0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Attenuation of DNA-Dependent Protein Kinase Activity and Its
Catalytic Subunit by the Herpes Simplex Virus Type 1
Transactivator ICP0
SUSAN P. LEES-MILLER,1MELISSA C. LONG,2M. ALISON KILVERT,2VIVIAN LAM,2
STEPHEN A. RICE,2ANDCHARLOTTE A. SPENCER2*
Department of Biological Sciences, University of Calgary, Calgary, Alberta,
Canada T2N 1N4,1and Department of Biochemistry, University
of Alberta, Edmonton, Alberta, Canada T6G 2H72
Received 24 April 1996/Accepted 22 July 1996
The DNA-dependent protein kinase (DNA-PK) is involved in several fundamental nuclear processes, in-cluding DNA double-strand break repair, V(D)J recombination, and transcription by RNA polymerases I and II. In this study, we show that infection of mammalian cells with herpes simplex virus type 1 attenuates DNA-PK activity by specifically depleting the PKcs catalytic subunit. The half-life of the p350/DNA-PKcs protein decreases from greater than 24 h to less than 4 h following infection. The depletion of DNA-PK activity and p350/DNA-PKcs abundance is dependent on expression of the viral immediate-early protein ICP0. As ICP0 acts as a promoter-independent transactivator of gene expression, these data suggest that ICP0 may function by directly or indirectly targeting the p350/DNA-PKcs subunit of DNA-PK, thereby altering the inhibitory effects of DNA-PK on RNA polymerase II transcription.
The DNA-dependent protein kinase (DNA-PK) is com-posed of a large polypeptide of approximately 460 kDa (DNA-PKcs, formerly known as p350) (12, 35) which contains the putative catalytic domain (29) and a dimeric protein of 70 and 80 kDa, known as Ku, which targets DNA-PK to DNA (18, 27). Recent evidence shows that DNA-PK is involved in DNA double-strand break repair and V(D)J recombination (3, 31, 50). Radiation-sensitive, V(D)J recombination-deficient mu-rine cell lines are defective in DNA end binding activity and lack the p80Ku subunit of DNA-PK (7, 23, 25, 44, 45, 54, 58). Repair and recombination activities are restored when Ku-defective cells are transfected with a p80Ku gene or when extracts are complemented with purified Ku. In addition, mu-rine SCID mice lack the p350/DNA-PKcs subunit of DNA-PK and DNA-PK activity (6, 32, 43). The DNA repair and V(D)J recombination defects in SCID cells are complemented by chromosome fragments expressing p350/DNA-PKcs (6, 32, 43). Also, we have shown that p350/DNA-PKcs and DNA-PK activity are absent in a radiosensitive human cell line which is also defective in DNA double-strand break repair (36).
DNA-PK may also be involved in the regulation of transcrip-tion from both RNA polymerase I and II (RNAP I and II) promoters. In vitro, DNA-PK acts as a serine/threonine pro-tein kinase that phosphorylates many transcription factors and DNA-binding proteins, such as p53, serum response factor, c-Fos, c-Jun, simian virus 40 large T antigen, and the carboxy-terminal domain of RNAP II (for reviews, see references 4 and 5). DNA-PK represses transcription initiation by RNAP I, apparently by phosphorylating a component of the RNAP I transcription initiation complex (33, 34). Recent evidence in-dicates that DNA-PK also represses RNAP II transcription. Giffin et al. have shown that DNA-PK, when bound at the mouse mammary tumor virus negative regulatory element 1,
represses glucocorticoid-induced transcription from the mouse mammary tumor virus promoter (26). This repression may operate via DNA-PK-induced phosphorylation of the gluco-corticoid receptor and Oct-1. In addition, DNA-PK activity is stimulated by some RNAP II transcription activators in vitro (42). Both Ku and p350/DNA-PKcs appear to be components of a human RNAP II complex that also contains a subset of RNAP II basal transcription factors (36b).
We were initially interested in examining DNA-PK because of the potential role of DNA-PK in transcription regulation and its ability to hyperphosphorylate the carboxy-terminal do-main of the large subunit of RNAP II. In cells infected with herpes simplex virus type 1 (HSV-1), the hyperphosphorylated form of RNAP II is lost and an aberrantly phosphorylated, transcriptionally active form appears (47, 48, 57). The shift in RNAP II phosphorylation coincides with major transcriptional shifts that occur in infected cells; after infection with HSV-1, cellular gene transcription is repressed while viral gene tran-scription is strongly induced.
