Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Expression of the Pseudorabies Virus Latency-Associated Transcript
Gene during Productive Infection of Cultured Cells
LING JIN
ANDGAIL SCHERBA*
Department of Veterinary Pathobiology, University of Illinois, Urbana, Illinois 61802
Received 19 May 1999/Accepted 25 August 1999
Like other alphaherpesviruses, pseudorabies virus (PrV) exhibits restricted gene expression during latency.
These latency-associated transcripts (LATs) are derived from the region located within 0.69 to 0.77 map units
of the viral genome. However, the presence of such viral RNAs during a productive infection has not been
described. Although several transcripts originating between 0.706 to 0.737 map units have been detected in
PrV-infected cultured cells, their relationship to the LATs has not been examined. Therefore, to determine if
any correlation exists between PrV LAT gene expression in the natural and laboratory systems, transcription
from the LAT gene region during lytic infection of cultured neuronal and nonneuronal cells was evaluated. A
Northern blot assay using single-stranded RNA probes complementary to the spliced in vivo 8.4-kb largest
latency transcript (LLT) detected , 2.0-, and 8.0-kb poly(A) RNAs in all PrV-infected cells lines. The
1.0-and 8.0-kb transcripts partially overlapped the first 1.0-and second exons of the LLT, respectively. In contrast,
portions of both LLT exons comprised the 2.0-kb RNA sequence, which lacked the same intron as the LLT.
Generation of this transcript began about 243 bp downstream of the LLT initiation site and terminated near
the junction of BamHI fragments 8
ⴕ
and 8. Its synthesis was inhibited by cycloheximide but not by cytosine

-
D-arabinofuranoside, which suggests that the 2.0-kb RNA is not an immediate-early gene product. Thus,
although the PrV LAT gene is transcriptionally active during a productive infection of cultured cells, the
resulting RNAs are distinctive from the LLT.
Porcine herpesvirus 1, or pseudorabies virus (PrV), is
clas-sified as a member of the
Alphaherpesvirinae
subfamily within
Herpesviridae
(6, 26, 27). This virus produces neurological,
respiratory, and reproductive disease (Aujeszky’s disease) in
swine, its natural host (1, 6). As with other alphaherpesviruses,
a primary PrV infection can progress into latency in its natural
host (14, 15, 19). During the PrV latent infection, transcription
of the viral genome is restricted to an inverted repeat region in
a manner similar to herpes simplex virus type 1 (HSV-1) (2–4,
10, 13, 21, 30). RNAs, termed latency-associated transcripts
(LATs), are expressed from the region between 0.69 and 0.77
map units (from
Bam
HI fragments 14 to 8) of the viral genome
and are synthesized in the opposite direction relative to IE180
and EP0 gene transcription (10, 21). Several sizes of LATs (8.4,
4.5 to 5.0, 2.0, and 0.95 kb) have been detected in latently
infected porcine trigeminal ganglia (8, 10, 20–22). The largest
latency transcript (LLT; 8.4 kb) expressed from this region is
polyadenylated and spliced to remove a 4.6-kb intron between
nucleotides 1510 (within
Bam
HI fragment 14) and 6164
(with-in
Bam
HI fragment 8
⬘
) relative to the transcriptional start site.
The first and second exon coding sequences of the LLT are
located between
Bam
HI fragments 6-14 and 8
⬘
-5, respectively,
and thus overlap head to tail with either the EP0 or IE180 gene
(Fig. 1). The origins of the other LATs have not been
defini-tively characterized. However, the available data indicate that
all of the LATs detected in latently infected porcine trigeminal
ganglia are complementary to the 3
⬘
end of the IE180 gene (10,
19–21). Therefore, it is possible that these LATs are smaller
processed products from the primary transcript that also gives
rise to the LLT.
HSV-1 (2–4, 26, 30) and bovine herpesvirus 1 (BHV-1) (18,
23–25) have been shown to produce LATs during infection of
cultured mammalian cells; similar expression by PrV has not
been demonstrated. Although several RNAs (2.0, 4.4, 8.2, and
9.5 kb) originating primarily from the LAT intron region have
been detected during a productive infection of cultured cells
(9), a possible correlation(s) between these lytic cycle viral
transcripts and LATs has not been determined. Therefore,
LAT gene expression during a lytic infection was examined by
using reverse transcription (RT)-PCR and Northern
hybridiza-tion with single-stranded RNA (ssRNA) probes
complemen-tary to the in vivo-processed LLT and its intron region. To
investigate possible host effects on LAT gene expression, this
phenomenon was examined in both neuronal and nonneuronal
cell lines.
MATERIALS AND METHODS
Cell lines and virus.Four cell lines permissive for a PrV productive infection were used: Crandall-Rees feline kidney (CRFK); porcine kidney (PK-15); Ma-din-Darby bovine kidney (MDBK); and N1E, a murine neuroblastoma cell line (ATCC C1300; American Type Culture Collection, Rockville, Md.). All four cell lines were maintained in Eagle’s minimum essential medium (MEM) supple-mented with 10% calf serum (HyClone Laboratories, Inc., Logan, Utah), peni-cillin (100 U/ml), streptomycin (100g/ml), and gentamicin (0.25 mg/ml) (Sigma, St. Louis, Mo.).
The Becker (Be) strain of PrV was propagated and titrated in CRFK cells maintained in MEM supplemented with 2% calf serum and antibiotics as de-scribed above. PrV LAT gene expression during a productive infection was investigated in each cell line infected with 5 to 6 PFU/cell of PrV-Be. The effect of protein synthesis inhibition on PrV LAT gene expression was examined by adding 100g/ml of cycloheximide (Sigma) to the inoculum and again in the medium after the adsorption period. To investigate whether PrV LAT gene expression was dependent on DNA synthesis, cytosine-D-arabinofuranoside
(araC; 50g/ml; Sigma) was added to the medium after adsorption as described previously (5).
RNA isolation.Total RNA was isolated from virus-infected or uninfected monolayers (105cells) by using a Trizol RNA extraction kit (Life Technologies,
Inc., Gaithersburg, Md.) according to manufacturer’s instructions. The extracted RNA was suspended in 50l of distilled H2O treated with diethyl pyrocarbonate
(DEPC; Sigma) and then incubated with 1 U of DNase (RNase free; Promega Corp., Madison, Wis.) in 50l of DNase buffer (50 mM Tris [pH 7.2], 10 mM MgCl2, 1 mM dithiothreitol, 5 U of RNase inhibitor) for 30 min at 37°C.
