
posttransfec-tion, similar to the transformation observed with Ha-ras (Fig.
2). All of the v-src-transformed cells tested had the ability to
grow as soft agar colonies, including the rare cells transformed
by v-src alone (Table 1). This cooperative transformation was
not abrogated by mutations in the second exon of E1A,
indi-cating that immortalization by E1A (which requires the second
exon) is not necessary for v-src transformation, in contrast to
E1A cooperation with Ad E1B (14, 15, 81). However,
muta-tions that have been shown to interfere with E1A’s ability to
stimulate cell cycle progression in quiescent cells also
inter-fered with v-src cotransformation. Specifically, deletion of the
NH2 terminus and CR1 (NTdl814, containing amino acids 83
to 243 [88]) or deletion of CR2 (dl891-1339, lacking amino
acids 110 to 174 [88]) of E1A abrogated cooperative
transfor-mation by v-src. Interestingly, pm563, which has been shown to
be unable to cooperate with Ha-ras (88), was still able to
cooperate with v-src (Fig. 2). pm563 does not form stable
complexes with the transcriptional coactivator p300 (16, 89).
However, this mutant does have the ability to stimulate cell
cycle progression, although the proliferation cannot be
main-tained (67). Thus, the ability of E1A to synergize with v-src to
transform primary epithelial cells is contained in the first exon
and correlates with activation of the cell cycle by E1A.
Expression and kinase activity of v-src is not consistently
altered by E1A.
E1A has been shown to affect expression of a
variety of cellular genes (for a review, see reference 2). To
investigate whether E1A had an effect on the overexpression of
v-src, cell lines were isolated from the transfections on BRK
cells and Western blot analysis was performed (Fig. 3). No
significant differences were observed in the expression of v-src
in the presence of WT or mutated 12S. Thus, the enhanced
transformation in the presence of E1A was not due to
differ-ential expression of v-src.
Some DNA tumor virus proteins have been demonstrated to
alter the activities of specific cellular kinases, such as
mitogen-activated protein (MAP) kinases (28) and c-Src (1). However,
the mechanisms for these effects are not clear. To test whether
the differential transformation efficiencies were due to changes
in v-Src activity, total cellular proteins were analyzed by
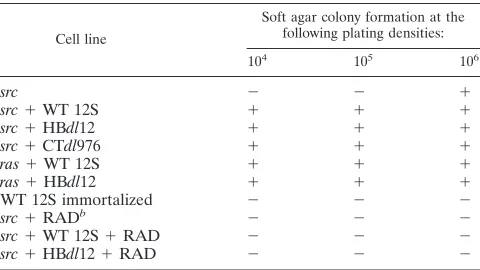
West-TABLE 1. Soft agar colony formation by transformed primary
epithelial cells
aCell line
Soft agar colony formation at the following plating densities:
104 105 106
src
2
2
1
src
1
WT 12S
1
1
1
src
1
HBdl12
1
1
1
src
1
CTdl976
1
1
1
ras
1
WT 12S
1
1
1
ras
1
HBdl12
1
1
1
WT 12S immortalized
2
2
2
src
1
RAD
b2
2
2
src
1
WT 12S
1
RAD
2
2
2
src
1
HBdl12
1
RAD
2
2
2
aCells were inoculated into soft agar cultures as described in Materials and Methods. Primary epithelial cells immortalized by WT 12S alone were included as a negative control. Colony formation was assayed by vital staining and mi-croscopy from two separate experiments.
bRAD, 0.1mg of radicicol per ml maintained in medium during the assay.
2816
FISCHER AND QUINLAN
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
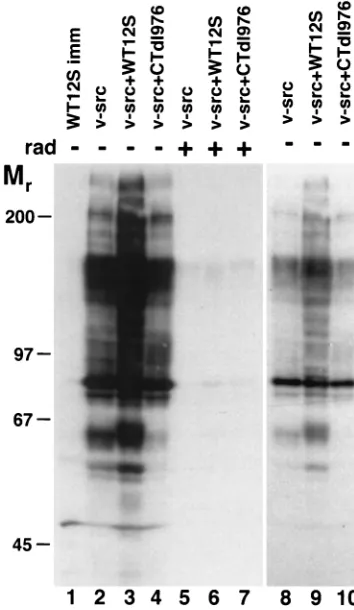
[image:2.612.50.290.89.224.2]ern blotting (Fig. 4). The presence of v-Src resulted in a large
number of proteins being tyrosine phosphorylated, compared
to BRK cells expressing only WT 12S. While the total protein
phosphotyrosine content was higher in the WT
12S-plus-v-src-transformed cells, compared to the v-src-12S-plus-v-src-transformed cells, a
similar increase was not observed with CTdl976, which was as
competent as WT 12S in the transformation assays.
Inconsis-tent increases in c-Src tyrosine kinase activity in the presence
of DNA tumor virus oncogenes have been previously reported,
which also did not correlate with cellular transformation (1). In
the presence of the specific Src kinase inhibitor radicicol (45),
the levels of tyrosine phosphorylation were significantly
dimin-ished in all of the v-src-transformed cell lines tested. These
data suggest that v-Src is active in transformed BRK cells but
that levels of activity may not correlate with observed
trans-formation efficiencies. Kinase activity nonetheless seems to be
required for transformation, since incubation of or
v-src-plus-WT 12S-transformed BRK cells in the presence of
radi-cicol inhibits growth in soft agar (Table 1).
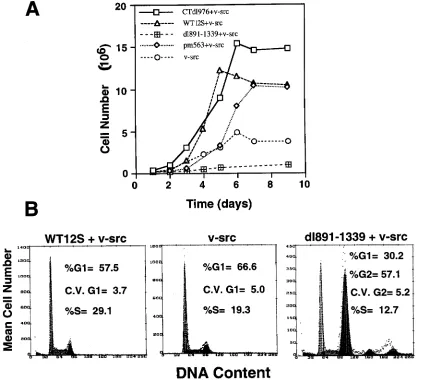
E1A enhances the proliferative properties of
v-src-trans-formed epithelial cells.
