0022-538X/11/$12.00
doi:10.1128/JVI.05584-11
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Compensation by the E138K Mutation in HIV-1 Reverse Transcriptase
for Deficits in Viral Replication Capacity and Enzyme Processivity
Associated with the M184I/V Mutations
䌤
Hong-Tao Xu,
1Eugene L. Asahchop,
1Maureen Oliveira,
1Peter K. Quashie,
1Yudong Quan,
1Bluma G. Brenner,
1and Mark A. Wainberg
1,2,3*
McGill University AIDS Centre, Lady Davis Institute for Medical Research, Jewish General Hospital, Montreal, Quebec, Canada,
1and Departments of Medicine
2and Microbiology and Immunology,
3McGill University, Montreal, Quebec, Canada
Received 1 July 2011/Accepted 8 August 2011
Recently, several phase 3 clinical trials (ECHO and THRIVE) showed that E138K and M184I were the most
frequent mutations to emerge in patients who failed therapy with rilpivirine (RPV) together with two
nucle-os(t)ide reverse transcriptase inhibitors, emtricitabine (FTC) and tenofovir (TDF). To investigate the basis for
the copresence of E138K and M184I, we generated recombinant mutated and wild-type (WT) reverse
trans-criptase (RT) enzymes and HIV-1
NL4-3infectious clones. Drug susceptibilities were determined in cord blood
mononuclear cells (CBMCs). Structural modeling was performed to analyze any impact on deoxynucleoside
triphosphate (dNTP) binding. The results of phenotyping showed that viruses containing both the E138K and
M184V mutations were more resistant to each of FTC, 3TC, and ETR than viruses containing E138K and
M184I. Viruses with E138K displayed only modest resistance to ETR, little resistance to efavirenz (EFV),
and no resistance to either FTC or 3TC. E138K restored viral replication capacity (RC) in the presence of
M184I/V, and this was confirmed in cell-free RT processivity assays. RT enzymes containing E138K, E138K/
184I, or E138K/184V exhibited higher processivity than WT RT at low dNTP concentrations. Steady-state
kinetic analysis demonstrated that the E138K mutation resulted in decreased
K
ms for dNTPs. In contrast,
M184I/V resulted in an increased
K
mfor dNTPs compared to those for WT RT. These results indicate that the
E138K mutation compensates for both the deficit in dNTP usage and impairment in replication capacity by
M184I/V. Structural modeling shows that the addition of E138K to M184I/V promotes tighter dNTP binding.
The reverse transcriptase (RT) of human immunodeficiency
virus type 1 (HIV-1) is a multifunctional enzyme possessing
both RNA- and DNA-dependent DNA polymerase (RDDP
and DDDP, respectively) activities, as well as an RNase H
activity (12). Due to its crucial role in viral replication, HIV-1
RT is an important target for anti-HIV drugs that currently
include nucleoside reverse transcriptase inhibitors (NRTIs),
i.e., zidovudine (AZT or ZDV), didanosine (ddI), stavudine
(d4T), zalcitabine(ddC), lamivudine(3TC), emtricitabine (FTC),
and abacavir (ABC) and a nucleotide reverse transcriptase
inhibitor, tenofovir disoproxil fumarate (TDF), as well as the
nonnucleoside reverse transcriptase inhibitors (NNRTIs)
nevirapine (NVP), delavirdine (DLV), efavirenz (EFV), and
etravirine (ETR). Both NRTIs and NNRTIs are key
compo-nents of highly active antiretroviral therapy (HAART), which
has led to significant declines in HIV-associated morbidity and
mortality (23, 33). However, the development of HIV drug
resistance is a formidable obstacle to the long-term success of
antiretroviral treatment (36, 40), and resistance mutations
have been described for all antiretroviral drugs currently in use
(21).
The rapid replication rate of HIV-1 and the error-prone
nature of its RT drive the development of drug resistance (38).
Resistance mutations arise prior to therapy due to errors in
HIV-1 replication and also can be selected during viral
repli-cation in the presence of incompletely suppressive drug
regi-mens. In the case of NRTIs and NNRTIs, drug resistance can
be due to either single mutations or the accumulation of
mu-tations specific for each individual drug in the HIV-1 RT
genes. First-generation NNRTIs have a low genetic barrier for
resistance, as only a single mutation, such as K103N, is
suffi-cient to confer diminished susceptibility. The same is true for
certain NRTIs, since high-level resistance to FTC and 3TC can
be conferred by the M184V and M184I mutations (39).
Etravirine (ETR) and rilpivirine (RPV) are
second-genera-tion NNRTIs. ETR is a component of therapy for
treatment-experienced patients, while RPV was recently approved by the
Food and Drug Administration for use in drug-naive patients.
ETR is active against HIV-1 containing RT mutations that
confer resistance to the first-generation NNRTIs (8). ETR also
has a high genetic barrier for resistance, requiring the
accu-mulation of several NNRTI-associated mutations for
high-level resistance to become manifest (4). The DUET clinical
trials identified 17 resistance-associated mutations (RAMs),
including V90I, A98G, L100I, K101E/H/P, V106I, E138A,
V179D/F/T, Y181C/I/V, G190A/S, and M230L, that are
asso-ciated with diminished susceptibility to ETR (16, 27, 37). Cell
culture selection experiments with ETR showed that E138K
was the first mutation to emerge, and it conferred low-level
resistance to ETR; E138K also was found to result in lower
viral replication capacity (3). The International AIDS
Society-* Corresponding author. Mailing address: McGill AIDS Centre,
Lady Davis Institute for Medical Research, Jewish General Hospital,
3755 Co
ˆte-Ste-Catherine Rd., Montreal, Quebec H3T 1E2, Canada.
Phone: (514) 340-8260. Fax: (514) 340-7537. E-mail: mark.wainberg
@mcgill.ca.
䌤
Published ahead of print on 17 August 2011.
11300
on November 7, 2019 by guest
http://jvi.asm.org/
USA (IAS-USA) drug resistance mutation list includes both
E138K and E138G for ETR resistance (17). Recently, two
phase 3 clinical trials (ECHO and THRIVE) on the use of
RPV/TDF/FTC in drug-naïve patients showed that E138K
confers cross-resistance to RPV and ETR and that the
com-bination of the E138K/M184I mutations was common in
pa-tients at treatment failure (20). These results indicate that
E138K is a signature mutation of relevance for the
second-generation NNRTIs ETR and RPV.
In this study, we employed both enzymatic and cell-based
assays to assess the impact of the E138K mutation in
combi-nation with M184I or M184V on enzyme processivity, viral
replication capacity, and phenotypic drug susceptibility. We
also have assessed why E138K/M184I was favored over E138K/
M184V in clinical trials with RPV/FTC/TDF.
MATERIALS AND METHODS
Chemicals, cells, and nucleic acids.Etravirine (ETR) was a gift of Tibotec (Titusville, NJ). Emtricitabine (FTC) was kindly provided by Gilead Sciences (Foster City, California). Lamivudine (3TC) was a gift of Glaxo-SmithKline (Greenford, United Kingdom). Efavirenz (EFV) was obtained from Bristol-Myers Squibb (Princeton, NJ).