* Corresponding author. Mailing address: Department of
Veteri-nary Pathobiology, College of VeteriVeteri-nary Medicine, University of
Illinois, 2001 South Lincoln, Urbana, IL 61802. Phone: (217) 244-0929.
Fax: (217) 244-7421. E-mail: [email protected].
9781
on November 9, 2019 by guest
http://jvi.asm.org/
Following incubation, the DNase-treated RNA was extracted once with phenol-chloroform (1:1) and then precipitated in 1 volume of 2-propanol at⫺20°C for at least 1 h. RNA pellets were washed once with cold 75% ethanol, suspended in 1 mM EDTA (DEPC treated, pH 7.5), and stored at⫺80°C. Poly(A) RNAs were isolated from 0.5 to 1.0 mg of total RNA by using PolyATract isolation systems (Promega) as instructed by the manufacturer. The poly(A) RNA was eluted in DEPC-treated distilled H2O and then stored at⫺80°C.
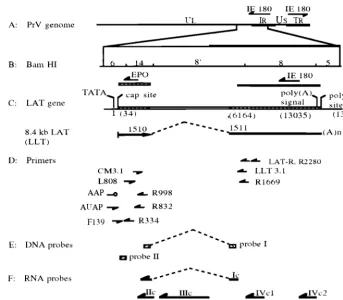
Plasmids.Plasmid pAC38 (provided by A. K. Cheung, National Animal Dis-ease Center, Ames, Iowa) contains 656 bp of the PrV LLT, which consists of a 422-bp upstream and a 234-bp downstream sequence adjacent to the intron splicing sites. Plasmids pB14, pB8⬘, and pB8 were constructed by inserting the PrVBamHI 14, BamHI 8⬘, andBamHI 8 fragments, respectively, into the BamHI site of pBluescript SK II (⫹) (Stratagene, La Jolla, Calif.). Plasmids pBK8⬘and pBN8 were constructed by ligating the remainder of pB8⬘and pB8 after digestion withKpnI andNotI, respectively.
Primers.Oligonucleotides were synthesized by the standard phosphoramidite procedure on an automated synthesizer (Operon Technologies, Inc., Alameda, Calif.). The DNA sequence data necessary for primer construction were ob-tained from the published sequences of PrV genes (GenBank accession no. M57505 [10]). All primer locations relative to the PrV LAT gene region are shown in Fig. 1. Primers R2280, LAT-R, R998, and R832 extend from nucleo-tides 2280 to 2260 (GTCCTCCTCCTCCTCTGCGT), 1613 to 1597 (TGGTGG GAGGTGGACG), 998 to 978 (TCGATACGCTGTTTGACAT), and 832 to 812 (GGTGCACACGGAGGATCTGA), respectively, on the LAT gene anticoding strand. Primers R1669 (GGTGGGAAGAAGTAGAAGGT) and L808 (CTCC GTCAGATCCTCCGTGT) extend from nucleotides 1669 to 1649 and 808 to 828 on the LAT anticoding and coding strands, respectively. Primers LLT3.1 and CM3.1 extend from nucleotides 1578 to 1558 and 1326 to 1346 on the LAT anticoding and coding strands, respectively (10, 11). Primers R334 (ACTTCGG CTCCCGCTC) and F139 (CAGCCATAGAAGACACCGGG) extend from nu-cleotides 334 to 318 and 139 to 158 on the LAT anticoding and coding strands, respectively.
RT-PCR.First-strand cDNA was synthesized from 1 to 3g of total or 50 to 100 ng of poly(A) RNA by using 10 pmol of primer LAT-R or R2280 and Superscript reverse transcriptase II (Life Technologies) according to the manu-facturer’s recommendations. PCR was performed with a volume of 50l con-sisting of 50 mM KCl, 10 mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 0.001% (wt/vol)
gelatin, 200 M each deoxynucleoside triphosphate, 0.6 M each primer (LLT3.1 and CM3.1 or L808 and R1669), 0.25 U ofTaqpolymerase (Perkin-Elmer, Branchburg, N.J.), and 2l of the completed RT reaction mixture. The mixture was subjected to 31 cycles of 94°C for 1 min, 60°C for 45 s, and 72°C for 1 min and then incubated at 72°C for 4 min in a DNA thermal cycler (MJ Research, Inc., Watertown, Mass.). Ten microliters (one-fifth) of each RT-PCR sample was analyzed in an 1.5% agarose gel in TBE buffer (45 mM Tris-borate, [pH 8.0], 1 mM EDTA) and then visualized by UV illumination after staining with ethidium bromide (1g/ml). Commercially available øX174 replicative-form DNA/HaeIII fragments (Life Technologies) served as size markers.
5ⴕRACE.The 5⬘ends of total or poly(A) RNAs were obtained by a 5⬘RACE (amplification of 5⬘cDNA ends) system as instructed by the manufacturer (Life Technologies). Briefly, the first-strand cDNA was synthesized with gene-specific primers LAT-R or R998 as indicated in Fig. 1. Approximately 1 to 3g of total or 500 ng of poly(A) RNA perl was used as the template in a 25-l RT reaction. After purification of the first-strand cDNA, its 5⬘end was tailed with dCTP by using terminal deoxynucleotidyltransferase. The oligo(dC) cDNA was then amplified with a second gene-specific primer, R832, and the abridged anchor primer (AAP) specific for the 5⬘dC tail. The primary PCR products were then reamplified with the heminested gene-specific primer R334 and the abridged universal amplification primer (AUAP) under conditions recom-mended by the manufacturer. The 5⬘RACE products were analyzed as described for the RT-PCR amplimers.