As shown above, of the five functional
regions of E1A, only the two regions required for cell cycle
activation are required to cooperate with v-src. Therefore, the
growth rates of BRK cells transformed with v-src or v-src plus
E1A (WT or mutant) were analyzed. As shown in Fig. 5A, cells
transformed by mutant or WT 12S and v-src had higher growth
rates and saturation densities, compared to primary epithelial
cells transformed by v-src alone. In normally cycling
popula-tions, the percentage of cells found in S phase was
approxi-mately 50% higher among BRK cells transformed by WT 12S
and v-src, compared to v-src alone (Fig. 5B). In the case of
dl891-1339 plus v-src, the resultant cells were not effectively
established into a cell line, such that they failed to proliferate
beyond early passages, which was exhibited in growth curves
(Fig. 5A). The dl891-1339-plus-v-src-transformed cells
ap-peared to experience a block in G2/M and/or G1
(Fig. 5B). This
was not a clonal aberrance, since the experiment was done with
a pool of multiple, independently arising foci. This may have
been due to loss of Src expression in these cells, since Src has
been shown to be required for G2
progression (70). It has been
previously shown that deletions of CR1 lead to a dominant
negative effect on the establishment of primary cells by WT
12S (50). In a similar manner, dl891-1339 may not allow the
stable establishment of primary cells, but this remains to be
determined. Thus, the ability to activate the cell cycle not only
is required for the initial transformation enhancement but also
has a significant effect on the growth properties of
v-src-ex-pressing cells.
E1A WT 12S is unable to modulate v-src transformation.
Functions encoded by the second exon of E1A have been
shown to down-modulate Ha-ras transformation of primary
epithelial cells, such that they retain some of their epithelial
morphology and characteristics (25). Deletion of the regions
encoding these functions leads to a marked increase in
trans-formation with ras (13, 80), but not E1B (14). This
hypertrans-formation is concomitant with dramatic alterations in cell
growth rates, adhesion, and actin cytoskeleton organization
(20a). To assess whether v-src transformation was subject to
such modulation imposed by E1A second-exon sequences, cell
morphologies and growth rates among cells transformed with
WT 12S plus v-src, CTdl976 plus v-src, and v-src alone were
compared. As shown in Fig. 6, none of the v-src transformants
maintained epithelial cell-style morphology or cell-cell
junc-tions; instead, they exhibited a star-shaped morphology, even
in the presence of WT 12S. Deletion of some or all of the
second exon did result in cells that were somewhat smaller and
more refractile and enabled them to grow to higher saturation
FIG. 1. Properties of E1A 12S mutants. The mutants have been described previously (64, 88) and are summarized here. Functional regions are indicated at the top. Protein binding indicates the ability of the protein to bind either Rb or p300 as well as WT E1A (1) or a level of binding that is less than that of WT (2). Data for the columns headed immort. (immortalized) and activation are derived from references 67 and 64; data for pRb and p300 are derived from references 89 and 24; data for ras were derived from references 88 and 13; data for E1B were derived from reference 74; and data for v-src were derived from the present study. aa, amino acids.
on November 9, 2019 by guest
http://jvi.asm.org/
densities (Fig. 5A), which is similar to what has been observed
in E1A cooperation with Ha-ras.
As shown in Fig. 5A, expression of E1A first-exon sequences
allowed cells to achieve much higher growth rates and
satura-tion densities than the cells transformed with v-src alone.
While deletion of the second exon did allow a modest
enhance-ment of saturation density over the WT 12S, the difference was
less than 50%. No differences were observed in the adhesive
properties of v-src-transformed cells, in the presence of either
WT or mutant 12S (data not shown). This is in contrast to
Ha-ras-transformed BRK cells, which are much more adhesive
in the presence of WT 12S expression, compared to
second-exon mutants of 12S (20a). These results indicate that
v-src-mediated cotransformation cannot be down-modulated in a
manner similar to Ha-ras cotransformation in primary
epithe-lial cells. The differences in the requisite functional regions of
E1A and in the potential for E1A to impact the extent of
transformation probably reflect the fundamental differences in
the signals generated by src, ras, and E1B.
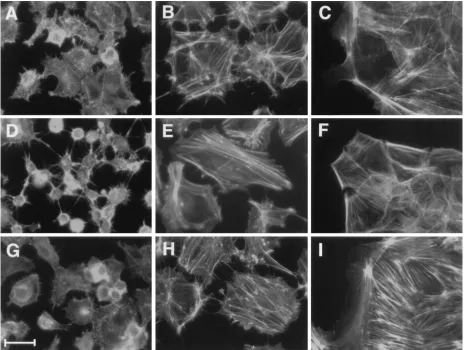
v-src transformation of primary epithelial cells leads to a
loss of actin stress fibers, which are maintained in
[image:4.612.373.482.70.186.2]E1A-im-mortalized BRK cells.
c-Src has been shown to be involved in
[image:4.612.88.252.74.538.2]FIG. 2. Transformation of BRK cells by v-src and E1A mutants. Cells were transfected with plasmids expressing the various mutants of E1A and v-src, as described in Materials and Methods. The plates were fixed at 14 days posttrans-fection and stained with Giemsa and represent four or more dishes in at least two independent experiments.
FIG. 3. Analysis of v-src overexpression. Cells were cloned from the indi-cated transfections, and normalized lysates were processed for Western blotting and probed with an anti-Src antibody. 10BRK, primary BRK cells.