Cord blood mononuclear cells (CBMCs) were obtained through the Depart-ment of Obstetrics, Jewish General Hospital, Montreal, Canada. The HEK293T cell line was obtained from the American Type Culture Collection. The following reagents and cells were obtained through the NIH AIDS Research and Refer-ence Reagent Program: the infectious molecular clone pNL4-3 was from Mal-colm Martin, and the TZM-bl (JC53-bl) cells were from John C. Kappes, Xiaoyun Wu, and Tranzyme Inc.
The pNL4.3PFB proviral DNA is a generous gift from Tomozumi Imamichi, National Institutes of Health, Bethesda, MD. pRT6H-PROT is a generous gift from Stuart F. J. Le Grice, National Institutes of Health, Bethesda, MD.
A 497-nucleotide (nt) HIV-1 PBS RNA template spanning the 5⬘untranslated
region (UTR) to the primer binding site (PBS) wasin vitrotranscribed from ACC
I-linearized pHIV-PBS DNA (2) by using a T7-MEGA shortscript kit (Ambion, Austin, TX) as described previously (42). The oligonucleotides used in this study were synthesized by Integrated DNA Technologies Inc. (Coralville, IA) and
purified by 6% polyacrylamide–7 M urea gel electrophoresis. For 5⬘-end labeling
of oligonucleotides with [␥-32
P]ATP, the Ambion KinaseMax kit was used, fol-lowed by purification through Ambion NucAway spin columns according to protocols provided by the supplier (Applied Biosystems, Streetsville, Canada). The names and sequences of the oligonucleotides used in this study are the
following: NLBalF, 5⬘-TGGCCATTGACAGAAGAAAAAATAAAAGCA-3⬘;
NLPFLMR, 5⬘-ATAATACACTCCATGTACtGGTTC-3⬘; and D25, 5⬘-GGAT
TAACTGCGAATCGTTCTAGCT-3⬘.
Site-directed mutagenesis and preparation of virus stocks. To construct
HIV-1 RT expression plasmids and HIV-1NL4-3variants harboring desired
mu-tations in the RT gene, site-directed mutagenesis reactions were carried out using a QuikChange II XL site-directed mutagenesis kit (Stratagene, La Jolla, CA) on HIV-1 RT expression plasmid pbRT6H-PROT (42), which contains the
RT (p66) coding region of HIV-1NL4-3(GenBank accession number AF324493)
with a C-terminal His tag and was constructed from pRT6H-PROT (32). To
make recombinant HIV-1NL4-3viruses containing the desired RT mutations, we
first amplified fragments spanning RT amino acids (aa) 25 to 314 from the pbRT6H-PROT variants by PCR using primers NLBalF and NLPFLMR. After digestion with restriction enzymes MscI and PflMI, the resultant 871-bp mutant DNAs were used to replace the corresponding 871-bp fragment of pNL4.3PFB proviral DNA. DNA sequencing was performed to verify the absence of spurious
mutations and the presence of any desired mutation. Recombinant HIV-1NL4-3
wild-type and mutant viruses were generated by the transfection of the corre-sponding proviral plasmid DNAs into HEK293T cells using Lipofectamine 2000 (Invitrogen, Burlington, Canada) according to the manufacturer’s instructions. Viral supernatants were harvested at 48 h posttransfection, centrifuged for 5 min
at 800⫻gto remove cellular debris, filtered through a 0.45-m-pore size filter,
aliquoted, and stored at⫺80°C. Levels of p24 in viral supernatant were measured
by a Perkin-Elmer HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) kit. Virion-associated RT activity was measured as described previously
(12) with 50l of RT reaction mixture containing 10l of culture supernatants;
0.5 U/ml of poly(rA)/p(dT)12-18template/primer (T/P) (Midland Certified
Re-agent Company, Midland, TX) in 50 mM Tris-HCl, pH 7.8; 75 mMKCl; 5 mM
dithiothreitol; 5 mM MgCl2; 0.05% Triton X-100; 2% ethylene glycol; 0.3 mM
reduced glutathione; and 5Ci of [3H]dTTP (70 to 80 Ci/mmol; 2.5 mCi/ml).
Following a 240-min incubation at 37°C, the reaction mixture was quenched by adding 0.2 ml of 10% cold trichloroacetic acid (TCA) and 20 mM sodium pyrophosphate and incubated for at least 30 min on ice. The precipitated prod-ucts were filtered onto Millipore 96-well MutiScreen HTS FC filter plates
(MSFCN6B) and sequentially washed with 200l of 10% TCA and 150l of
95% ethanol. The radioactivity of incorporated products was analyzed by liquid scintillation spectrometry using a Perkin-Elmer 1450 MicroBetaTriLux micro-plate scintillation and luminescence counter.
Replication capacity in TZM-bl cells.The relative replicative capacities of
recombinant wild-type HIV-1NL4-3and E138K, M184I, M184V, E138K/M184I,
and E138K/M184V mutants were evaluated in a noncompetitive short-term infectivity assay using TZM-bl cells as previously described (3, 43). Twenty thousand cells per well were added in triplicate into a 96-well culture plate in 100
l of Dulbecco’s modified Eagle medium (DMEM) (Invitrogen) supplemented
with 10% fetal bovine serum (Gibco), 1% penicillin-streptomycin, and 1%L
-glu-tamine (Invitrogen). Viral stocks for both wild-type and mutant viruses were normalized by p24, and recombinant viruses were serially diluted 2-fold from
viral stock suspensions. After 4 h, 50l of DMEM was removed from the wells
and replaced by 50l of virus dilution; a control well did not contain virus. Virus
and cells were cocultured for 48 h, after which 100l of Bright-Glo reagent was
added and luciferase activity measured in a luminometer as described in the manufacturer’s instructions (Promega). The viral replication level was expressed as a percentage of relative light units (RLU) with reference to wild-type virus.
Phenotypic drug susceptibility assays.Phenotypic susceptibility analysis of the RT inhibitors ETR, FTC, EFV, and 3TC was performed with recombinant
HIV-1NL4-3clones in a cell-basedin vitroassay as described previously (3).
Briefly, CBMCs were infected for 2 h with either wild-type recombinant virus or mutants, washed with cold Dulbecco’s phosphate-buffered saline to remove un-bound virus, and plated onto 96-well plates in the presence of each RT inhibitor. After 3 days in culture, the culture wells were refreshed with media containing the corresponding drug dilutions. After 7 days, the culture supernatants were collected and analyzed for RT activity to determine the dose-response curve. The
EC50(50% effective concentration) was calculated using the program GraphPad
Prism (GraphPad Software, San Diego, CA).