HSV-1 is a prevalent human pathogen which replicates lyti-cally at the site of infection and establishes latent infection in sensory neurons (49). It is a linear double-stranded DNA virus that replicates in the host cell nucleus and utilizes the host’s RNAP II transcription machinery to express its genome (55). During a lytic infection, viral genes are expressed in a temporal cascade and are regulated at both the transcriptional and post-transcriptional levels. Transcription of the five immediate-early (IE) genes is stimulated by the virion protein, VP16, that binds in complexes with cellular proteins (including Oct-1) to TAA TGARAT sequences, present in the promoters of the IE genes. Four of the five IE gene products (ICP0, ICP4, ICP22, and ICP27) are regulatory proteins that are involved in the subsequent expression of the delayed early (DE) and late (L) genes (49). Unlike the IE gene promoters, DE and L gene
promoters lack known binding sites for virus-encoded
cis-act-ing transcription activators that would direct RNAP II in the appropriate temporal manner. Therefore, the mechanisms by which the DE and L genes are transcriptionally activated in the
* Corresponding author. Mailing address: Department of Biochem-istry, University of Alberta, Edmonton, Alberta, Canada T6G 2H7. Phone: (403) 492-7737. Fax: (403) 492-0886. Electronic mail address: [email protected].
7471
on November 9, 2019 by guest
http://jvi.asm.org/
proper temporal order are still unknown, but they may involve modifications to the RNAP II transcription machinery (47, 48, 55). In the latent state, viral DNA remains dormant in the nucleus, as a nonintegrated episome, and gene expression is restricted to transcription of the latency-associated transcript region of the genome (22).
In this report, we demonstrate that DNA-PK activity and the p350/DNA-PKcs catalytic subunit are rapidly depleted follow-ing infection with HSV-1. This depletion is dependent on ex-pression of the virus IE protein ICP0. As ICP0 is necessary during lytic infection at low multiplicities of infection (MOIs) to activate gene expression, and as ICP0 contributes to early gene expression during reactivation from latency, these data suggest a mechanism for the action of ICP0—one that involves DNA-PK and the stimulation of RNAP II transcription during virus infection.
MATERIALS AND METHODS
Cells, viruses, and infections.HeLa S3, a human epithelioid cervical carci-noma cell line, was used for infections. Cells were obtained from the American Type Culture Collection, Rockville, Md., and were grown as monolayer cultures in Dulbecco modified Eagle’s medium with 10% heat-inactivated newborn calf serum (GIBCO). HSV-1 strain KOS1.1 (wild-type) was obtained from M. Levine (University of Michigan). The ICP4 mutant virusd120 was obtained from Neal DeLuca (University of Pittsburgh School of Medicine). The ICP6 mutant virus ICP6D was obtained from Sandy Weller (University of Connecticut Health Center). The ICP0 mutant virusn212 and the ICP22 mutant virus 22/n199 were obtained from Priscilla Schaffer (Dana-Farber Cancer Institute and Harvard Medical School). The ICP27 mutant virusd27-1 has been described elsewhere (46). Virus strains were propagated and titers were determined as described previously (48). A stock of wild-type HSV-1 KOS1.1 was UV irradiated and inactivated as described previously (48). Virion proteins remained active after UV irradiation, as assayed by the ability of the UV-inactivated stock to transac-tivate an IE gene promoter (48). In all experiments, cells were infected with HSV-1 strains at an MOI of 10 PFU per cell.
Western blot (immunoblot) analysis.Whole cell extracts were prepared and analyzed as described previously (47, 48). In brief, at various times postinfection, cells were scraped, washed with phosphate-buffered saline containing protease inhibitors (50mg ofNa-p-tosyl-L-lysine chloromethyl ketone [TLCK] and 25mg
of phenylmethylsulfonyl fluoride per ml) and lysed directly into sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) buffer. Proteins were analyzed by SDS-PAGE and Western blotting by the enhanced chemilumines-cence (Amersham) method. Antibodies used for Western analysis were DPK1, a polyclonal antibody raised to amino acids 2018 to 2136 of p350/DNA-PKcs (14, 36), Ku24.2, a polyclonal antibody recognizing the p70 and p80 subunits of Ku (13), and H1101, a monoclonal antibody recognizing ICP4 (Goodwin Institute, Plantation, Fla.). Secondary antibodies were horseradish peroxidase-conjugated goat anti-mouse or horseradish peroxidase-conjugated goat anti-rabbit (Jackson ImmunoResearch). Bands on Western blots were quantitated by using a Joyce-Loebl Chromoscan 3 densitometer, and peak values were expressed as peak integrals.