FIG. 4. Expression of v-src leads to increased phosphorylation of cellular proteins and is inhibited by radicicol. Total cell lysates were prepared from the cells indicated, which had been incubated in the presence (1[lanes 5 to 7]) or absence (2[lanes 1 to 4 and 8 to 10]) of radicicol (rad; 0.1mg/ml) for 12 h prior to lysate preparation. The blot was probed with antiphosphotyrosine antibody. Lanes 8 to 10, lower exposure of lanes 2 to 4.
2818
FISCHER AND QUINLAN
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.338.515.371.676.2]the assembly of focal adhesion complexes and activation of
integrin-mediated signals, primarily due to its ability to form
stable complexes with FAK (40, 74, 75). c-Src kinase activity is
involved in the regulation of epidermal growth factor
(EGF)-dependent, actin cytoskeleton rearrangement via
phosphoryla-tion of rhoGAP p190 (7). v-Src, on the other hand, has been
demonstrated to cause disassembly of actin stress fibers and
focal adhesions in fibroblasts (20, 42). Similarly, other viral
oncoproteins have been observed to affect actin stress fibers
(59, 86). E1A has been shown to induce and maintain
epithe-lial cell differentiation and morphology (Fig. 7C) (8, 22, 25, 64,
66). Therefore, the organization of F actin in
src-plus-E1A-transformed versus E1A-immortalized BRK cells was
ana-lyzed.
Actin stress fiber organization was observed in BRK cells
transformed with v-src and E1A via phalloidin-FITC staining,
followed by immunofluorescence. As shown in Fig. 7, cells
expressing v-src exhibited a significant loss of actin stress fibers,
compared to either the primary BRK cells or
E1A-immortal-ized cell lines, as well as increased amounts of actin
micro-spikes at the cell periphery. In contrast, both the primary cells
and E1A-immortalized epithelial cells maintained prominent
actin stress fibers, as well as anti-paxillin-staining focal
adhe-sion plaques, which were also lost in v-src-transformed cells
(data not shown). Actin microspikes were not observed in
primary or immortalized epithelial cells. When
E1A-plus-v-src-transformed BRK cells were exposed to radicicol prior to
phal-loidin staining, prominent actin stress fibers were again
ob-served, although complete epithelial morphology was not
regained. In addition, while all v-src-expressing cells grew in
soft agar assays, cells maintained in radicicol did not form soft
agar colonies (Table 1). Thus, the cytoskeletal alterations
ob-served in v-src-plus-E1A-transformed BRK cells correlate with
substrate independence. Furthermore, these alterations in
ac-tin organization are due entirely to v-Src kinase actions, not to
functions provided by E1A. This differs from E1A plus Ha-ras
transformation of BRK cells, in which case the morphology
and actin cytoskeleton of the resulting transformed cells can be
affected by specific mutations in E1A (13, 20a, 25). In the case
of Ad E1B-transformed epithelial cells, the actin cytoskeleton
is not disrupted in the presence of WT or mutated E1A (data
not shown). Thus, alterations of the actin cytoskeleton of
v-src-transformed primary epithelial cells are not subject to
modu-FIG. 5. (A) Cell growth curves. Cells from the indicated transfections were seeded at 105cells per dish, and growth rates were measured as described in Materials and Methods. Each data point represents an average of four counts. (B) FACS analysis of cells transformed with v-src, v-src plus dl891-1339, and WT 12S plus v-src. Cells were stained with propidium iodide and sorted as described in Materials and Methods.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.89.510.69.449.2]FIG. 6. Cell morphologies of transformed or immortalized (imm) BRK cells. Photomicrographs were taken at equal magnifications. Bar, 100mm; 10BRK, primary BRK cells.
2820
FISCHER AND QUINLAN
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
lation by WT 12S, but direct inhibitors of Src kinase can
mod-ulate such actin reorganization.
DISCUSSION
The results presented here show that transformation of
mammalian primary epithelial cells by v-src alone is inefficient,
in contrast to primary fibroblasts (34) or established fibroblast
lines such as NIH 3T3 cells. However, coexpression of E1A
leads to efficient transformation of these cells, such that they
are able to grow indefinitely and in an anchorage-independent
fashion. This synergistic cotransformation ability mapped to
the sequences contained in the first exon, in particular, CR1
and CR2 of E1A 12S. Interestingly, a deletion mutant lacking
CR2 (dl891-1339) produced few transformed foci, which did
not produce stable cell lines. Furthermore, an NH2-terminal
deletion mutant lacking CR1 (NTdl814) did not produce
formed foci with v-src. That the latter did not enable src
trans-formation was not surprising, since our lab has previously
found NTdl814 to actually inhibit WT 12S functions (50).