Recombinant reverse transcriptase expression and purification.Recombinant RTs in heterodimeric forms were expressed from plasmid pbRT6H-PROT and purified as described previously, with minor modifications (42). In brief, RT
expression inEscherichia coliM15 (pREP4) (Qiagen, Mississauga, Canada) was
induced with 1 mM isopropyl--D-thiogalactopyranoside (IPTG) at room
tem-perature. The pelleted bacteria were lysed under native conditions with Bug-Buster protein extraction reagent containing benzonase (Novagen, Madison, WI) according to the manufacturer’s instructions. After clarification by high-speed centrifugation, the clear supernatant was subjected to the batch method of nickel-nitrilotriacetic acid (Ni-NTA) metal-affinity chromatography (QIAexpres-sionist) (Qiagen). All buffers contained Complete protease inhibitor cocktail (Roche). Histidine-tagged RT was eluted with an imidazole gradient. RT-con-taining fractions were pooled, passed through DEAE-Sepharose (GE Health-care), and further purified using SP-Sepharose (GE Healthcare, Mississauga, Canada). Fractions containing purified RT were pooled, dialyzed against storage buffer (50 mM Tris-HCl [pH 7.8], 50 mM NaCl, and 50% glycerol), and con-centrated to 4 to 8 mg/ml with Centricon Plus-20 MWCO30 kDa (Millipore).
Aliquots of proteins were stored at⫺80°C. Protein concentration was measured
by a Bradford protein assay kit (Bio-Rad Laboratories), and the purity of the recombinant RT preparations was verified by SDS-PAGE. The polymerase ac-tivity of each recombinant RT preparation was evaluated in duplicate as de-scribed previously (29) using various amounts of RTs and a synthetic
homopoly-meric poly(rA)/(dT)12-18T/P (Midland Certified Reagent Company).
Processivity assays.The processivity of recombinant RT proteins was analyzed
under various dNTP concentrations (0.5 and 200M) using the heteropolymeric
RNA template in the presence of a heparin enzyme trap to ensure a single processive cycle, i.e., a single round of binding and of primer extension and dissociation. The T/P was prepared by annealing the 497-nt HIV PBS RNA with
the32P-5⬘end-labeled 25-nt DNA primer D25 at a molar ratio of 1:1, denatured
at 85°C for 5 min, and then slowly cooled to 55°C for 8 min and 37°C for 5 min to allow for the specific annealing of primer to the template. Reactions were performed essentially as described previously (42). RT enzymes with equal amounts of activity and 40 nM T/P were preincubated for 5 min at 37°C in a
buffer containing 50 mMTris-HCl (pH 7.8), 50 mM NaCl, and 6 mM MgCl2.
Reactions were initiated by the addition of dNTPs and heparin trap (final concentration, 3.2 mg/ml) and were incubated at 37°C for 30 min. Two volumes
on November 7, 2019 by guest
http://jvi.asm.org/
of stop solution (90% formamide, 10 mM EDTA, and 0.1% each of xylene cyanol and bromophenol blue) were added to stop the reaction. Each reaction was run in duplicate. Reaction products were denatured by heating at 95°C and analyzed using 6% denaturing polyacrylamide gel electrophoresis and phosphorimaging. The effectiveness of the heparin trap to limit polymerization on the RNA tem-plate was verified in control reactions in which the heparin trap was preincubated with substrate before the addition of RT enzymes and dTTP.
RNA-dependent DNA polymerase activity assay.The 497-nt RNA and 5⬘-end
32
P-labeled D25 primers described above were used to assess the polymerization rate of recombinant RT enzymes in time course experiments. The final reaction mixtures contained 20 nM T/P, 400 nM RT enzyme, 50 mM Tris-HCl (pH 7.8),
and 50 mM NaCl. Reactions were initiated by adding 6 mM MgCl2and dNTPs
at various concentrations as described above and sampled at variable time points, i.e., 30 s, 60 s, 5 min, and 20 min, respectively, and mixed with two volumes of stop solution. Reaction products were separated by 6% denaturing polyacryl-amide gel electrophoresis and analyzed as described above.
Steady-state kinetic analysis.Kinetic studies were carried out as described
previously (30) using homopolymeric poly(rA)/p(dT)12-18and complementary
dTTP as the nucleotide substrate, with modifications. The reaction mixture (10
l) contained 50 mM Tris-HCl (pH 7.8), 60 mM KCl, 6 mM MgCl2, 5 mM DTT,
50g/ml poly(rA)/p(dT)12-18, 8.5 nM RT enzymes, and various concentrations of
tracer [3H]dTTP and cold dTTP (0.2 to 200M). Reactions were run at 37°C and
quenched by adding 0.2 ml of 10% cold TCA and 20 mM sodium pyrophosphate; products were filtered onto Millipore 96-well MutiScreen HTS FC filter plates
(MSFCN6B) and sequentially washed with 200l of 10% TCA and 150l of
95% ethanol. The radioactivity of incorporated products was analyzed by liquid scintillation spectrometry using a Perkin-Elmer 1450 MicroBetaTriLux micro-plate scintillation and luminescence counter. The steady-state kinetic parameter
(Km) for nucleotide substrates was determined by the nonlinear regression
anal-ysis of the substrate concentration and initial velocity data using the Michaelis-Menten equation with the program GraphPad Prism according to the manufac-turer’s instructions.
Structural modeling of E138K/M184V and E138K/M184I double mutants.
Homology structural models of E138K/M184V and E184K/M184I double
mu-tants were obtained by means of multiple template modeling (MTM) andab
initiosimulations from the I-TASSER 3D protein prediction server (31) using the lead PDB template of 3KK2 (19), a crystal structure of wild-type HIV-1 RT in complex with template DNA, and dNTP bound in the dNTP binding pocket. Initial model assessment was based on root mean square deviations (RMSDs) of the global monomeric homology structure from the monomeric lead template, and the resulting structures were aligned to appropriate crystal structural files using the RCSB PDB protein comparison tool (28) to verify structural homology as well as to ensure that all structural files used have similar orientations. To mimic the binding of dNTPs into ternary RT complexes, dNTP was removed from the 3KK2 structure and docked using high stringency into the dNTP binding pocket of the various structures using AUTODOCK Tools and AUTODOCK Vina. (34) PyMol (available at http//www.pymol.org; accessed 20 March 2011) was used for structural visualization, alignment, and image pro-cessing.
RT-catalyzed RNase H activity.RNase H activity was assayed using a 41-mer
5⬘-32
P heteropolymeric RNA template, kim40R, that was annealed to a comple-mentary 32-nucleotide DNA oligomer, termed kim32D, at a 1:4 molar ratio as described previously (18). Reactions were conducted at 37°C in mixtures con-taining an RNA-DNA duplex substrate with equal activities of RT enzymes in
assay buffer, 50 mM Tris-HCl, pH 7.8, 60 mM KCl, and 5 mM MgCl2in the
presence of a heparin trap (2 mg/ml). Aliquots were removed at different time points after the initiation of reactions and quenched by using an equal volume of formamide loading dye. The samples were heated to 90°C for 3 min, cooled on ice, and electrophoresed through 6% polyacrylamide–7 M urea gels. The gels were analyzed by phosphorimaging. The efficacy of the heparin trap was verified by preincubation experiments done by 10-min preincubation of enzymes with substrate and various concentrations of heparin trap, followed by the initiation of RNase H activities with magnesium and dNTPs.