RNA and DNA probes.All ssRNA probes were labeled with [␣-32P]UTP (400
[image:2.612.129.473.71.371.2]Ci/mmol; Amersham Corp., Arlington Heights, Ill.) and generated with known specificity as runoff RNA transcripts from recombinant Bluescript plasmids by using an RNA transcription kit (Stratagene). Probes Ic, IIc and IIa, IIIc, IVc1,
FIG. 1. Schematic of the PrV genome with locations of primers used in RT-PCR and 5⬘RACE as well as probes used for hybridization relative to the LAT gene region. (A) Diagram of the PrV genome and locations of the two IE180 gene copies, with the arrows showing their direction of transcription. (B) Expanded diagram ofBamHI fragments 6, 14, 8⬘, 8, and 5; locations of the IE180 and EP0 genes, with arrows showing their direction of transcription. (C) Location of the LAT gene in relation to PrV-BeBamHI fragments, direction of its transcription (arrow), intron boundaries (dashed line), map units after intron splicing, and the resultant in vivo spliced transcript (LLT, 8.4-kb LAT). (D) Relative locations and orientations of genomic primers CM3.1, F139, L808, LAT-R, LLT3.1, R1669, R2280, R334, R832, and R998 as well as nongenomic primers AAP and AUAP. (E) Relative locations of DNA probes I and II used in Southern hybridization. (F) Relative positions and orientations of ssRNA probes Ic, IIc, IIIc, IVc1, IVc2, and IIa. Probe Ic extends from first to the second exon over the intron region. All probes are drawn in an approximate scale with respect to their relative location in the PrV genome shown in panel B.
on November 9, 2019 by guest
http://jvi.asm.org/
and IVc2 were derived from sequences in pAC38, pB14, pBK8⬘, pB8, and pBN8, respectively (Fig. 1). Probes having the “a” or “c” designation are either the same sense as (a) or antisense to (c) the LATs. Probe Ic is homologous to the entire 656 nucleotides of the processed LLT present in pAC38 and was used to detect LAT gene-specific transcripts. Probes IIc and IIa, produced from the complete PrVBamHI 14 fragment, were used for the detection of the LLT first exon coding sequence and the EP0 gene mRNA, respectively. Probe IIIc, which was used to detect the intron sequence spliced from the LLT, was generated from the PrVBamHI 8⬘sequence located between an internalKpnI site and theBamHI site adjacent to theBamHI 14 fragment. Probe IVc1 was transcribed from the BamHI 8 sequence between an internalSmaI site and theBamHI site distal to theBamHI 8⬘fragment. Probe IVc2 contains theBamHI 8 sequence between an internalNotI site and theBamHI site adjoining theBamHI 5 region. Both probes IVc1 and IVc2 were used for the detection of the LLT second exon coding region.
DNA probe I was derived from the 252-bp PCR product that was amplified from pAC38 with primers CM3.1 and LLT3.1. Primer sequences at both ends were removed neatly by digestion withSmaI andMaeIII, resulting in a 174-bp fragment (Fig. 1), which was used to verify the RT-PCR products made with primer pairs CM3.1 and LLT3.1 or R1669 and L808. DNA probe II was gener-ated from the 200-bp PCR product amplified from the 5⬘end of the first exon of the LLT coding sequence, using primers R340 and F139 with PrV genomic DNA as the template (Fig. 1). The sequence representative of primer R340 was removed byHaeIII digestion of the PCR product, and the remaining portion was used as a probe to verify the 5⬘RACE products. The DNA probes were gel purified and labeled with [␣-32P]dCTP (Amersham Corp.), using a random
priming labeling system (Life Technologies).
Northern blot analysis.Approximately 2 to 5g of poly(A) RNA or 15 to 20 g of total RNA was resolved by electrophoresis in an 1% agarose-formaldehyde gel (2.2 M formaldehyde, 40 mM morpholinepropanesulfonic acid [pH 7.0], 1 mM EDTA, 10 mM sodium acetate). The resolved RNAs were then transferred to a Zeta-Probe membrane (Bio-Rad Laboratories, Richmond, Calif.) in 10⫻ SSC (1.5 M NaCl, 0.15 M sodium citrate [pH 7.0]) overnight. The membrane was rinsed in 2⫻SSC, air dried, and then exposed in a UV cross-linker unit (Bio-Rad Laboratories) for 50 s. Prehybridization was performed at 60°C for at least 4 h in 50% formamide–5⫻Denhardt’s solution (0.1% bovine serum albumin, 0.1% polyvinylpyrrolidone, 0.1% Ficoll)–0.5 M NaCl–10 mM Tris-HCl (pH 8.0)–10 mM EDTA–100g of denatured sonicated salmon sperm DNA per ml–100g of yeast RNA per ml–1% sodium dodecyl sulfate (SDS). Prior to its addition for overnight hybridization at 60°C, the RNA probe was denatured in 50% form-amide at 70°C for 5 min. After hybridization, the blot was sequentially washed once in 1⫻SSC–0.1% SDS at 60°C, once in 1⫻SSC–0.1% SDS at 68°C, and twice in 0.1⫻SSC–0.1% SDS at 68°C for 30-min intervals and finally placed on Cronex 4⫻film (Dupont Corp., Boston, Mass.) at⫺70°C. The blot was reprobed after removal of the previous probe by boiling the membrane twice for 30 min in 0.1% SDS. RNA sizes were estimated by comparison to their mobilities to those of commercially available RNA markers (Ambion, Inc., Austin, Tex.).
Southern hybridization analysis.The RT-PCR and 5⬘RACE products were verified by hybridization with DNA probes I and II, respectively (Fig. 1). The products were separated in an 1.5% agarose gel and then transferred to a Zeta-Probe membrane in the presence of 0.4 N NaOH for a period of at least 4 h. Conditions for prehybridization and hybridization have been previously de-scribed (28).
RT-PCR and 5ⴕRACE product sequence analysis.Gel-purified RT-PCR prod-ucts amplified from total and poly(A) RNA templates were sequenced at the Biotechnology Center (University of Illinois, Urbana) by an automated fluores-cent sequencing method. Gel-purified 5⬘RACE products were separately cloned into the pCR II Vector (Invitrogen Corp., San Diego, Calif.) according to the manufacturer’s instructions and then sequenced as described above. Alignments to previously published PrV LAT sequences (10) were performed with the BLAST function provided by the National Center for Biotechnology Information (19a).
RESULTS
Expression of lytic cycle viral RNAs from the PrV LAT gene.
Expression of the PrV LAT gene during a productive infection
of cultured cells was first investigated by using RT-PCR.