These observations underscore the requirement for CR1 and
CR2 functions for v-src transformation. An NH2-terminal
point mutant, pm563, which lacks the ability to bind p300 (89),
was able to enhance v-src transformation, indicating that the
requirements for v-src cooperation are less stringent than those
[image:7.612.67.534.69.419.2]for E1B or Ha-ras cotransformation (14, 79, 88). Indeed,
nei-ther E1B, Ha-ras, nor pmT, which activates c-src (5, 10, 39), is
able to transform primary cells alone, but each requires
mul-tiple but different functions encoded by E1A (14, 71, 79, 81,
88). Nevertheless, v-src alone is able to transform primary
epithelial kidney cells, albeit inefficiently. Previous studies have
shown that primary rodent (but not human) fibroblasts can be
transformed by v-src alone (34). Susceptibility to
transforma-tion by v-src in primary cells appears to be partly dependent on
cellular senescence and aging (82). Also, it has been shown
that adrenocortical cells can be inefficiently transformed by
v-src and that v-myc can cooperate with v-src to efficiently
transform such cells (48). The inefficient transformation by
v-src alone in this study as well as previous studies could rely on
the acquisition of mutations in cell cycle regulatory genes
po-tentially reproducing the effects of E1A expression. While this
hypothesis remains unconfirmed, heterogeneity in primary
cul-tures may be relevant to the observation of rare v-src
transfor-mants (83). It seems likely that the inefficiency of
transforma-tion observed here may be due at least in part to the fact that
BRK epithelial cells undergo senescence and apoptosis after
24 h in culture, which E1A is able to rescue (63). In addition,
the ability of E1A, via CR1- and CR2-mediated binding of the
pRB family (6, 89), to activate the cell cycle probably enables
FIG. 7. F-actin organization of E1A-immortalized epithelial cells versus E1A-plus-v-src-transformed epithelial cells. Cells were stained with phalloidin-FITC, as described in Materials and Methods. (A and B) WT 12S-plus-v-src-transformed cells; (C) WT 12S-immortalized cells; (D and E) HBdl12-plus-v-src-transformed cells; (F) HBdl12-immortalized cells; (G and H) v-src-transformed cells; (I) primary BRK cells; (B, E, and H) cells exposed to 0.1mg of radicicol per ml for 24 h prior to fixation. Bar, 30mm.
on November 9, 2019 by guest
http://jvi.asm.org/
the somewhat higher growth rates observed in the E1A- and
src-transformed cells, compared with the cells transformed by
src alone. The ability of E1A to activate quiescent cells into the
cell cycle seems crucial for the enhancement of v-src
transfor-mation of primary epithelial cells.
Hyperphosphorylation of cellular proteins by v-Src seems to
be enhanced by WT 12S, but not by mutants lacking the second
exon, such as CTdl976. The significance of this is not clear at
present, since CTdl976 is as competent in transformation
as-says with v-src as WT 12S. Furthermore, the enhanced kinase
activity does not correlate with substrate-independent growth
or growth rate in vitro (Fig. 5; Table 1). Other investigators
have observed inconsistent increases in tyrosine
phosphoryla-tion by src in cells expressing E1A (1), but the data neither
indicate the significance of nor suggest a mechanism for the
increase. We find here that the mechanism does not involve
increased expression of the v-src oncogene. However,
differ-ences in localization, stability, or specific activity have not been
established at present.
Requirements for transformation of primary epithelial cells
by v-src and E1A are different from those of other cooperating
oncogenes (Fig. 1). For efficient transformation of primary
epithelial cells by v-src and E1A, the ability of E1A to induce
cell cycle via CR1 and CR2 is apparently the only requirement.
This is interesting, because previous studies have suggested
that immortalization of primary cells might be required for
efficient v-src transformation (38, 48), while we have observed
here that mutants incapable of immortalization by themselves
are able to cooperate with v-src to transform primary epithelial
cells (Fig. 1). Thus, E1A mutants lacking the ability to establish
cell lines alone can cooperate with v-src to establish
trans-formed lines. In fact, the requirements for E1A to cooperate
with v-src in transforming primary epithelial cells are less
strin-gent than those for Ad E1B or even Ha-ras. This is further
reflected in the ability of v-src to transform such primary cells
alone, albeit inefficiently, while neither Ad E1B nor Ha-ras is
able to do so (47, 71).
The role of p300 binding by E1A in cell transformation has
been found to involve changes in gene expression (for reviews,
see references 2 and 54). Similarly, changes in cellular gene
expression can also be induced by v-src transformation (27, 52,
76). Furthermore, it has been shown that p300 binding by E1A
can contribute to cell proliferation (36, 92), by interfering with
the expression of p21
WAF(3, 12, 53, 87). p27
KIPand p21
WAFare cyclin-dependent kinase inhibitors with many targets in
common (9, 17, 19, 31, 41, 56, 60, 61, 84). However, p27
KIPcan
be inhibited by E1A directly, apparently independently of
tran-scription alterations (49). It is possible that primary kidney
epithelial cells (as opposed to the PC12 cells used in the
afore-mentioned studies) are prevented from proliferation primarily
by p27
KIPand not p21
WAF, although this has not been formally
tested. Since p300 binding does not seem to be required for
E1A’s cooperative transformation with v-src but is required for
ras cotransformation (for a review, see reference 2), it seems
possible that v-src may allow cells to escape a proliferation
block by a p21
WAF-dependent mechanism. Given that p27
KIPand p21
WAFhave been shown to be involved in
attachment-dependent growth (19) and that inhibition of Src kinase activity
prevents soft agar growth (Table 1), it is possible that Src
kinase activity allows cells to circumvent such cell cycle
regu-lation steps, as has been suggested elsewhere (72).
The data presented here also show that while E1A is able to
enhance transformation efficiency, v-src-mediated
morpholog-ical alterations are not affected dramatmorpholog-ically by E1A. These
alterations include loss of cell-cell contact, actin stress fibers
and/or microfilaments, and focal adhesion plaques. Radicicol,
a potent and reversible inhibitor of Src kinase activity (44, 45,
58), is able to reverse the loss of actin stress fibers induced by
v-Src, although cells do not regain cell-cell contacts. Although
radicicol has been shown to be specific to Src kinase (45), it can
inhibit mos and ras transformation as well (93). The fact that
v-src-transformed primary epithelial cells exhibit loss of stress
fibers is hardly surprising, given the wealth of data showing this
phenomenon in other cell types (for a review, see reference 57
and references therein). However, c-Src has a positive effect on
cell spreading and focal adhesion formation on fibronectin, but
this role of c-Src does not seem to require its kinase activity
(40). However, it is interesting that E1A is not able to suppress
v-src-mediated morphological transformation of epithelial
cells, given E1A’s ability to do so with an activated ras gene
(13, 20a, 25).