RESULTS
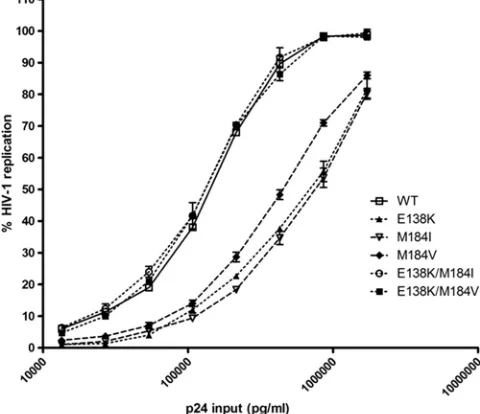
The E138K mutation compensates for the impaired viral
replication capacity (RC) of HIV containing either M184I or
M184V.
We previously showed that the replication capacity of
HIV-1 containing the E138K mutation was impaired by 2- to
3-fold compared to that of wild-type virus (3). Here, we wanted
to investigate the impact of the E138K, M184I, M184V,
E138K/M184I, and E138K/M184V mutations on RC by
infect-ing TZM-bl cells with serially diluted viral stocks normalized
by p24 antigen. The infectivities of the WT and mutant
vi-ruses were determined by measuring luciferase activity at 48 h
postinfection. The relative RC of viruses containing M184V
was decreased by
⬃
2-fold compared to that of the WT, while
the replication capacity of viruses containing either E138K or
M184I were decreased by
⬃
3-fold (Fig. 1). Interestingly, the
replication capacity of the E138K/M184I and E138K/M184V
double mutant viruses was equal to that of the WT virus. Thus,
the addition of E138K to either M184I or M184V compensates
for the impaired RC observed with each of the singly mutated
viruses. The order of replication was the following: WT
⫽
E138K/M184I
⫽
E138K/M184V
⬎
M184V
⬎
E138K
⫽
M184I. We did not observe an advantage of E138K/M184V in
RC over E138K/M184I.
[image:3.585.300.540.74.281.2]Drug susceptibilities monitored in cell culture phenotyping
assay.
Previous cell culture assays showed that HIV-1 clones
containing E138K displayed phenotypic resistance to ETR
(3.8-fold) (3). In this study, we determined the impact of the
interactions of E138K and the M184I/V mutations in HIV-1 on
susceptibility to each of the RT inhibitors FTC, 3TC, ETR, and
EFV. The results are summarized in Table 1. E138K conferred
no resistance on its own to the NRTIs FTC and 3TC and had
only minor impact on sensitivity to EFV but conferred
low-level resistance to ETR (3.2-fold-change in EC
50), which is
consistent with our previous report (3). For FTC and 3TC, all
FIG. 1. E138K mutation in HIV-1 reverse transcriptase
compen-sates for the impaired viral replication capacity of both M184I and
M184V. Viral stocks of wild-type virus and viruses containing E138K,
M184I, M184V, E138K/M184I, and E138K/M184V were normalized
for p24 and used to infect TZM-bl cells. Luciferase activity was
mea-sured at 48 h postinfection as an indication of viral replication. The
relative infectivity of WT compared to mutant viruses is shown on the
y
axis, while the
x
axis denotes the input of p24. Each experiment was
performed in triplicate. The figure is representative of two
indepen-dent experiments. These values translate to a 2-fold virus replication
disadvantage for M184V virus and a 3-fold disadvantage for the M184I
and E138K viruses compared to the replication of the WT virus.
on November 7, 2019 by guest
http://jvi.asm.org/
of the viruses containing the M184I or M184V mutation, either
alone or in combination with E138K, exhibited high-level
re-sistance. We also observed that the M184V-containing viruses
showed a higher level of resistance to FTC and 3TC than the
M184I-containing viruses. Neither M184I nor M184V
dramat-ically altered susceptibility to the NNRTIs ETR and EFV. For
ETR, the E138K/M184I double mutation resulted in a 2.3-fold
change in EC
50and a 2.7-fold change for the E138K/M184V
virus. These results indicate that the high prevalence of E138K/
M184I rather than E138K/M184V in clinical samples cannot
be explained by levels of drug resistance.
Purification of recombinant HIV-1 RT enzymes.
Recombi-nant WT heterodimeric (p66/p51) RT and RT enzymes
con-taining each of E138K, E138K/M184I, M184I, E138K/M184V,
and M184V substitutions were purified to
⬎
95% homogeneity;
the RT p66 and p51 subunits were processed to similar molar
ratios based on SDS-PAGE analysis (data not shown). The
mutations introduced into the recombinant HIV-1 RT did not
interfere with either heterodimer formation or enzyme
purifi-cation. Consistently with previously published data, the E138K
mutation did not have an effect on RT dimerization (1, 25).
[image:4.585.42.541.82.171.2]E138K restores the enzyme processivity of RT containing
either M184I or M184V at low dNTP concentrations.
The
pro-cessivity of a polymerase is defined as the number of
nucleo-tides incorporated in a single round of binding, elongation, and
dissociation. Earlier studies showed that diminished HIV-1 RT
processivity is the major determinant of impaired viral
repli-cation capacity associated with M184I/V mutations, especially
at low dNTP concentrations (5, 24). We wished to determine
whether the compensatory effect of E138K on the viral
repli-cation capacity of M184I/V viruses was due to a restoration of
enzyme processivity. To this end, we performed single-cycle
processivity assays with recombinant RT enzymes at both low
and high dNTP concentrations (Fig. 2). Assays performed with
TABLE 1. Drug susceptibilities for recombinant HIV-1
NL4-3WT and site-directed mutant viruses assayed in CBMC cultures
aVirus EC50⫾SD (M) (fold change in resistance) EC50⫾SD (nM) (fold change in resistance)
FTC 3TC ETV EFV
WT
0.008
⫾
0.005
0.041
⫾
0.029
1.0
⫾
0.3
1.2
⫾
0.6
E138K
0.018
⫾
0.006 (2.2)
0.024
⫾
0.025 (0.6)
3.3
⫾
0.4 (3.2)
2.2
⫾
0.4 (1.8)
E138K/M184I
2.1
⫾
0.9 (250)
26.5
⫾
14.0 (
⬎
500)
2.4
⫾
0.1 (2.3)
2.0
⫾
0.9 (1.6)
E138K/M184V
⬎
50 (
⬎
1,000)
⬎
100 (
⬎
1,000)
2.8
⫾
0.9 (2.7)
2.5
⫾
0.9 (2.1)
M184I
1.5
⫾
0.4 (185)
14.0
⫾
2.7 (350)
1.2
⫾
0.5 (1.2)
1.2
⫾
0.5 (1.0)
M184V
⬎
50 (
⬎
1,000)
⬎
100 (
⬎
1,000)
1.2
⫾
0.5 (1.2)
1.4
⫾
1.0 (1.2)
aEC
50s (50% drug effective concentrations) were determined by RT assays using culture fluids of CBMCs. Data represent the averages from two to three
representative experiments performed in duplicate. Values in parentheses represent the fold change in EC50for mutated viruses compared to that of the WT virus.