DNA-PK activity assays.Cells were harvested and lysed by single freeze-thaw in low-salt buffer as described previously (5). After addition of protease inhibi-tors (0.5 mM phenylmethylsulfonyl fluoride, 2mg of leupeptin per ml, and 2mg of pepstatin per ml), extracts were made to 0.5 M NaCl, 10 mM magnesium chloride, and 0.5 mM dithiothreitol. Supernatants were collected by centrifuga-tion at 10,0003gand assayed as described previously (2, 14) except that the substrate peptide was PESQEAFADLWKK (4). Control reactions were per-formed with the nonsubstrate peptide EPPLSEQAFADLWKK. Sonicated calf thymus DNA was present as activator at 10mg/ml. One unit of activity is defined as the amount of enzyme required to incorporate 1 nmol of phosphate (after subtraction of nanomoles of phosphate incorporated into a nonsubstrate pep-tide) per minute under standard assay conditions. Incorporation of phosphate into nonsubstrate peptide was less than 5% of that incorporated into substrate peptide. Protein concentrations were determined with the Bio-Rad protein assay. p350/DNA-PKcs add-back assays were performed as described previously (36) except that the PESQEAFADLWKK peptide was used. Both p350/DNA-PKcs and Ku were purified to homogeneity from human placenta as described previ-ously (14). Assay mixtures contained purified p350/DNA-PKcs alone (0.06mg), Ku alone (0.06mg), p350/DNA-PKcs plus Ku (0.06mg of each), or extracts (2 to 3mg of total protein) in the presence or absence of 0.06mg of purified p350/ DNA-PKcs.
DNA-PK half-life determination.Approximately 33106cells were labeled with [35S]methionine-cysteine (100mCi) for 2 h, washed, and then either mock infected or infected with HSV-1 KOS1.1. After infection, the cells were incu-bated at 378C in unlabeled medium supplemented with 200mM nonradioactive methionine. At various times postinfection, cells were lysed in
radioimmunopre-cipitation assay buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS) including protease inhibitors (20mg of benzamidine, 2mg of TLCK, 10mg of leupeptin, and 200mg of phenylmethyl-sulfonyl fluoride per ml) and 148 U of DNase I at 48C. Supernatants were precleared, and p350/DNA-PKcs was immunoprecipitated with 12ml of DPK1 polyclonal antibody for 2.5 h followed by protein A-Sepharose for 45 min at 48C. Sepharose was washed four times with radioimmunoprecipitation assay buffer including protease inhibitors. p350/DNA-PKcs was eluted with 23 cracking buffer (120 mM Tris [pH 6.8], 2% SDS, 2%b-mercaptoethanol, 20% glycerol, 0.02% bromophenol blue). Eluted proteins were visualized by SDS-PAGE and autoradiography after fixation of the gel in 1 M sodium salicylate. The relative radioactivity in each band was measured by phosphorimage analysis using a Fujix BAS100 bioimaging analyzer with MacBAS imaging software.
Artwork for Fig. 1 to 3.Autoradiographs were scanned on an Abaton 300 color scanner, with Adobe Photoshop software, and images were saved in TIFF. Images were assembled and labeled with QuarkXPress 3.0 software on a Macin-tosh IIfx.
RESULTS
DNA-PK activity and the p350/DNA-PKcs subunit decline following HSV-1 infection. To examine the fate of DNA-PK during infection with HSV-1, HeLa S3 cells were infected with the wild-type HSV-1 strain, KOS1.1. Cells were harvested at various times postinfection, and whole cell extracts were pre-pared for DNA-PK assays and immunoblotting. Although p350/DNA-PKcs remained abundant in mock-infected cells (Fig. 1A, lanes 1 to 3), it was rapidly depleted in KOS1.1-infected cells between 2 and 6 h postinfection (Fig. 1A, lanes 4 to 6, and Table 1). In contrast, the abundance of Ku subunits remained unchanged up to 6 h postinfection (Fig. 1B, lanes 4 to 6). In this particular experiment, the Ku antibody reacted weakly with the p70 subunit. In other experiments (Fig. 3 and data not shown), the reaction with the p70 subunit was stron-ger.