Ini-tially, the cDNAs generated by using primer LAT-R were
subjected to PCR with the primer pair CM3.1 and LLT3.1,
designed to amplify a 252-bp sequence spanning the intron
region of the LLT. When PrV-infected neuronal (N1E) and
nonneuronal (MDBK, CRFK, and PK-15) cells were screened
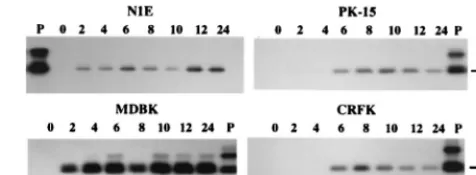
for the presence of a spliced transcript, a 252-bp product could
be generated from the RNA of N1E and MDBK cells starting
at 2 h postinfection (p.i.) and from the RNA of CRFK and
PK-15 cells by 6 h p.i. (Fig. 2). Thereafter, a spliced transcript
was detected in all four cell lines up to 24 h p.i. Amplification
did not occur when the reverse transcriptase was omitted from
the RT reaction or with RNA from mock-infected cells (data
not shown). The correctly sized amplimers also annealed to
DNA probe I, which corresponds to the PrV genomic regions
that flank the LLT intron but is positioned internal to the
primer binding sites (Fig. 1). Thus, at least a portion of the
LAT gene is expressed early in both neuronal and nonneuronal
infected cells. Moreover, either transcription of the LAT gene
is continuous or the transcript remains stable throughout the in
vitro productive infection.
To verify that the transcripts detected in the infected cell
lines are spliced like the LLT, total and poly(A) RNAs from
PrV-infected N1E and MDBK cells were subjected to RT-PCR
using an additional pair of amplification primers (R1669 and
L808). In this case, the predicted size of the amplimer would
increase from 252 to 860 bp. For the first-strand cDNA
syn-thesis, gene-specific primer R2280 (2,280 nucleotides
down-stream of the 5
⬘
end of the LLT) was used (Fig. 1). Both a
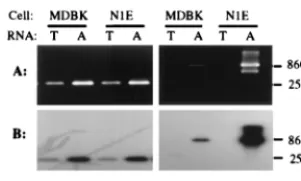
252-bp and an 860-bp product could be amplified from poly(A)
RNA obtained from PrV-infected N1E and MDBK cells at 6 h
p.i., using primers pairs CM3.1-LLT 3.1 and R1169-L808,
re-spectively (Fig. 3A). Once again, both products were
comple-mentary to DNA probe I (Fig. 3B). Therefore, a transcript that
maintained the same polarity and displayed the same splicing
pattern as LLT was detected during a productive infection of
cultured cells.
Sequence analysis of the RT-PCR products.
To determine
the degree of homology between the spliced lytic cycle viral
RNA and the LLT, the 252- and 860-bp RT-PCR products
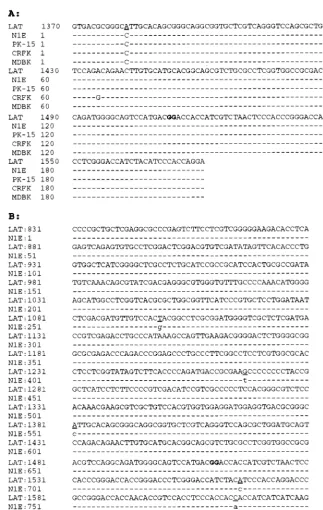
were sequenced. The 252-bp amplimers generated from the
total RNA of each infected cell line were found to have 99%
homology with the corresponding region of the processed LLT
(Fig. 4A). Likewise, the 860-bp amplimer generated from the
poly(A) RNA of infected N1E cells had greater than 98%
homology with the corresponding region of the LLT (Fig. 4B).
The intron boundary in the 252- and 860-bp products is exactly
the same as that of the LLT. Thus, both the LLT and some of
the viral RNAs present during a productive infection of
cul-tured mammalian cells undergo the same posttranscriptional
modification.
[image:3.612.313.551.75.162.2]As to the discrepancies between the nucleotide sequences,
the underlined transversion (A-C) and transition (T-G and
G-T) mutations reflect the genetic differences between the Be
strain used in this study and the Indiana Funkhauser strain
from which the published in vivo LLT was derived (10). In
contrast, the unique transition mutation in the LAT-like RNA
FIG. 2. Autoradiogram of RT-PCR amplicons generated by using total RNA from PrV-Be-infected cells. The identity of the cell line is indicated above each blot. The products were generated by PCR with primers LLT3.1 and CM3.1 and cDNA synthesized from the total RNA extracted from cells at 0 to 24 h p.i. (as indicated above each lane) with primer LAT-R as templates. The resultant amplicons were resolved in an agarose gel, transferred to a nylon membrane, and then hybridized with32P-labeled DNA probe I. The positive control (P) is theresult of PCR amplification of pAC38 using primers CM3.1 and LLT3.1. The expected size of the amplicon arising from a spliced LAT-like transcript is shown on the right.
on November 9, 2019 by guest
http://jvi.asm.org/
from virus-infected CRFK cells (Fig. 4A) and the transversion
in the 830-bp sequence (Fig. 4B) may have occurred during
RT-PCR.
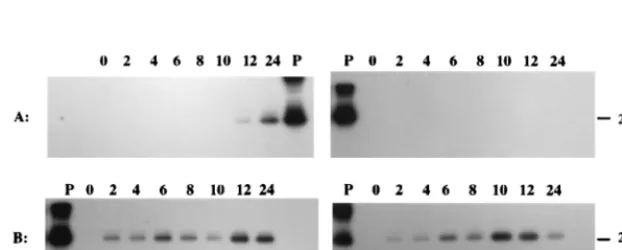
Northern hybridization analysis.
The initial RT-PCR results
and sequence comparisons demonstrated that RNAs having
undergone splicing identical to that of the LLT could be
de-tected in PrV-infected cells at 6 h p.i. (Fig. 2). To explore a
possible relationship between these two types of transcripts,
Northern hybridization was performed with total and poly(A)
RNAs from PrV-infected neuronal (N1E) and nonneuronal
(CRFK, PK-15, and MDBK) cells, using LLT-specific ssRNA
probes. When the ssRNA probe Ic, which contains a 422-bp
upstream and 234-bp downstream contiguous sequence
adja-cent to the intron junction in the processed LLT, was used,
poly(A) transcripts of approximately 1.0, 2.0, and 8.0 kb in size
were detected in infected N1E (Fig. 5B, lane 2), MDBK (Fig.