FIG. 2. E138K mutation in HIV-1 RT restores enzyme processivity of both the M184I- and M184V-containing enzymes at low dNTP
concentrations. The processivity of purified recombinant RT enzymes was analyzed using 5
⬘
-end-labeled DNA primer (D25) annealed to a 471-nt
RNA template as the substrate; the resulting full-length DNA (FL DNA) is 471 nt in size. Processivities were determined by the size distribution
of DNA products in fixed-time experiments at two different concentrations of dNTPs in the presence of heparin trap, a low dNTP concentration
(0.5
M) (A), and a high dNTP concentration (200
M) (B). Each reaction was performed in duplicate and terminated at 30 min. The sizes of
some fragments of the
32P-labeled 25-bp DNA ladder (Invitrogen) in nucleotide bases are indicated on the left side of the panel. All reaction
products were resolved by denaturing 6% polyacrylamide gel electrophoresis and visualized by phosphorimaging. Positions of
32P-labeled D25
primer (
32P-D25) and the 471-nt full-length extension DNA (FL DNA) product are indicated on the right.
on November 7, 2019 by guest
http://jvi.asm.org/
0.5
M each dNTP showed that both M184I and M184V RTs
have lower processivity than wild-type RT, whereas the three
E138K-containing mutant enzymes had higher processivity,
in-dicating that E138K compensates for the diminished enzyme
processivity associated with M184I/V (Fig. 3A). We did not
observe a processivity advantage for E138K/M184I over
E138K/M184V. At 200
M each dNTP, all enzymes displayed
similar processivity based on the production of full-length
products (Fig. 3B); the distortion of running samples in each
lane was from blotting the gel onto Whatman paper. This is in
agreement with previous studies showing that the processivity
defect of the M184I/V RT mutants is minimized at high dNTP
concentrations (5, 11) and implies that the restoration of
pro-cessivity by E138K is due the compensation of dNTP usage
associated with M184I/V.
Effects of dNTP concentrations on polymerization rates of
recombinant RT enzymes.
To measure the rate of DNA
polymerization at specific dNTP concentrations, we performed
RNA-dependent DNA polymerase reactions in time course
experiments for 30 s, 60 s, 5 min, and 20 min using a 497-nt
HIV-1 PBS RNA template (Fig. 3). RT molecules were used at
an
⬃
20-fold excess above the substrate, so that any RTs that
dissociated from the primer terminus during synthesis would
be rapidly replaced. In this case, the rate-limiting step would be
nucleotide addition (11). Polymerase reactions were carried
out at two different dNTP concentrations, 0.5 and 200
M. The
rate of polymerization was calculated as the number of
nucle-otide additions divided by reaction time. The longest extension
products made after 60 s were used to calculate the
polymer-ization rate. At 0.5
M each dNTP, the rate of synthesis of
reaction product for WT, E138K, E138K/M184I, and E138K/
M184V RTs was
⬃
0.9 nt/s. However, for M184I and M184V,
the
32P-labeled primer D25 was hardly extended (Fig. 4B).
When reactions were performed at 200
M dNTPs, all of the
RT variant enzymes except E138K showed similar
polymeriza-tion rates of
⬃
3.2 nt/s, while the rate for E138K was
⬃
2.0 nt/s
(Fig. 4B), which is consistent with previously reported
maxi-mum polymerization rates for WT HIV-1 RT on RNA
tem-plates (10, 11). These data confirm that the M184I/V mutants
have defects in dNTP usage (5, 11), and that the E138K
mu-tation can compensate for this deficit, even though the E138K
enzyme itself has lower catalytic efficiency at high dNTP
con-centrations.
Steady-state kinetic analyses of WT and mutant RTs.
To
assess the impact of the E138K mutation in the context of
M184I/V on dNTP binding affinity, steady-state kinetic
analy-ses were carried out using a homopolymeric poly(rA)/
p(dT)
12-18template/primer to determine the steady-state
ki-netic constants
K
mand
V
maxfor WT and mutant HIV-1
recombinant RTs; the results are summarized in Table 2. In
the case of RTs containing M184V or M184I compared to WT
RT, increased
K
mvalues for dTTP of 1.4-fold and 2.4-fold,
respectively, were observed. For E138K RT, the value was
0.46-fold of that of the WT. For the double mutants E138K/
M184I and E138K/M184V, the fold change in
K
mvalue was
1.25 and 1.1, respectively. Although
K
mis not a direct measure
of dNTP binding, changes in
K
mvalues suggest that the
mu-tations affect dNTP binding affinity. These results indicate that
the deficit in dNTP binding affinity associated with the HIV-1
RT M184I/V mutations was compensated by the copresence of
E138K; this is further demonstrated in structural modeling as
described below.
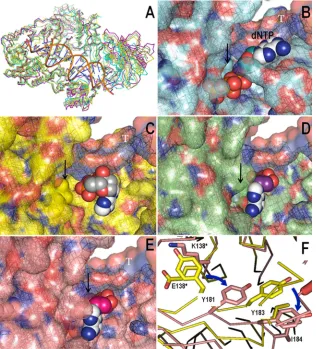
Homology modeling shows that the E138K/M184V and
E138K/M184I double mutants promote tighter dNTP binding.
Individual models of RT molecules containing E138K/M184I
that were generated from either WT, E138K, or M184V RT
FIG. 3. Time course experiments showing the effects of dNTP concentrations on rates of DNA synthesis by recombinant HIV-1 RT enzymes.
The
32P-labeled D25 primer (
32P-D25) was annealed to the 497-nt RNA template, and extension assays were performed at an excess of
recombinant RT enzymes at dNTP concentrations of 0.5
M (A) and 200
M (B). The reactions were stopped at 30 s (30
⬙
), 60 s (60
⬙
), 5 min (5
⬘
),
and 20 min (20
⬘
), respectively. The sizes of some fragments of the
32P-labeled 25-bp DNA ladder (Invitrogen) in nucleotide bases are indicated
on the left side of the panel. The longest extension products generated at 60 s were identified by arrows and indicate differences in rates of
polymerization.
on November 7, 2019 by guest
http://jvi.asm.org/
did not differ significantly from WT RT in global structure.
This attests to the accuracy of the modeling method and the
reliability of the models produced. E138K/M184I and E138K/
M184V homology structures that were formed using either
WT, M184I, or E138K as the lead template were in agreement
with wild-type and single mutant structures with mean RMSDs
in the range of 0.3 to 2.3 Å (Fig. 4A).There did not appear
to be large variations between the structures of the E138K/
M184V and E138K/M184I double mutants (RMSD,
⬍
0.5 Å).