The ability of v-src alone (present study and reference 34)
but not Ha-ras (present study and references 47 and 71) to
transform primary cells suggests further differences between
the signals mediated by ras and src. Both ras and src activation
cause stimulation of the ERK-MAP kinase cascade via c-raf
activation (18, 51, 91). The mechanisms employed by Src and
Ras to accomplish this seem to be different (78). The activation
of raf, however, does not lead to full transformation of either
NIH 3T3 cells (43, 62) or primary epithelial cells, even with
E1A (20a), and activation of rho-related proteins is required in
combination with the activation of raf for full transformation.
The activation of Rho family proteins by ras is well
docu-mented (for a review, see reference 68). src is also able to
transduce signals through the Rho family G proteins via their
GAPs (7, 33). However, activated ras and activated src have
been shown to have opposite effects on actin stress fibers in
fibroblast cells, presumably by their actions on Rho activity (7,
69). In BRK cells, however, cotransformation by either Ha-ras
or v-src leads to a loss of stress fibers, again suggesting common
cellular effects for these oncogenes, although the mechanisms
may be different. Perhaps the differences in their abilities to
transform primary epithelial cells alone, or in conjunction with
E1A, can be explained by the observation that src can act
upstream of ras, as some signal transduction cascade models
would suggest (73). This might indicate that the activities or
signals induced by activated ras are but a subset of those
induced by activated src in epithelial cells. In addition, since
Src mediates important alterations in epithelial cell
transfor-mation directly by its kinase activity (29), it may be a more
potent oncogene than ras in epithelium-derived cells (48).
ACKNOWLEDGMENTS
We thank A. Reynolds for the gift of the pJH v-src plasmid and S.
Sharma for the radicicol. The technical expertise provided by M.
Dock-ter and J. Hermann and graphics assistance of T. Higgins of the UT
Molecular Resource Center are also appreciated.
This work was supported by the National Institutes of Health, and
R.S.F. was supported by the Regan Memorial Fellowship.
REFERENCES
1. Amini, S., A. J. Lewis, M. A. Israel, J. S. Butel, and J. B. Bolen. 1986. Analysis of pp60c-src protein kinase activity in hamster embryo cells trans-formed by simian virus 40, human adenoviruses, and bovine papillomavirus 1. J. Virol. 57:357–61.
2. Bayley, S. T., and J. S. Mymryk. 1994. Adenovirus E1A proteins and trans-formation (review). Int. J. Oncol. 5:325–444.
3. Billon, N., L. van Grunsven, and B. B. Rudkin. 1996. The CDK inhibitor p21WAF1/Cip1 is induced through a p300-dependent mechanism during NGF-mediated neuronal differentiation of PC12 cells. Oncogene 13:2047– 2054.
4. Bishop, J. M. 1991. Molecular themes in oncogenesis. Cell 64:235–248. 5. Bolen, J. B., C. J. Thiele, M. A. Israel, W. Yonemoto, L. A. Lipsich, and J. S.
Brugge.1984. Enhancement of cellular src gene product tyrosyl kinase ac-tivity following polyoma virus infection and transformation. Cell 38:767–777. 6. Branton, P. E., S. T. Bayley, and F. L. Graham. 1985. Transformation by
2822
FISCHER AND QUINLAN
J. V
IROL.
on November 9, 2019 by guest
http://jvi.asm.org/
human adenoviruses. Biochim. Biophys. Acta 780:67–94.
7. Chang, J.-H., S. Gill, J. Settleman, and S. J. Parsons. 1995. c-Src regulates the simultaneous rearrangement of actin cytoskeleton, p190RhoGAP, and p120RasGAP following epidermal growth factor stimulation. J. Cell Biol. 130:355–368.
8. Chinnadurai, G. 1992. Adenovirus E1a as a tumor-suppressor gene. Onco-gene 7:1255–1258.
9. Coats, S., W. M. Flanagan, J. Nourse, and J. M. Roberts. 1996. Requirement of p27Kip1 for restriction point control of the fibroblast cell cycle. Science 272:877–880.
10. Courtneidge, S. A., and A. E. Smith. 1983. Polyoma virus transforming protein associates with the product of the c-src gene. Nature 303:435–439. 11. Cuzin, F. 1984. The polyoma virus oncogenes. Coordinated functions of
three distinct proteins in the transformation of rodent cells in culture. Bio-chim. Biophys. Acta 781:193–204.
12. Datto, M. B., P. P. Hu, T. F. Kowalik, J. Yingling, and X. F. Wang. 1997. The viral oncoprotein E1A blocks transforming growth factorb-mediated induc-tion of p21/WAF1/Cip1 and p15/INK4B. Mol. Cell. Biol. 17:2030–2037. 13. Douglas, J. L., S. Gopalakrishnan, and M. P. Quinlan. 1991. Modulation of
transformation of primary epithelial cells by the second exon of the Ad5 E1A 12S gene. Oncogene 6:2093–2103.
14. Douglas, J. L., and M. P. Quinlan. 1995. Efficient nuclear localization and immortalizing ability, two functions dependent on the adenovirus type 5 (Ad5) E1A second exon, are necessary for cotransformation with Ad5 E1B but not with T24 ras. J. Virol. 69:8061–8065.
15. Douglas, J. L., and M. P. Quinlan. 1994. Efficient nuclear localization of the Ad5 E1A 12S protein is necessary for immortalization but not cotransfor-mation of primary epithelial cells. Cell Growth Differ. 5:475–483. 16. Egan, C., T. N. Jelsma, J. A. Howe, S. T. Bayley, B. Ferguson, and P. E.