5C), and PK-15 (Fig. 5C) cells. Since probe Ic is specific for the
LLT region that lacks the intervening sequence present in the
LAT gene, it is possible that the RNA species detected in this
study have homology to only one of the flanking exons in the
LLT. To examine this possibility, RNA probes complementary
to the first (IIc) or second (IVc1) flanking exon were used in a
subsequent hybridization. While a 2.0-kb and 1.0-kb poly(A)
RNA species hybridized with probe IIc (Fig. 5B, lane 4), only
an 8.0-kb and a 2.0-kb poly(A) RNA annealed to probe IVc1
(Fig. 5B, lane 8). Similar results were obtained in
hybridiza-tions of RNAs from PrV-infected nonneuronal cells with
probes IIc and IVc1 (data not shown). Therefore, the 1.0- and
8.0-kb poly(A) RNAs transcribed from the LAT gene during a
lytic viral infection contain at least a portion of the first and
second exons of the LLT, respectively. The 2.0-kb poly(A)
RNA appears to be comprised of part of both exons.
[image:4.612.98.249.72.162.2]Since the 2.0-kb poly(A) RNA annealed to the ssRNA
probes complementary to sequences that are separated by a
distance of 4.8 kb (
Bam
HI fragments 14 [probe IIc] and 8
[probe IVc1]), this lytic cycle viral RNA must be the processed
transcript detected by RT-PCR (Fig. 1). To confirm that this
2.0-kb RNA is the spliced product having an intervening
se-quence between
Bam
HI fragments 14 and 8 removed, the
PrV-infected N1E cellular RNA blot was hybridized with
probe IIIc. This probe is specific for the intron region of the
unprocessed LLT. As shown in Fig. 5B (lane 6), four poly(A)
RNAs (0.9, 1.5, 6.0, and 8.0 kb) having sizes other than 2.0 kb
were detected.
Since the 8.0-kb poly(A) RNA identified by probe Ic did not
hybridize to probe IIc, it is possible that this transcript starts
within the
Bam
HI 8
⬘
fragment downstream of the first exon of
the LLT and extends through
Bam
HI fragments 8 and 5. To
address this possibility, RNA from PrV-infected N1E cells was
hybridized with a probe (IVc2) specific for the region at the
end of the
Bam
HI 8 fragment adjoining the
Bam
HI 5 region.
As shown in Fig. 5B (lane 10), only the 8.0-kb RNA annealed
with this probe. Since this transcript was also complementary
to probes representing the antisense regions of
Bam
HI
frag-ments 8 (IVc1 and IVc2) and 8
⬘
(IIIc) but not 14 (IIc) (Fig. 1),
it appears that transcription of the 8.0-kb RNA initiates within
the LLT intron or the
Bam
HI 8
⬘
fragment and continues into
the
Bam
HI 8
⬘
and 5 regions.
To verify that the probes and the detected RNAs were of the
described specificity and quality, the RNA blot used above
(Fig. 5B) was examined by using ssRNA probe IIa, which is
specific for the EP0 gene mRNA. As shown in Fig. 6, only one
RNA species of a size corresponding to the EP0 gene
tran-script (1.75 kb [10]) annealed to this probe.
Determination of the 5
ⴕ
end of the in vitro 2.0-kb RNA.
To
define the 5
⬘
end of the 2.0 kb RNA expressed during infection
of cultured cells, 5
⬘
RACE was used. Total and poly(A) RNAs
isolated from PrV-infected N1E and MDBK cells at 6 h p.i.
were analyzed. Primers R998 and LAT-R, complementary to
regions of the first or second exon of LLT, respectively, were
used separately for first-strand cDNA synthesis. Regardless of
which cDNA served as the template, an approximately 600- or
100-bp product was generated with primer pair R832-AAP or
AUAP-R334 (heminested primer), respectively (Fig. 7A). The
larger amplimer was produced from both poly(A) and total
RNAs from infected N1E and MDBK cells but could be
visu-alized only after hybridization with DNA probe II, which is
specific for the region of the PrV genome located between the
binding sites of primers R334 and R832 (Fig. 7A, lanes 1 to 4).
However, the 100-bp 5
⬘
RACE product could be readily
ob-served when the primary PCR product was reamplified with
AUAP and the heminested primer R334. This smaller
am-plimer has about 98% sequence homology with the region
extending between nucleotides 243 and 334 of the PrV-Be
LLT. Therefore, synthesis of the 2.0-kb RNA appears to
ini-tiate about 243 bp downstream of the start site of the LLT (Fig.
7B).
Determination of alpha or beta gene origin of the 2.0-kb
lytic cycle viral RNA.
To investigate the effects of protein
synthesis inhibition on the transcription of the 2.0-kb spliced
lytic cycle viral RNA, N1E and MDBK cells were infected with
PrV-Be in the presence of cycloheximide and then maintained
in MEM containing cycloheximide. Production of the 2.0-kb
RNA was monitored between 2 and 24 h p.i. by RT-PCR with
primers LAT-R, CM3.1, and LLT3.1. Amplification of a
spe-cific product from the total RNA isolated from infected N1E
cells between 2 and 24 h p.i. or from MDBK cells before 12 h
p.i. was not observed (Fig. 8A). Presumably, its presence in
MDBK cells after 12 h p.i. was due to an exhaustion of the
cycloheximide. Therefore, generation of this spliced transcript
during a productive infection requires protein synthesis,
indi-cating that it does not originate from an alpha
(immediate-early) gene.
The effect of the DNA synthesis inhibitor, araC, on the
transcription of the 2.0-kb RNA also was examined in both
PrV-infected MDBK and N1E cells. In this case, total RNA
from araC-treated, virus-infected cells was screened for the
presence of a spliced transcript as described above. As shown
in Fig. 8B, a product of the correct size (252 bp) could be
amplified from RNA isolated after 2 h p.i. from either infected
cell line but not from uninfected cells. Thus, the spliced 2.0-kb
RNA encoded by the LAT gene is not dependent on viral
FIG. 3. Electrophoregram and autoradiogram of RT-PCR products derivedfrom total (lanes T) and poly(A) (lanes A) RNAs isolated from PrV-infected N1E and MDBK cells at 6 h p.i. Products were generated by using cDNAs synthesized from RNAs with primer R2280. Primer pairs LLT3.1-CM3.1 and R1669-L808 were used for the specific amplification of a spliced region analo-gous to that of the LLT. The products were resolved in an 1.5% agarose gel (A), transferred to a nylon membrane, and hybridized with32P-labeled DNA probe I
(B). The anticipated amplicon sizes are shown on the right.
on November 9, 2019 by guest
http://jvi.asm.org/
DNA synthesis and therefore is probably a beta (early) gene
transcript.