In the WT RT, the finger subdomain aids in binding the
dNTP in the right conformation (Fig. 4B), while mutations at
M184 move this subdomain away and open up the dNTP
bind-ing pocket, thus causbind-ing dNTP to bind loosely (Fig. 4C). The
presence of the E138K mutation in association with the
M184I/V mutations (Fig. 4D and E) caused a series of steric
interferences with several p66 residues (Y181 and Y183) at the
interface between p66 and p51. These changes translate into a
FIG. 4. Modeling and docking studies showing the concerted effect of the E138K and M184I/V double mutations. (A) The overlay of wild-type
(blue) and M184I (yellow) crystal structures with homology models E138K/M184I (pink) and E138K/M184V (green) shows minimal differences
in global structure. (B to E) dNTP docked into identical binding sites/coordinates in all structures with residue 184 is indicated by a black arrow,
and template DNA is indicated in white (T). (B) The finger subdomain which assists in binding and orienting bound dNTP in the wild-type RT
is altered when the M184I mutation is present. (C) M184I also opens up the dNTP binding pocket and prevents the tight association of dNTP with
RT. (D) The E138K/M184V double mutant has a less open dNTP binding site than M184I, thus contributing to tighter dNTP binding. (E) The
E138K/M184I double mutant appears to display the partial recovery of the finger subdomain and shows tight binding similar to that of
E138K/M184V RT. (F) Ribbon overlay of the M184I crystal structure with the E138K/M184I homology model. The blue arrows indicate the
downward movement of residues and the subsequent closing up of the dNTP binding site in the p66 unit induced by the repositioning of the side
chain containing residue 138 in the p51 subunit.
TABLE 2. Kinetic parameters of recombinant RT enzymes as
determined by steady-state kinetic analysis
Virus
Parametera
Vmax(pMg⫺ 1
min⫺1
) Km(M) (FC
b
)
WT
139.3
⫾
5.1
5.63
⫾
0.1 (1)
E138K
112.9
⫾
3.2
2.59
⫾
0.1 (0.46)
E138K/M184I
177.9
⫾
4.3
7.0
⫾
0.15 (1.25)
M184I
193.2
⫾
7.1
14.6
⫾
0.3 (2.6)
E138K/M184V
221.3
⫾
6.4
6.2
⫾
0.3 (1.1)
M184V
192.2
⫾
5.6
7.88
⫾
0.4 (1.4)
a
The steady-state kinetic parametersVmaxandKmfor dTTP of WT HIV-1 RT
and its mutant derivatives were measured using poly(rA)/p(dT)12-18template/
primer. The recombinant RT enzymes were purified in heterodimeric forms, and
the mutations were introduced into both subunits. Values are averages⫾
stan-dard deviations from representative experiments performed in triplicate.
b
Fold change (FC) ofKmof mutant RT variants compared to that of the WT.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.42.283.587.676.2]downward shift of residue 184 (Fig. 4F) and partially close up
the dNTP binding site (Fig. 4D and E). Thus, the E138K/
M184I and E138K/M184V double mutants appear to have
tighter dNTP binding than the M184I/V single mutants.
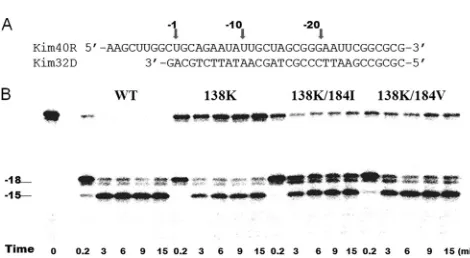
RNase H activity.
We previously showed that M230L in HIV
RT impairs RNase H activity and contributes to reductions in
viral replication capacity (43). We wished to test the impact of
the E138K mutation alone and in combination with M184I/V
on intrinsic RNase H activity. Therefore, we performed an
RNase H time course study in the presence of a heparin trap
by using a recessed 32-mer DNA primer hybridized to a 5
⬘
-end-labeled 40-mer RNA to monitor 3
⬘
-DNA-directed RNase
H activity (Fig. 5.). The presence of the heparin trap permits
the analysis of cleaved products from a single binding event of
RT to the substrate. The results show that the RNase H activity
of the E138K mutant RT was lower than that of the WT RT,
and that the M184I/V mutations can restore the RNase H
activity of E138K, albeit not to the WT level (Fig. 5). Thus, the
E138K mutation also impairs RNase H activity despite the fact
that it also results in a defect in regard to initial rates of
polymerization.
DISCUSSION
The diarylpyrimidine (DAPY) compounds etravirine (ETR;
TMC125) and rilpivirine (RPV; TMC278) are representative
potent second-generation NNRTIs. ETR has been licensed by
the FDA and is recommended for use in salvage therapy in
combination with other active antiretrovirals (ARVs). Most
clinical data sets regarding ETR resistance are based on the
DUET studies (16, 37), and little information is available on
the roles of individual resistance mutations on RT enzyme
properties, viral replication, and diminished sensitivity to ETV.
Even less information is available for RPV in this regard (4).
We previously reported that E138K in RT is usually the first
resistance mutation to emerge in tissue culture with ETR
se-lection. Recently, the ECHO and THRIVE phase 3 clinical
trials showed that E138K/M184I was the most frequent
mu-tation combination in patients who failed regimens of RPV
combined with FTC/TDF. We believe that E138K possesses
clinical relevance to ETR and RPV. Our current findings
demonstrate that the E138K mutation can restore the enzyme
processivity of the RT and viral replication capacity of viruses
containing the M184I/V mutation. This renders the doubly
mutated viruses containing E138K/M184I and E138K/M184V
highly competent in replication capacity and resistant to each
of FTC, 3TC, and ETR.
Our cell culture-based phenotypic data show that
recombi-nant HIV-1
NL4-3viruses harboring E138K and M184I/V
dou-ble mutations possessed high-level resistance to FTC and 3TC,
as does the M184I/V single mutation, and possessed modest
resistance to ETR, as does the E138K single mutation. None of
the mutations tested in this study significantly affected EFV
susceptibility. We did not observe that the E138K/M184I
combination conferred higher levels of resistance to ETR
and 3TC compared to those of an E138K/M184V double
mutant. Therefore, the high prevalence of E138K/M184I
in-stead of E138K/M184V among patients who failed
RPV/FTC-containing therapy in the ECHO and THRIVE clinical trials
cannot be explained by differences in levels of drug resistance
(20). Although we did not test FTC and ETR in combination,
it is unlikely that significant differences would have been
ob-tained. Nonetheless, this experiment is being planned. It is
possible, of course, that M184I has an advantage over M184V
when both ETR and FTC are present, since both the M184I
and E138K mutations were selected in the ECHO and
THRIVE clinical trials that employed these two drugs.
Of note, we observed that recombinant HIV-1 virus
harbor-ing E138K confers 3.2-fold resistance to ETR, which is
consis-tent with our previous report (3), while the combination of
E138K and either M184I or M184V conferred 2.4- or 2.8-fold
resistance to ETR, respectively. The published lower clinical
cutoff (CCO) for ETR is 3.0 (37); it is conceivable that
addi-tional clinical data will be necessary to reevaluate the role of
ETR mutations such as E138K and G190A in regard to the
durability of ETR-based therapy (41). Although we did not
have access to rilpivirine (RPV) for these studies, we believe
that findings similar to those obtained with ETR would have
been obtained with this compound.