Branton.1988. Mapping of cellular protein-binding sites on the products of early-region 1A of human adenovirus type 5. Mol. Cell. Biol. 8:3955–3959. 17. El-Deiry, W. S., T. Tokino, V. E. Velculescu, D. B. Levy, R. Parsons, J. M. Trent, D. Lin, W. E. Mercer, K. W. Kinzler, and B. Vogelstein.1993. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825. 18. Fabian, J. R., I. O. Daar, and D. K. Morrison. 1993. Critical tyrosine residues
regulate the enzymatic and biological activity of Raf-1 kinase. Mol. Cell. Biol. 13:7170–7179.
19. Fang, F., G. Orend, N. Watanabe, T. Hunter, and E. Ruoslahti. 1996. De-pendence of cyclin E-CDK2 kinase activity on cell anchorage. Science 271: 499–502.
20. Felice, G. R., P. Eason, M. W. Nermut, and S. Kellie. 1990. pp60v-src asso-ciation with the actin cytoskelton induces actin reorganization without af-fecting polymerization status. J. Cell Biol. 52:47–59.
20a.Fischer, R. S., Y. Zheng, and M. P. Quinlan. Primary epithelial cell trans-formation by E1A and ras requires Rac1 and ERK activation, while progres-sion can be modulated by E1A and Rac1. Cell Growth Differ., in press. 21. Franza, B. R., Jr., K. Maruyama, J. I. Garrels, and H. E. Ruley. 1986. In vitro
establishment is not a sufficient prerequisite for transformation by activated ras oncogenes. Cell 44:409–418.
22. Frisch, S. M. 1994. E1A induces the expression of epithelial characteristics. J. Cell Biol. 127:1085–1096.
23. Gilmer, T. M., L. A. Annab, M. Oshimura, and J. C. Barret. 1985. Neoplastic transformation of normal and carcinogen-induced preneoplastic Syrian ham-ster embryo cells by the v-src oncogene. Mol. Cell. Biol. 5:1707–1713. 24. Gopalakrishnan, S., J. L. Douglas, and M. P. Quinlan. 1997.
Immortaliza-tion of primary epithelial cells by E1A 12S requires late, second exon-encoded functions in addition to complex formation with pRb and p300. Cell Growth Differ. 8:541–551.
25. Gopalakrishnan, S., and M. P. Quinlan. 1995. Modulation of E-cadherin localization in cells expressing wild type E1A 12S or hypertransforming mutants. Cell Growth Differ. 6:985–998.
26. Graham, F. L., J. Smiley, W. C. Russell, and R. Nairn. 1977. Characteristics of a human cell line transformed by DNA from adenovirus type 5. J. Gen. Virol. 36:59–72.
27. Gu, H., and N. Oliver. 1995. Transcriptional repression of fibronectin gene expression in v-src transformation. Exp. Cell Res. 217:428–439.
28. Gu, Z., and G. Matlashewski. 1995. Effect of human papillomavirus type 16 oncogenes on MAP kinase activity. J. Virol. 69:8051–8056.
29. Hamaguchi, M., N. Matsuyoshi, Y. Ohnishi, B. Gotoh, M. Takeichi, and Y. Nagai.1993. p60v-src causes tyrosine phosphorylation and inactivation of the N-cadherin-catenin cell adhesion system. EMBO J. 12:307–314.
30. Hara, E., R. Smith, D. Parry, H. Tahara, S. Stone, and G. Peters. 1996. Regulation of p16CDKN2expression and its implications for cell immortal-ization and senescence. Mol. Cell. Biol. 16:859–867.
31. Harper, J. W., G. R. Adami, N. Wei, K. Keyomarsi, and S. J. Elledge. 1993. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75:805–816.
32. Herzog, M., A. Dreager, E. Ehler, and J. V. Small. 1994. Immunofluores-cence microscopy of the cytoskeleton: double and triple immunofluores-cence, p. 355–374. In J. E. Celis (ed.), Cell biology: a laboratory handbook, 1st ed., vol. 2. Academic Press, San Diego, Calif.
33. Hildebrand, J. D., J. M. Taylor, and J. T. Parsons. 1996. An SH3
domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol. Cell. Biol. 16:3169–3178.
34. Hjelle, B., E. Liu, and J. M. Bishop. 1988. Oncogene v-src transforms and establishes embryonic rodent fibroblasts, but not diploid human fibroblasts. Proc. Natl. Acad. Sci. USA 85:4355–4359.
35. Houweling, A., P. J. van der Elsen, and A. J. Van der Eb. 1980. Partial transformation of primary rat cells by the leftmost 4.5% fragment of ade-novirus 5 DNA. Virology 105:537–550.
36. Howe, J. A., J. S. Mymryk, C. Egan, P. E. Branton, and S. T. Bayley. 1990. Retinoblastoma growth suppressor and a 300-kilodalton protein both appear to regulate cellular DNA synthesis. Proc. Natl. Acad. Sci. USA 87:5883– 5887.
37. Hunter, T. 1991. Cooperation between oncogenes. Cell 64:249–270. 38. Inoue, H., N. Tavoloni, and H. Hanafusa. 1995. Suppression of v-Src
trans-formation in primary rat embryo fibroblasts. Oncogene 11:231–238. 39. Kaplan, D. R., D. C. Pallas, W. Morgan, B. Schaffhausen, and T. M. Roberts.
1989. Mechanism of transformation by polyoma virus middle T antigen. Biochim. Biophys. Acta 948:345–368.
40. Kaplan, K. B., J. R. Swedlow, D. O. Morgan, and H. E. Varmus. 1995. c-Src enhances the spreading of src2/2fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 9:1505–1517.
41. Kato, J. Y., M. Matsuoka, K. Polyak, J. Massague, and C. J. Sherr. 1994. Cyclic AMP-induced G1 phase arrest mediated by an inhibitor (p27Kip1) of cyclin-dependent kinase 4 activation. Cell 79:487–496.