DISCUSSION
Expression of the PrV LAT gene during a productive
infec-tion of cultured cells has not been previously documented.
Although it has been reported that RNAs with the same
po-larity as the LATs were produced from the PrV
Bam
HI 8
⬘
[image:5.612.133.456.66.576.2]region, an association between these lytic cycle viral transcripts
and the RNAs expressed during in vivo latency was not
estab-lished (9). Our study demonstrated that the PrV LAT gene is
actively transcribed during a productive infection of various
mammalian cell lines. At least three lytic cycle viral poly(A)
RNAs originate from that portion of the LAT gene extending
from the
Bam
HI 14 to 8 region of the PrV genome. They are
difficult to detect unless first resolved on the basis of
polyad-enylation (Fig. 5). Of these poly(A) RNA species, only the
FIG. 4. Sequence alignment of the in vitro PrV LAT RT-PCR product with the reported in vivo cDNA sequence of the PrV LLT (GenBank accession no. M57505 [9]). (A) The sequence sources (in vivo LLT cDNA and the 208-bp RT-PCR products from the corresponding PrV-infected cells) are shown on the left. (B) LAT, the cDNA sequence of the LLT expressed during latency; N1E, the sequence of the 831-bp RT PCR products generated by using poly(A) RNA from PrV-infected N1E cells at 6 h p.i. with primers R2280, R1669, and L808. Dashes, identity to the published sequence; underlined nucleotides, transversions in the PrV-Be strain genome. The juncture between the first and second exons of the processed LLT is between the boldface nucleotides.
on November 9, 2019 by guest
http://jvi.asm.org/
2.0-kb one was determined to have the same intron removed as
the LLT. Moreover, apparently the entire nucleotide sequence
of the 2.0-kb RNA is contained within that of the LLT. Since
its transcriptional start site is about 243 bp downstream of the
one for the LLT, synthesis of the 2.0-kb RNA may be regulated
by the closely positioned LAP2 promoter, located about 40 to
50 nucleotides upstream of the predicted start site (10, 12).
This conclusion is supported by our LAT promoter studies in
which removal of the LAP1 promoter abolished LLT
produc-tion but did not alter expression of the 2.0-kb RNA
(unpub-lished data). Moreover, it should be noted that the analogous
herpes simplex virus LAP2 promoter is a LAT gene
transcrip-tional regulatory element (7). Although the PrV-encoded
2.0-kb RNA species could be detected as early as 2 h p.i. in
infected cell cultures, inhibition of its synthesis by
cyclohexi-mide but not by araC indicated that its source is a beta class
gene.
[image:6.612.130.471.72.424.2]The 2.0-kb lytic cycle viral RNA can be considered to be
either a special product made only during a productive
infec-tion or a homolog to the one made during latency. Although
clear distinction between these two possibilities awaits
se-quencing of the in vivo spliced 2.0-kb LAT, its similarity in size
FIG. 5. Northern hybridization of total and poly(A) RNAs from PrV-infected cells. (A) Polarity and relationship of probes and transcripts from the LAT gene region during latency (in vivo) and productive infection of cultured cell lines. The arrow above transcripts represents the direction of transcription of the LAT gene. (B) Results of Northern hybridization of total (lanes T) and poly(A) (lanes A) RNAs from PrV-infected neuronal cells at 6 h p.i. RNAs were separated in a 2.2 M formaldehyde–1% agarose gel and then transferred to a nylon membrane. The blot was sequentially hybridized with32P-labeled ssRNA probes Ic, IIc, IVc1, IIIc, and
IVc2. Molecular size markers are indicated on the right. (C) At 6 h p.i., RNAs were isolated from PK-15 (lanes P), CRFK (lanes C), and MDBK (lanes M) cells. The RNAs [total (lanes T) and poly(A) (lanes A)] were separated in a 2.2 M formaldehyde–1% agarose gel and then transferred to a nylon membrane. The blot was hybridized with32P-labeled ssRNA probe Ic. Molecular size markers are indicated on the right.
FIG. 6. Northern hybridization of total and poly(A) RNAs from PrV-in-fected cells. RNAs from PK-15 (lanes P), MDBK (lanes M), CRFK (lanes C), and N1E (lanes N) cells were harvested at 6 h p.i., separated in a 2.2 M formaldehyde–1% agarose gel, and then transferred to a nylon membrane. RNAs were hybridized with32P-labeled ssRNA probe IIa, which is
complemen-tary to the EP0 mRNA and has the same polarity as LAT. Molecular size for the EP0 mRNA is indicated on the right.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.312.551.519.666.2]to the lytic cycle viral RNA may not be simply fortuitous. A
consensus polyadenylation signal (AATAAA) is found to be
located about 170 bp upstream of the junction between the
Bam
HI 8
⬘
and 8 fragments. This position is near the predicted
termination site for both the in vivo 2.0-kb LAT (8) and the
lytic cycle viral 2.0-kb transcript described in this study. Thus,
at least these two RNAs may have a common 3
⬘
terminus.
Based on the polyadenylation status of the 2.0-kb lytic cycle
viral RNA, this transcript could be translated. In this regard,
the RNA has two potential open reading frames (ORFs)
ca-pable of encoding a 20-kDa (ORF1, 163 amino acids) and a
47-kDa (ORF2, 393 amino acids) protein. The postulated
pro-tein encoded by ORF1 has 44% homology to the apoptosis
repressor ARC (17), 36% homology to a protein kinase C
substrate (29), and 30% homology to a serine/threonine
pro-tein kinase (16). In contrast, homology of the predicted ORF2
protein with any known proteins cannot be established. It
would be interesting to determine if either putative protein is
actually made during an in vivo productive or latent infection.