[image:7.585.42.277.62.191.2]Our replication capacity studies carried out in TZM-bl cells
show that recombinant viruses harboring E138K, M184I, or
M184V alone exhibited 2- to 3-fold lower replication capacity
compared to that of WT virus and that the copresence of
E138K with either M184I or M184V restored viral replication
capacity to WT levels. Previously, we observed that the HIV-1
E138K mutation results in lower replication capacity in several
HIV-1 subtypes, and M184I/V viruses also have impaired
rep-lication capacity, especially in cells with low dNTP
concentra-tions (15, 39). Although some compensatory mutaconcentra-tions in RT
might augment viral resistance levels and fitness (7, 22, 26), this
is the first report on the compensatory effects of E138K on the
enzymatic activity of RT containing M184V or M184I. Our
biochemical assays provide the first evidence that E138K RT
has a lower rate of polymerization and lower RNase H activity
than WT RT, which helps to explain the lower replication
capacity of the E138K virus. In our studies, the combination of
E138K/M184I did not confer a replication advantage over
FIG. 5. RNase H activity of WT and E138K-containing
recombi-nant RTs. (A) Graphic representation of the RNA/DNA (kim40R/
kim32D) duplex substrate used to monitor the cleavage efficiency of
recombinant RT. The 40-mer RNA kim40R was labeled at its 5
⬘
terminus with
32P and annealed to the 32-mer DNA oligonucleotide
kim32D. (B) RNase H activity was analyzed by monitoring substrate
cleavage in time course experiments in the presence (right) of a
hep-arin trap. The positions of cleaved products are indicated on the left.
The uncleaved substrate indicates variations in RNase H activity
among different RT enzymes. All reactions were resolved by
denatur-ing 6% polyacrylamide gel electrophoresis.
on November 7, 2019 by guest
http://jvi.asm.org/
E138K/M184V in the absence of drugs, which means that the
high prevalence of E138K/M184I instead of E138K/M184V in
the ECHO and THRIVE clinical trials cannot be explained by
differences in viral replication capacity. Others have shown
that the combination of E138K/M184I was more fit than
E138K/M184V in a sensitive growth competition assay (14,
14a). It is possible that the different cell types and assay
sys-tems used have affected results obtained.
Our enzyme assays with purified recombinant RT proteins
provide evidence at a molecular level for the compensatory
effects between E138K and M184I/V. M184V RT is known to
have lower enzyme processivity than WT RT, especially at low
dNTP concentrations (5, 9, 35), and M184I is even more
im-paired than M184V in terms of processivity (11, 15). Deficits in
M184I/V RT primer elongation rate, processivity, and strand
transfer may all be attributable to defective dNTP utilization
(9, 11, 15, 35, 39). Here, we have shown that E138K
compen-sates for the defect in dNTP usage associated with M184I/V,
thus restoring enzyme processivity and viral replication
capac-ity. Indeed, RT enzymes containing E138K alone, E138K/
M184I, or E138K/M184V exhibited higher processivity than
WT RT at low dNTP concentrations. Steady-state kinetic
anal-ysis also demonstrated that the E138K mutation resulted in
decreased
K
mvalues. This was further supported by structural
modeling showing that the addition of E138K to M184I/V
promotes tighter dNTP binding.
Under selection pressure with 3TC or FTC, either
in vitro
or
in vivo
, M184I usually emerges first as a resistance mutation
and is rapidly replaced by M184V because of its fitness
advan-tage over M184I (6, 9, 13, 18, 35). The reason that M184I
(ATG
3
ATA) appears before M184V (ATG
3
GTG) is that
G
3
A is the most frequent mutation to occur during HIV-1
replication (6, 13).The fact that M184I is more impaired than
M184V RT in dNTP usage and processive DNA synthesis
contributes to the
in vivo
instability of the M184I mutation and
its supplementation by M184V (5, 11, 15, 39). What has
prob-ably happened in the ECHO and THRIVE clinical trials is that
E138K stabilized M184I as a result of the compensatory effects
described here. In clinical studies on the use of ETR or RPV
and FTC or 3TC, it is not known whether E138K or M184I
emerges first. The clonal analysis of clinical samples from
pa-tients undergoing treatment failure should be able to answer
this question.
ACKNOWLEDGMENTS
We thank Stuart Le Grice for providing the pRT6H-PROT DNA,
Tomozumi Imamichi for the pNL4.3PFB plasmid DNA, and Daniela
Moisi for technical assistance in DNA sequencing reactions.
This work was supported by research grants from the Canadian
Institutes of Health Research (CIHR) and by the International
Part-nership on Microbicides.
We have no conflicts of interest to declare.
REFERENCES
1.Ambrose, Z., et al.2009. The human immunodeficiency virus type 1 non-nucleoside reverse transcriptase inhibitor resistance mutation I132M confers
hypersensitivity to nucleoside analogs. J. Virol.83:3826–3833.
2.Arts, E. J., et al. 1994. Comparison of deoxyoligonucleotide and tRNA(Lys-3) as primers in an endogenous human immunodeficiency virus-1 in vitro reverse transcription/template-switching reaction. J. Biol. Chem.
269:14672–14680.
3.Asahchop, E. L., et al.2011. Characterization of the E138K resistance mu-tation in HIV-1 reverse transcriptase conferring susceptibility to etravirine in
B and non-B HIV-1 subtypes. Antimicrob. Agents Chemother.55:600–607.
4.Azijn, H., et al.2010. TMC278, a next-generation nonnucleoside reverse transcriptase inhibitor (NNRTI), active against wild-type and
NNRTI-resis-tant HIV-1. Antimicrob. Agents Chemother.54:718–727.
5.Back, N. K., et al.1996. Reduced replication of 3TC-resistant HIV-1 variants in primary cells due to a processivity defect of the reverse transcriptase
enzyme. EMBO J.15:4040–4049.
6.Cheynier, R., S. Gratton, J. P. Vartanian, A. Meyerhans, and S. Wain-Hobson.1997. G3A hypermutation does not result from polymerase chain
reaction. AIDS Res. Hum. Retrovir.13:985–986.
7.Chunduri, H., D. Rimland, V. Nurpeisov, C. S. Crumpacker, and P. L. Sharma.2011. A Leu to Ile but not Leu to Val change at HIV-1 reverse transcriptase codon 74 in the background of K65R mutation leads to an
increased processivity of K65R⫹L74I enzyme and a replication competent
virus. Virol. J.8:33.
8.Das, K., et al.2009. Structural basis for the role of the K65R mutation in HIV-1 reverse transcriptase polymerization, excision antagonism, and
teno-fovir resistance. J. Biol. Chem.284:35092–35100.
9.Diallo, K., M. Gotte, and M. A. Wainberg.2003. Molecular impact of the M184V mutation in human immunodeficiency virus type 1 reverse
transcrip-tase. Antimicrob. Agents Chemother.47:3377–3383.
10.Fuentes, G. M., L. Rodriguez-Rodriguez, C. Palaniappan, P. J. Fay, and R. A. Bambara.1996. Strand displacement synthesis of the long terminal
repeats by HIV reverse transcriptase. J. Biol. Chem.271:1966–1971.