The DNA-PK activity in the infected extracts declined in parallel with the decrease in p350/DNA-PKcs levels (Table 1). DNA-PK activity at 6 h postinfection was approximately 14% of that in mock-infected cells, and p350/DNA-PKcs protein declined to approximately 9% of mock levels by 6 h
postinfec-FIG. 1. Depletion of p350/DNA-PKcs and DNA-PK activity following HSV-1 infection. (A) Western blot of p350/DNA-PKcs. HeLa S3 cells were mock infected (lanes 1 to 3) or infected with wild-type HSV-1 strain KOS1.1 (lanes 4 to 6). Cells were harvested at the times indicated and lysed directly into SDS-PAGE buffer. Proteins were separated by SDS-SDS-PAGE and analyzed for p350/ DNA-PKcs by using the polyclonal antibody DPK1. (B) Western blot of Ku p70 and p80. The same samples as used for panel A were analyzed for Ku p70 and p80 subunits by using Ku polyclonal antibody Ku24.2. The p70 subunit of Ku cross-reacted weakly on this particular blot but was visible on longer exposures and in other experiments analyzing mock-infected and KOS1.1-infected cells (see Fig. 3).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.326.546.72.242.2]tion. We have observed similar declines in p350/DNA-PKcs protein following HSV-1 infection of human embryonic lung cells and HeLaCCL2 cells and following HSV-2 infection of HeLa cells (data not shown).
No proteolytic breakdown products were observed on these immunoblots, or on blots probed with two monoclonal anti-bodies against p350/DNA-PKcs (data not shown), suggesting that the subunit is rapidly degraded to small fragments follow-ing infection.
The half-life of p350/DNA-PKcs decreases following virus infection.One possible explanation for the decrease in p350/ DNA-PKcs following infection is that the protein has a short half-life in HeLa cells and protein levels simply decline follow-ing the virus-induced shutoff of host cell translation that occurs rapidly after HSV-1 infection (55). However, the half-life of p350/DNA-PKcs in human erythroleukemia cells has been re-ported to be greater than 5 days (1).
To confirm that the decline in p350/DNA-PKcs was due to decreased half-life of the protein in infected cells, we deter-mined the half-life of p350/DNA-PKcs in mock-infected and
HSV-1-infected HeLa cells. Cells were labeled with [35
S]me-thionine-cysteine for 2 h, washed, mock infected or infected with wild-type virus, and chased for up to 24 h. The p350/DNA-PKcs protein was immunoprecipitated with DPK1 antibody, and the labeled protein was visualized by SDS-PAGE. Phos-phorimage analysis of these pulse chase experiments showed that the half-life of p350/DNA-PKcs was greater than 24 h in mock-infected HeLa cells (Fig. 2). In contrast, the half-life was less than 4 h in HSV-1-infected cells. Therefore, the rapid elimination of p350/DNA-PKcs seen after virus infection (within 3 to 4 h) suggests that an active p350/DNA-PKcs deg-radation process is induced in HSV-1-infected cells.
Degradation of cellular proteins is not a general effect of HSV-1 infection, as the abundance of HeLa cell proteins such as RNAP II large subunit (48), TATA-binding protein, protein kinase C, protein phosphatase 1, cdk2, tubulin, and several basal RNAP II transcription factors did not change signifi-cantly up to 8 h postinfection (57).
Depletion of p350/DNA-PKcs requires expression of the vi-ral IE genes.One explanation for the decline in p350/DNA-PKcs levels is that a component of the virion particle could alter the protein’s half-life. The HSV-1 virion contains several regulatory proteins, including VP16, which is required for
tran-scription activation of the viral IE genes and thevhs-encoded
protein, which is instrumental in effecting a shutoff of host cell translation and a decline in many host RNAs (55).
To investigate whether the depletion of p350/DNA-PKcs was a consequence of functions introduced with the virion particle, we examined DNA-PK activity and p350/DNA-PKcs abundance in cells infected with UV-inactivated virus. Short-wave UV treatment of HSV-1 virus stocks cross-links viral DNA and prevents transcription of all viral genes but leaves virion proteins intact and active (48). This stock of UV-inac-tivated virus has been tested previously and contains active virion proteins, as measured by the ability of the virions to transactivate an IE promoter (48). HeLa cells were mock in-fected or inin-fected with either KOS1.1 or UV-inactivated virus. Cells were harvested and analyzed for p350/DNA-PK or Ku by Western blotting. Infection of cells with UV-inactivated virus resulted in no changes to p350/DNA-PKcs levels or to the abundance of p70Ku or p80Ku (Fig. 3A). Assays of DNA-PK activity in extracts prepared from cells infected with UV-inac-tivated virus showed no changes up to 8 h postinfection (Fig. 4A). These results indicate that the expression of the viral genome is necessary for loss of both the p350/DNA-PKcs and DNA-PK activity.