Two other lytic cycle viral poly(A) RNAs transcribed from
the LAT gene region were also determined to be partially
colinear with the LLT. The first, an 1.0-kb RNA, seems to
overlap only the first exon sequence of the LLT, since it was
complementary only to those ssRNA probes (Ic and IIc) which
are specific for this region (Fig. 1). The second, an 8.0-kb RNA
species, is also different from the LLT since (i) it hybridizes
with a ssRNA probe (IIIc) specific for the intron sequence
within the Bam HI 8
⬘
fragment and (ii) it did not anneal with
probe IIc, which is specific for the 3
⬘
half of the first exon as
well as the first 200 nucleotides of the intron. Thus, since this
transcript was also complementary to ssRNA probes IVc1 and
Ivc2 with specificity for either the 5
⬘
or 3
⬘
end of the
Bam
HI 8
fragment, it is reasonable to conclude that transcription of this
RNA initiates within the
Bam
HI 8
⬘
region and contains most
of the LLT second exon coding sequence. Based on the
ap-parent weak hybridization signal obtained between probe Ic
and an 8.0-kb RNA derived from infected CRFK cells,
pro-duction of this 8.0-kb species seems to be relatively low in this
cell line (Fig. 5C).
[image:7.612.57.292.72.242.2]In addition to the three viral lytic cycle RNA products
de-tected by the ssRNA probes specific for the LLT, three poly(A)
RNA species (approximately 0.9, 1.5, and 6.0 kb) were found
to be transcribed from the LAT gene within the
Bam
HI 8
⬘
region. They could be detected by hybridization with a ssRNA
probe (IIIc) specific for the intron region of the LLT but not by
probes IIc and IVc2, which are complementary to the first and
second LAT exons, respectively. Therefore, they either have
different initiation sites or are alternative splicing products
from transcripts giving rise to the 2.0-kb lytic cycle viral RNA.
Furthermore, they appear to be different from the previously
documented
Bam
HI 8
⬘
and 8 fragment-derived RNAs (2.0, 4.4,
8.2, and 9.5 kb) in PrV-infected MDBK cells (9). The presence
of those transcripts was detected by ssRNA and
double-stranded DNA probes derived from the second exon coding
sequence of LLT. It was shown that a 220-nucleotide ssRNA
probe (representing the 3
⬘
end of the
Bam
HI 8
⬘
fragment) and
a 1,250-nucleotide ssRNA probe (comprised of 550
nucleo-tides from the 3
⬘
end of the
Bam
HI 8
⬘
fragment and 700 bases
of the 5
⬘
end of the
Bam
HI 8 region) could anneal to
similar-sized viral RNA products in an S1 nuclease protection assay
(9). However, a ssRNA probe derived from the entire
Bam
HI
FIG. 7. Autoradiogram and sequence analysis of 5⬘RACE productgener-ated by using RNAs from PrV-infected N1E and MDBK cells. The cDNA for 5⬘ RACE analysis was generated by using total RNA (MDBK [MT] and N1E [NT]) or poly(A) RNA (MDBK [MA] and N1E [NA]) from infected cells 6 h p.i. with primer R998 (for MT and NT) or LAT-R (for MA and NA). (A) Autoradiogram of 5⬘RACE products amplified only with primers R832 and AAP (lanes 1 to 4) or subsequently with primers R334 and AUAP (lanes 5 to 8) and then hybridized to DNA probe II. (B) Sequence alignment of the 100-bp 5⬘RACE product with the reported cDNA sequence of the PrV LLT (GenBank accession no. M57505 [9]). 5⬘RACE, product generated by using total RNA from infected N1E cells with primers AUAP and R334; LAT, published cDNA sequence of the LLT. Nucleotides in lowercase are insertions that occurred in the PrV-Be genome. The underlined nucleotides represent the genetic differences between PrV-Be and the published in vivo PrV-Funkhauser LLT cDNA sequence.
FIG. 8. Autoradiograms of the LAT RT PCR amplicons generated by using total RNA from PrV-infected cells treated with cycloheximide or araC. Virus-infected MDBK and N1E cells were treated with either cycloheximide (A) or araC (B) prior to total RNA extraction at 0 to 24 h p.i. (as indicated above each blot). RT-PCR products were synthesized from total RNA by using primers LAT-R (RT) and CM3.1 plus LLT3.1 (PCR). The products were resolved in an 1.5% agarose gel, transferred to a nylon membrane, and then hybridized with32P-labeled DNA probe I. The positive control (P) is the PCR product obtained with pAC38 and primers
CM3.1 and LLT3.1. The expected size of the amplicon arising from a spliced LAT-like transcript is shown on the right.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.148.459.559.684.2]8
⬘
fragment could protect only 550 to 1,000 nucleotides of
RNAs obtained from infected MDBK cells (9). Thus, the
pre-viously described 2.0- and 8.2-kb
Bam
HI 8
⬘
-derived RNAs may
be homologues of the 2.0- and 8.0-kb transcripts detected in
this study.
A 4.0- and 5.0-kb poly(A) RNAs (Fig. 5C) and an apparently
nonpolyadenylated 9.5-kb RNA (Fig. 5B, lanes 2 and 7; Fig.
5C) were detected in PrV-infected PK-15 and MDBK cells by
hybridization with probe Ic. The inability to always detect the
largest transcript may be attributed to variability in its relative
amount. The 4.0-kb transcript may be homologous to the
4.4-kb
Bam
HI 8
⬘
-derived RNA detected in MDBK cells by
Cheung (9). The nature of the other two transcripts is
un-known, but these three appear to be preferentially expressed in
nonneuronal cell lines.
In conclusion, the PrV LAT gene region is far from being
quiescent during a lytic infection of cultured mammalian cells.
Its activity during an in vivo productive infection of the natural
host awaits examination. Uncovering the nature of LAT gene
expression during both a productive and a latent infection of
the natural host may aid in understanding the mechanism by
which herpesvirus latency is established.
ACKNOWLEDGMENTS
We are grateful to W. M. Schnitzlein, M. H. Vodkin, and D. C.
Bloom for helpful discussions and review of the manuscript. We also
thank A. K. Cheung for providing plasmid pAC38.
This work was supported in part by a grant from the USDA Animal
Health and Disease (ILLU-70-0989) and in part by a grant from the
University of Illinois Campus Research Board, log no. 95126.