11.Gao, L., et al.2008. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse
trans-criptase derive solely from a dNTP utilization defect. J. Biol. Chem.283:
9196–9205.
12.Goff, S. P.1990. Retroviral reverse transcriptase: synthesis, structure, and
function. J. Acquir. Immune Defic. Syndr.3:817–831.
13.Gu¨nther, S., et al. 1997. Naturally occurring hepatitis B virus genomes
bearing the hallmarks of retroviral G3A hypermutation. Virology235:104–
108.
14.Hu, Z., and D. R. Kuritzkes.2011. Interaction of reverse transcriptase (RT) mutations conferring resistance to lamivudine and etravirine: effects on fit-ness and RT activity of human immunodeficiency virus type 1. J. Virol.
85:11309–11314.
14a.Hu, Z. X., J. Li, S. Gallien, and D. R. Kuritzkes.2011. Impact of the interactions of rilpivirine (E138K) and lamivudine/emtrictabine (M184V/I) resistance mutations on viral DNA synthesis and fitness of HIV-1. Antiviral
Ther.16(Suppl.):A20.
15.Jamburuthugoda, V. K., et al.2008. Reduced dNTP binding affinity of 3TC-resistant M184I HIV-1 reverse transcriptase variants responsible for
viral infection failure in macrophage. J. Biol. Chem.283:9206–9216.
16.Johnson, L. B., and L. D. Saravolatz.2009. Etravirine, a next-generation
nonnucleoside reverse-transcriptase inhibitor. Clin. Infect. Dis.48:1123–
1128.
17.Johnson, V. A., et al.2010. Update of the drug resistance mutations in
HIV-1: December 2010. Top. HIV Med.18:156–163.
18.Keulen, W., N. K. Back, A. van Wijk, C. A. Boucher, and B. Berkhout.1997. Initial appearance of the 184Ile variant in lamivudine-treated patients is caused by the mutational bias of human immunodeficiency virus type 1
reverse transcriptase. J. Virol.71:3346–3350.
19.Lansdon, E. B., et al.2010. Visualizing the molecular interactions of a nucleotide analog, GS-9148, with HIV-1 reverse transcriptase-DNA
com-plex. J. Mol. Biol.397:967–978.
20.Macarthur, R. D.2011. Clinical trial report: TMC278 (rilpivirine) versus efavirenz as initial therapy in treatment-naive, HIV-1-infected patients.
Curr. Infect. Dis. Rep.13:1–3.
21.Martinez-Cajas, J. L., and M. A. Wainberg.2008. Antiretroviral therapy:
optimal sequencing of therapy to avoid resistance. Drugs68:43–72.
22.Martinez-Picado, J., and M. A. Martinez.2008. HIV-1 reverse transcriptase inhibitor resistance mutations and fitness: a view from the clinic and ex vivo.
Virus Res.134:104–123.
23.Mocroft, A., et al.2003. Decline in the AIDS and death rates in the
Euro-SIDA study: an observational study. Lancet362:22–29.
24.Naeger, L. K., N. A. Margot, and M. D. Miller.2001. Increased drug sus-ceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovudine-associated mutations: analysis of enzyme processivity,
chain-ter-minator removal and viral replication. Antivir. Ther.6:115–126.
25.Nissley, D. V., et al.2007. Characterization of novel non-nucleoside reverse transcriptase (RT) inhibitor resistance mutations at residues 132 and 135 in
the 51 kDa subunit of HIV-1 RT. Biochem. J.404:151–157.
26.Olivares, I., et al.1999. Second-site reversion of a human immunodeficiency virus type 1 reverse transcriptase mutant that restores enzyme function and
replication capacity. J. Virol.73:6293–6298.
27.Poveda, E., et al.2008. Phenotypic impact of resistance mutations on etra-virine susceptibility in HIV patients with prior failure to nonnucleoside
analogues. AIDS22:2395–2398.
28.Prlic, A., et al.2010. Pre-calculated protein structure alignments at the
RCSB PDB website. Bioinformatics26:2983–2985.
29.Quan, Y., et al.2003. Drug resistance profiles of recombinant reverse
on November 7, 2019 by guest
http://jvi.asm.org/
criptases from human immunodeficiency virus type 1 subtypes A/E, B, and C.
AIDS Res. Hum. Retrovir.19:743–753.
30.Quan, Y., et al.1996. Endogenous reverse transcription assays reveal
high-level resistance to the triphosphate of (-)2⬘-dideoxy-3⬘-thiacytidine by
mu-tated M184V human immunodeficiency virus type 1. J. Virol.70:5642–5645.
31.Roy, A., A. Kucukural, and Y. Zhang.2010. I-TASSER: a unified platform
for automated protein structure and function prediction. Nat. Protoc.5:725–
738.
32.Schatz, O., J. Mous, and S. F. Le Grice.1990. HIV-1 RT-associated
ribo-nuclease H displays both endoribo-nuclease and 3⬘–5⬘ exonuclease activity.
EMBO J.9:1171–1176.
33.Torres, R. A., and M. Barr.1997. Impact of combination therapy for HIV
infection on inpatient census. N. Engl. J. Med.336:1531–1532.
34.Trott, O., and A. J. Olson.2010. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and
multithreading. J. Comput. Chem.31:455–461.
35.Turner, D., B. Brenner, and M. A. Wainberg.2003. Multiple effects of the M184V resistance mutation in the reverse transcriptase of human
immuno-deficiency virus type 1. Clin. Diagn. Lab. Immunol.10:979–981.
36.Turner, D., and M. A. Wainberg.2006. HIV transmission and primary drug
resistance. AIDS Rev.8:17–23.
37.Vingerhoets, J., et al.2010. Resistance profile of etravirine: combined anal-ysis of baseline genotypic and phenotypic data from the randomized,
con-trolled phase III clinical studies. AIDS24:503–514.
38.Wainberg, M. A.2003. HIV resistance to nevirapine and other non-nucleo-side reverse transcriptase inhibitors. J. Acquir. Immune Defic. Syndr.
34(Suppl. 1):S2–S7.
39.Wainberg, M. A. 2004. The impact of the M184V substitution on drug
resistance and viral fitness. Expert Rev. Anti Infect. Ther.2:147–151.
40.Wainberg, M. A., and G. Friedland.1998. Public health implications of
antiretroviral therapy and HIV drug resistance. JAMA279:1977–1983.
41.Xu, H., et al.2009. Human immunodeficiency virus type 1 recombinant reverse transcriptase enzymes containing the G190A and Y181C resistance mutations remain sensitive to etravirine. Antimicrob. Agents Chemother.
53:4667–4672.
42.Xu, H. T., et al.2010. Comparative biochemical analysis of recombinant reverse transcriptase enzymes of HIV-1 subtype B and subtype C.
Retrovi-rology7:80.
43.Xu, H. T., et al. 2010. The M230L nonnucleoside reverse transcriptase inhibitor resistance mutation in HIV-1 reverse transcriptase impairs enzy-matic function and viral replicative capacity. Antimicrob. Agents
Che-mother.54:2401–2408.
on November 7, 2019 by guest
http://jvi.asm.org/