To test whether viral DNA replication, DNA packaging, or expression of DE and L gene products was necessary for de-pletion of p350/DNA-PKcs, we examined protein levels and kinase activity in cells infected with viruses containing a null mutation in the gene coding for ICP4. Expression of the ICP4 gene product is absolutely required for viral DE and L gene expression and hence for viral DNA replication and packaging. HeLa cells were mock infected or infected with the ICP4 mutant virus. Cells were harvested at various times postinfec-tion, and extracts were analyzed for p350/DNA-PKcs or Ku levels by Western blotting. Infection of cells with the ICP4 mutant virus resulted in decreases in p350/DNA-PKcs levels similar to those seen after infection with wild-type virus (Fig. 3B). Abundance of Ku subunits remained unchanged up to 8 h postinfection. Assays of DNA-PK activity in extracts prepared from ICP4 mutant infected cells showed rapid loss of kinase activity, paralleling decreases in p350/DNA-PKcs (Fig. 4A). These results indicate that expression of a viral IE protein other than ICP4 is necessary to induce the loss of p350/DNA-PKcs and kinase activity. In addition, because DE and L gene expression is required for viral DNA replication and packag-ing, this result suggests that neither of these functions is nec-essary for loss of DNA-PK activity or the p350/DNA-PKcs subunit.
The ICP0 gene product is required for modifications to DNA-PK and its catalytic subunit.We next sought to identify
[image:3.612.57.300.90.207.2]FIG. 2. Stability of p350/DNA-PKcs in mock-infected and HSV-1-infected HeLa S3 cells. Cells were pulse-labeled in the presence of [35 S]methionine-cysteine for 2 h, washed, infected or mock infected, and chased for the times indicated. Cells were lysed, p350/DNA-PKcs was immunoprecipitated with DPK1 antibody, and labeled p350/DNA-PKcs was visualized by SDS-PAGE and autoradiography. Radioactivity in each band was measured by phosphorimage analysis, and values are shown below each lane, expressed as percentage of the value for the mock-infected cells at 0 h.
TABLE 1. Depletion of DNA-PK activity and p350/DNA-PKcs following HSV-1 infection
Sample
DNA-PK activitya p350/DNA-PKcs level
U/mg of protein
% of mock,
6 h Integral
b % of mock, 6 h
Mock
0 h 8.26
4 h 9.88
6 h 9.09 100 2,949 100
KOS
2 h 7.28 80.1 2,601 88.2
4 h 2.27 24.9 485 16.4
6 h 1.27 13.9 261 8.9
a
Activity in extracts prepared from the same set of samples as analyzed in Fig. 1. Each value is the average of duplicate assays.
b
Integral under peak determined by densitometer scans of autoradiograph shown in Fig. 1A.