REFERENCES
1.Baskerville, A., J. B. McFerran and C. Dow.1973. Aujeszky’s disease in pigs. Vet. Bull.43:465–480.
2.Bennett, J. L., and D. H. Gilden.1996. The molecular genetics of herpes simplex virus latency and pathogenesis: a puzzle with many pieces still miss-ing. J. Neurovirol.2:225–229.
3.Black, D. L.1995. Finding splice sites within a wilderness of RNA. RNA 1:763–771.
4.Block, T. M., and J. M. Hill.1997. The latency associated transcript (LAT) of herpes simplex virus: still no end in sight. J. Neurovirol.3:313–321. 5.Bratanich, A. C., and J. Jones.1992. Localization ofcis-acting sequences in
the latency-related promoter of bovine herpesvirus 1 which are regulated by neuronal cell type factors and immediate-early genes. J. Virol.66:6099–6106. 6.Brown, F.1989. The classification and nomenclature of viruses: summary of results of meeting of the international committee on taxonomy of viruses in edmonton, Canada 1987. Intervirology30:181–186.
7.Chen, X., M. C. Schmidt, W. F. Goins, and J. C. Glorioso.1995. Two herpes simplex virus type 1 latency-acting promoters differ in their contributions to latency-associated transcript expression during lytic and latent infections. J. Virol.69:7899–7908.
8.Cheung, A. K.1989. Detection of pseudorabies virus transcripts in trigeminal ganglia of latently infected swine. J. Virol.63:2908–2913.
9.Cheung, A. K.1990. TheBamHI J fragment (0.76 to 0.737 map units) of pseudorabies virus is transcriptionally active during viral infection. J. Virol. 64:977–983.
10. Cheung, A. K.1991. Cloning of the latency gene and the early protein O gene of pseudorabies virus. J. Virol.65:5260–5271.
11. Cheung, A. K.1994. Detection of the large latency transcript of pseudorabies virus by RNA-PCR and its potential in diagnosis. J. Vet. Diagn. Investig. 6:483–486.
12. Cheung, A. K., and T. A. Smith.1999. Analysis of the latency-associated transcript/UL-3.5 gene cluster promoter complex of pseudorabies virus. Arch. Virol.144:381–391.
13. Enquist, L. W.1994. Infection of the mammalian system by pseudorabies virus (PRV). Semin. Virol.5:221–231.
14. Gutekunst, D. E.1979. Latent pseudorabies virus infection in swine detected by RNA-DNA hybridization. Am. J. Vet. Res.40:1568–1572.
15. Gutkunst, D. E., E. C. Pirtle, L. D. Miller, and W. C. Stewart.1980. Isolation of pseudorabies virus from trigeminal ganglia of a latently sow. Am. J. Vet. Res.41:1315–1316.
16. Leung-Tack, P., J. C. Audonnet, and M. Riviere.1994. The complete DNA sequence and genetic organization of the short unique region (Us) of the bovine herpesvirus type 1 (ST strain). Virology199:409–421.
17. Koseki, T., N. Inohara, S. Chen, and G. Nunez.1998. ARC, an inhibitor of apoptosis expressed in skeletal muscle an heart that interacts selectively with caspases. Proc. Natl. Acad. Sci. USA95:5156–5160.
18. Kutish, G., T. Mainprize, and D. L. Rock.1990. Characterization of the latency-related transcriptionally active region of the bovine herpesvirus 1 genome. J. Virol.64:5730–5737.
19. Lokensgard, J. R., D. G. Thawley, and T. W. Molitor.1990. Pseudorabies virus latency: restricted transcription. Arch. Virol.110:129–136.
19a.National Center for Biotechnology Information.17 May 1999, revision date. BLAST. [Online.] http://www.ncbi.nlm.nih.gov/Blast. National Center for Biotechnology Information, National Institutes of Health, Bethesda, Md. [4 June 1999, last date accessed.]
20. Priola, S. A., D. P. Gustafson, E. K. Wagner, and J. G. Stevens.1990. A major portion of the latent pseudorabies virus genome is transcribed in trigeminal ganglia of pigs. J. Virol.64:4755–4760.
21. Priola, S. A., and J. G. Stevens.1991. The 5⬘and 3⬘limits of transcription in the pseudorabies virus latency associated transcription unit. Virology182: 852–856.
22. Rock, D. L., W. A. Hagemoser, F. A. Osorio and H. A. McAllister.1988. Transcription from the pseudorabies virus genome during latent infection. Arch. Virol.98:99–106.
23. Rock, D. L., W. A. Hagesmoser, F. A. Osorio, and D. E. Reed.1986. Detec-tion of bovine herpesvirus type 1 RNA in trigeminal ganglia of latently infected rabbits by in situ hybridization. J. Gen. Virol.67:2515–2520. 24. Rock, D. L., S. L. Beam, and J. E. Mayfield.1987. Mapping bovine
herpes-virus type 1 latency-related RNA in trigeminal ganglia of latently infected rabbits by in situ hybridization. J. Virol.61:3827–3831.
25. Rock, D. L.1994. Latent infection with bovine herpesvirus type 1. Semin. Virol.5:233–240.
26. Roizman, B.1996. Herpesviridae, p. 2221–2230.InB. N. Fields, D. M. Knipe, P. M. Howley, et al (ed.), Fields virology, 3rd ed. Lippinocott-Raven Pub-lishers, Philadelphia, Pa.
27. Roizman, B., L. Denrosiers, B. Fleckenstein, C. Lopez, A. C. Minson, and M. J. Studdert.1992. The family herpesviridae: an update. Arch. Virol. 123:425–449.
28. Scherba, G., L. Jin, W. M. Schnitzlein, and M. H. Vodkin.1992. Differential polymerase chain reaction for detection of wild-type and a vaccine strain of Aujeszky’s disease. J. Virol. Methods38:131–144.
29. Stumpo, D. J., J. M. Graff, K. A. Albert, P. Greengard, and P. J. Blackshear. 1989. Molecular cloning, characterization, and expression of a cDNA encod-ing the ‘80- to 87-kDa’ myristoylated alanine-rich C kinase substrate: a major cellular substrate for protein kinase C. Proc. Natl. Acad. Sci. USA86:4012– 4016.
30. Wagner, E. K., and D. C. Bloom.1997. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev.10:419–443.
on November 9, 2019 by guest
http://jvi.asm.org/