42. Kellie, S., B. Patel, N. M. Wigglesworth, D. R. Critchley, and J. A. Wyke. 1986. The use of Rous sarcoma virus transformation mutants with differing tyrosine kinase activities to study the relationships between vinculin phos-phorylation, pp60v-src location and adhesion plaque integrity. Exp. Cell. Res. 165:216–228.
43. Khosravi-Far, R., P. A. Solski, G. J. Clark, M. S. Kinch, and C. J. Der. 1995. Activation of Rac1, RhoA, and mitogen-activated protein kinases is required for Ras transformation. Mol. Cell. Biol. 15:6443–6453.
44. Kwon, H. J., M. Yoshida, K. Abe, S. Horinouchi, and T. Beppu. 1992. Radicicol, an agent inducing the reversal of transformed phenotypes of src-transformed fibroblasts. Biosci. Biotech. Biochem. 56:6926–6930. 45. Kwon, H. J., M. Yoshida, Y. Fukui, S. Horinouchi, and T. Beppu. 1992.
Potent and specific inhibition of p60v-src protein kinase both in vivo and in vitro by radicicol. Cancer Res. 52:6926–6930.
46. Land, H., L. F. Parada, and R. A. Weinberg. 1983. Cellular oncogenes and multistep carcinogenesis. Science 222:771–778.
47. Land, H., L. F. Parada, and R. A. Weinberg. 1983. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature (London) 304:596–602.
48. MacAuley, A., and T. Pawson. 1988. Cooperative transforming activities of
ras, myc, and src viral oncogenes in nonestablished rat adrenocortical cells.
J. Virol. 62:4712–4721.
49. Mal, A., R. Y. C. Poon, P. H. Howe, H. Toyoshima, T. Hunter, and M. L. Harter.1996. Inactivation of p27Kip1by the viral E1A oncoprotein in TGFb -treated cells. Nature (London) 380:262–265.
50. Malladi, A., and M. P. Quinlan. 1997. Mutations in CR1 are dominant negative suppressors of E1A functions in immortalization and the lytic cycle. Virology 233:51–62.
51. Marais, R., Y. Light, H. F. Paterson, and C. J. Marshall. 1995. Ras recruits Raf-1 to the plasma membrane for activation by tyrosine phosphorylation. EMBO J. 14:3136–3145.
52. Meijne, A. M., L. Ruuls-Van Stalle, C. A. Feltkamp, J. B. McCarthy, and E. Roos.1997. v-src-induced cell shape changes in rat fibroblasts require new gene transcription and precede loss of focal adhesions. Exp. Cell. Res. 234:477–485.
53. Missero, C., E. Calautti, R. Eckner, J. Chin, L. H. Tsai, D. M. Liningston, and G. P. Dotto.1995. Involvement of the cell-cycle inhibitor Cip1/WAF1 and the E1A-associated p300 protein in terminal differentiation. Proc. Natl. Acad. Sci. USA 92:5451–5455.
54. Mymryk, J. S. 1996. Tumour suppressive properties of the adenovirus 5 E1A oncogene. Oncogene 13:1581–1589.
55. Newbold, R. F., and R. W. Overell. 1983. Fibroblast immortality is a prereq-uisite for transformation by the EJ cHa-ras oncogene. Nature 304:648–651. 56. Nourse, J., E. Firpo, W. M. Flanagan, S. Coats, K. Polyak, M. H. Lee, J. Massague, G. R. Crabtree, and J. M. Roberts.1994. Interleukin-2-mediated elimination of the p27Kip1 cyclin-dependent kinase inhibitor prevented by rapamycin. Nature (London) 372:570–573.
57. Parsons, J. T., and S. J. Parsons. 1997. Src family protein tyrosine kinases: cooperating with growth factor and adhesion signalling pathways. Curr. Opin. Cell Biol. 9:187–192.
58. Pillay, I., H. Nakano, and S. V. Sharma. 1996. Radicicol inhibits tyrosine phosphorylation of mitotic src substrate Sam68 and retards subsequent exit from mitosis of src-transformed cells. Cell Growth Differ. 7:1487–1499. 59. Pollack, R., M. Osborn, and K. Weber. 1975. Patterns of organization of
actin and myosin in normal and transformed cultured cells. Proc. Natl. Acad. Sci. USA 72:994–998.
60. Polyak, K., J. Y. Kato, M. J. Solomon, C. J. Sherr, J. Massague, J. M. Roberts, and A. Koff.1994. p27Kip1, a cyclin-Cdk inhibitor, links
on November 9, 2019 by guest
http://jvi.asm.org/
ing growth factor-beta and contact inhibition to cell cycle arrest. Genes Dev. 8:9–22.
61. Polyak, K., M. H. Lee, B. H. Erdjument, A. Koff, J. M. Roberts, P. Tempst, and J. Massague. 1994. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 78:59–66.
62. Qui, R.-G., J. Chen, D. Kirn, F. McCormick, and M. Symons. 1995. An essential role for rac in ras transformation. Nature 374:457–459. 63. Quinlan, M. P. 1993. E1A 12S in the absence of E1B or other cooperating
oncogenes enables cells to overcome apoptosis. Oncogene 8:3289–3296. 64. Quinlan, M. P., and J. L. Douglas. 1992. Immortalization of primary
epi-thelial cells requires first and second-exon functions of adenovirus type 5 12S. J. Virol. 66:2020–2030.
65. Quinlan, M. P., and T. Grodzicker. 1987. Adenovirus E1A 12S protein induces DNA synthesis and proliferation in primary epithelial cells in both the presence and absence of serum. J. Virol. 61:673–682.
66. Quinlan, M. P., N. Sullivan, and T. Grodzicker. 1987. Growth factor(s) produced during infection with an adenovirus variant stimulates prolifera-tion of nonestablished epithelial cells. Proc. Natl. Acad. Sci. USA 84:3283– 3287.