on November 9, 2019 by guest
http://jvi.asm.org/
the viral IE protein that was required for depletion of p350/ DNA-PKcs and DNA-PK activity, by infecting cells with vi-ruses mutant in individual IE genes. HeLa cells were infected with viruses mutant in the IE genes encoding ICP22, ICP27,
ICP0, and ICP6 (a DE gene product which is synthesized at IE times). Cells were harvested and extracts were analyzed by Western blotting. Levels of p350/DNA-PKcs declined in cells infected with viruses mutant in the genes encoding ICP22,
FIG. 3. Requirement for ICP0 in the decline of p350/DNA-PKcs protein levels. HeLa S3 cells were mock infected or infected with wild-type virus, UV-inactivated virus, or viruses mutant in IE genes. Cells were harvested, extracts were prepared for DNA-PK assays, and aliquots containing 10mg of protein were analyzed by immunoblotting and probed with polyclonal antibody DPK1 or Ku24.2 or a monoclonal antibody against ICP4. (A) Viral gene expression is required for loss of p350/DNA-PKcs. Lanes: 1, cells mock infected (m) for 6 h; 2 and 3, cells infected with wild-type virus KOS1.1 for 2 or 4 h; 4 and 5, cells infected with UV-inactivated virus for 6 and 8 h. (B) An IE gene product, other than ICP4, is required for loss of p350/DNA-PKcs. Lanes: 1, cells mock infected for 4 h; 2 to 5, cells infected with the ICP4 null mutantd120. (C) The IE gene product, ICP22, is not required for loss of p350/DNA-PKcs. Lanes: 1, cells mock infected for 2 h; 2, cells infected with wild-type virus KOS1.1 for 2 h; 3 to 5, cells infected with the ICP22 nonsense mutant 22/n199. (D) The IE gene product ICP27 is not required for loss of p350/DNA-PKcs. Lanes: 1, cells mock infected for 2 h; 2 to 5, cells infected with the ICP27 null mutantd27-1. (E) The DE gene product ICP6 is not required for loss of p350/DNA-PKcs. Lanes: 1, cells mock infected for 2 h; 2 to 5, cells infected with the ICP6 deletion mutant ICP6D. (F) The IE gene product ICP0 is required for loss of p350/DNA-PKcs. Lanes: 1, cells mock infected for 2 h; 2 to 5, cells infected with the ICP0 nonsense mutantn212. In the middle panel, the same blot that was probed for p350/DNA-PKcs was stripped and reprobed with monoclonal antibody (H1101) against ICP4.
on November 9, 2019 by guest
http://jvi.asm.org/
ICP27, and ICP6, with kinetics similar to those seen in cells infected with wild-type virus (Fig. 3C to E). However, cells infected with the ICP0 mutant virus retained p350/DNA-PKcs at uninfected levels (Fig. 3F). We confirmed that the ICP0 mutant virus infection occurred efficiently by probing the same Western blot for expression of the ICP4 protein (Fig. 3F, middle panel). Ku levels were unchanged in any of the infec-tions (Fig. 3A to F).
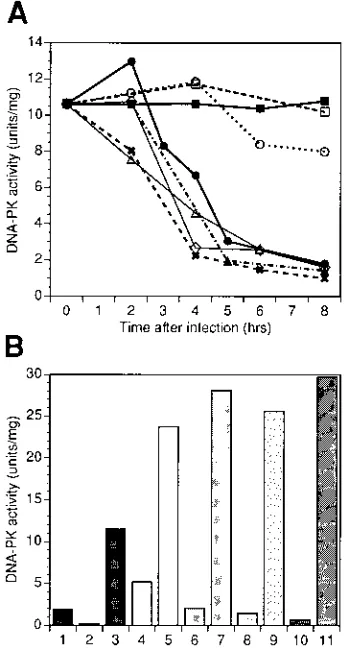
Extracts from the foregoing experiments were also assayed for DNA-PK activity. Infection of cells with viruses mutant in ICP22, ICP27, and ICP6 resulted in loss of DNA-PK activity, similar to losses seen after infection with wild-type virus (Fig. 4A). However, activity remained high in cells infected with the ICP0 mutant virus (Fig. 4A).
To test Ku function when DNA-PK activity was low, we performed add-back experiments (Fig. 4B). Extracts from cells infected with wild-type virus or viruses mutant in the IE gene encoding ICP4, ICP6, or ICP27 were reconstituted with
puri-fied p350/DNA-PKcs and assayed for DNA-PK activity. Puri-fied p350/DNA-PKcs restored DNA-PK activity in all extracts depleted of activity after infection, indicating that Ku remained functional following HSV-1 infection.
Therefore, we conclude that infection of human cells with HSV-1 specifically targets the p350/DNA-PKcs catalytic sub-unit of DNA-PK and attenuates DNA-PK kinase activity while maintaining the DNA binding function of Ku. Both the loss of p350/DNA-PKcs and the inactivation of DNA-PK require ex-pression of the viral IE gene product, ICP0.
DISCUSSION
ICP0 is a nuclear phosphoprotein of 775 amino acids with an apparent molecular mass of 110 to 120 kDa (21). ICP0 con-tains several structural features reminiscent of transcription activators, such as an acidic amino-terminal region, two central proline-rich regions, and an amino-terminal cysteine-rich zinc finger-like domain known as the RING finger, a motif which is shared by proteins involved in transcription, recombination, and signal transduction, as well as the PML oncogene product (19, 24). RING finger motifs are thought to interact with both DNA and other proteins; however, the mode of action of RING finger motifs is still unknown.