67. Quinlan, M. P., P. Whyte, and T. Grodzicker. 1988. Growth factor induction by the adenovirus type 5 E1A 12S protein is required for immortalization of primary epithelial cells. Mol. Cell. Biol. 8:3191–3203.
68. Ridley, A. J. 1995. Rho-related proteins: actin cytoskeleton and cell cycle. Curr. Opin. Genet. Dev. 5:24–30.
69. Ridley, A. J., and A. Hall. 1992. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell 70:389–399.
70. Roche, S., S. Fumagalli, and S. A. Courtneidge. 1995. Requirement for Src family protein tyrosine kinases in G2 for fibroblast cell division. Science 269:1567–1569.
71. Ruley, H. E. 1983. Adenovirus early region 1A enables viral and cellular transforming genes to transform primary cells in culture. Nature (London) 304:602–606.
72. Sabe, H., M. Hamaguchi, and H. Hanafusa. 1997. Cell to substratum adhe-sion is involved in v-Src-induced cellular protein tyrosine phosphorylation: implication for the adhesion-regulated protein tyrosine phosphatase activity. Oncogene 14:1779–1788.
73. Schieffer, B., W. G. Paxton, Q. Chai, M. B. Marrero, and K. E. Bernstein. 1996. Angiotensin II controls p21ras activity via pp60c-src. J. Biol. Chem. 271:10329–10333.
74. Schlaepfer, D. D., S. K. Hanks, T. Hunter, and P. Van der Geer. 1994. Integrin-mediated signal transduction linked to ras pathway by GRB2 bind-ing to focal adhesion kinase. Nature (London) 372:786–791.
75. Schlaepfer, D. D., and T. Hunter. 1996. Evidence for in vivo phosphorylation of the Grb2 SH2-domain binding site on focal adhesion kinase by Src-family protein-tyrosine kinases. Mol. Cell. Biol. 16:5623–5633.
76. Scholz, G., C. Martinerie, B. Perbal, and H. Hanafusa. 1996. Transcriptional
down regulation of a novel proto-oncogene in fibroblasts transformed by p60v-src. Mol. Cell. Biol. 16:481–6.
77. Serrano, M., E. Gomez-Lahoz, R. A. DePinho, D. Beach, and D. Bar-Sagi. 1995. Inhibition of ras-induced proliferation and cellular transformation by p16INK4. Science 267:249–252.
78. Stokoe, D., and F. McCormick. 1997. Activation of c-Raf-1 by ras and src through different mechanisms: activation in vivo and in vitro. EMBO J. 16:2384–2396.
79. Subramanian, T., M. Kuppuswamy, R. J. Nasr, and G. Chinnadurai. 1988. An N-terminal region of adenovirus E1A essential for cell transformation and induction of an epithelial cell growth factor. Oncogene 2:105–112. 80. Subramanian, T., M. La Regina, and G. Chinnadurai. 1989. Enhanced ras
oncogene mediated cell transformation and tumorigenesis by adenovirus 2 mutants lacking the c-terminal region of E1a protein. Oncogene 4:415–420. 81. Subramanian, T., S. E. Malstrom, and G. Chinnadurai. 1991. Requirement of the C-terminal region of adenovirus E1a for cell transformation in coop-eration with E1b. Oncogene 6:1171–1173.
82. Tavoloni, N., and H. Inoue. 1997. Cellular aging is a critical determinant of primary cell resistance to v-src transformation. J. Virol. 71:237–247. 83. Tavoloni, N., H. Inoue, H. Sabe, and H. Hanafusa. 1994. v-src transformation
of rat embryo fibroblasts. Inefficient conversion to anchorage-independent growth involves heterogeneity of primary cultures. J. Cell Biol. 126:475–483. 84. Toyoshima, H., and T. Hunter. 1994. p27, a novel inhibitor of G1 cyclin-Cdk
protein kinase activity, is related to p21. Cell 78:67–74.
85. Vogelstein, B., and K. W. Kinzler. 1993. The multistep nature of cancer. Trends Genet. 9:138–141.
86. Vollet, J. J., J. S. Brugge, C. A. Noonan, and J. S. Butel. 1977. The role of SV40 gene A in the alteration of microfilaments in transformed cells. Exp. Cell Res. 105:119–126.
87. Wang, H. G., E. Moran, and P. Yaciuk. 1995. E1A promotes association between p300 and pRB in multimeric complexes required for normal bio-logical activity. J Virol. 69:7917–7924.
88. Whyte, P., H. E. Ruley, and E. Harlow. 1988. Two regions of the adenovirus early region 1A proteins are required for transformation. J. Virol. 62:257– 265.
89. Whyte, P., N. M. Williamson, and E. Harlow. 1989. Cellular targets for transformation by the adenovirus E1A proteins. Cell 56:67–75.
90. Wigler, M., A. Pellicer, S. Silverstein, R. Axel, G. Urlaub, and L. Chasin. 1979. DNA-mediated transfer of the adenine phosphoribosyltransferase lo-cus into mammalian cells. Proc. Natl. Acad. Sci. USA 76:1373–1376. 91. Williams, N., T. M. Roberts, and P. Li. 1992. Both p21ras and pp60v-src are
required, but neither alone is sufficient, to activate the raf-1 kinase. Proc. Natl. Acad. Sci. USA 89:2922–2926.
92. Zerler, B., R. J. Roberts, M. B. Mathews, and E. Moran. 1987. Different functional domains of the adenovirus E1A gene are involved in regulation of host cell cycle products. Mol. Cell. Biol. 7:821–829.
93. Zhao, J. F., H. Nakano, and S. Sharma. 1995. Suppression of RAS and MOS transformation by radicicol. Oncogene 11:161–173.