The ICP0 protein forms multimers and interacts with a num-ber of viral and cellular proteins, including ICP4 and an un-identified 135 kDa cellular protein (38, 40, 59). During infec-tion, ICP0 becomes associated with nuclear structures which contain the PML protein, and this association is followed by disruption of these structures (19, 37). Both recombinant ICP0 and ICP0 in crude extracts bind nonspecifically to DNA-cellu-lose columns (20, 30). In addition, ICP0 is associated with chromatin in vivo (30).
In transient transfection assays, ICP0 acts as a potent trans-activator of gene expression from any RNAP II promoter which exhibits a basal level of transcription (10, 11). ICP0
appears to act independently of any cis-acting elements,
in-cluding the TATA box (15, 41). ICP0 interacts with the viral transcription activator, ICP4, to elicit synergistic stimulation of reporter genes during transient transfections (for a review, see reference 21). Despite evidence for transcriptional activation, only one known viral gene (the ICP6 gene, which encodes a subunit of ribonucleotide reductase) specifically requires ICP0 during a lytic infection in cultured cells (16).
The role of ICP0 during lytic infection is complicated and depends on the MOI used to infect cultured cells. ICP0 mutant
viruses grow poorly at low MOIs (,1 PFU per cell), exhibiting
reduced levels of DE and L RNAs and proteins. However, at high MOIs (5 to 10 PFU per cell), wild-type levels of gene expression and virus production occur (21, 53). Therefore, when cells are infected with ICP0 mutant viruses at a high MOI, efficient expression of viral genes must be independent of ICP0. It has been suggested that high levels of virion pro-teins such as VP16, present following high-MOI infections, stimulate IE gene expression to a degree that overcomes the requirement for ICP0. Therefore, ICP0 and VP16 may per-form complementary roles in initiating efficient viral gene ex-pression (11, 21).
[image:5.612.91.263.72.396.2]Further evidence that ICP0 may play a role in early viral gene expression comes from studies of activation of viral gene expression from transfected virus DNA. When cells are trans-fected with purified viral DNA mutant in ICP0, virus produc-tion is delayed by 2 days and titers are reduced by several orders of magnitude compared with transfection with purified wild-type DNA. Cotransfection of an ICP0 plasmid along with mutant viral DNA complements the defect (9). Therefore,
FIG. 4. Loss of DNA-PK activity and retention of Ku activity following in-fection with wild-type and mutant viruses. HeLa S3 cells were mock infected or infected with viruses as described in the legend to Fig. 3. (A) DNA-PK activity in infected cell extracts. Cells were harvested, extracts were prepared, and DNA-PK activity assays were performed as described in Materials and Methods. Cells were mock infected (filled squares) or infected with UV-inactivated virus (open squares), KOS1.1 (filled circles), ICP0 mutant (open circles), ICP4 mutant (crosses), ICP6 mutant (diamonds), ICP27 mutant (open triangles), or ICP22 mutant (closed triangles). (B) Add-back experiments. Add-back experiments were performed as described in Materials and Methods. Bars: 1, p350/DNA-PKcs alone; 2, Ku alone; 3, p350/DNA-PKcs plus Ku; 4, KOS-infected extract alone; 5, KOS-infected extract plus p350/DNA-PKcs; 6, ICP4-infected extract alone; 7, ICP4-infected extract plus p350/DNA-PKcs; 8, ICP6-infected extract alone; 9, ICP6-infected extract plus p350/DNA-PKcs; 10, ICP27-infected extract alone; 11, ICP27-infected extract plus p350/DNA-PKcs.
on November 9, 2019 by guest
http://jvi.asm.org/
ICP0 provides a critical transactivating function in the absence of virion components such as VP16.
In another study of viral gene expression in the absence of virion components, ICP0 appears to similarly provide impor-tant transactivating functions. In these experiments, HSV-2 is forced into a dormant, latent-like state by heat treatment of cells following infection. The viral DNA can be reactivated into a lytic cycle by superinfection with wild-type HSV-1, viruses mutant in any IE gene except ICP0, and an adenovirus recom-binant that expresses ICP0 (21, 28, 51, 52). Superinfections with UV-inactivated HSV-1, viruses mutant in ICP0, or adeno-viruses not expressing ICP0 are unable to reactivate the latent virus. Hence, ICP0 is both required and sufficient in this sys-tem, in the absence of any other HSV-1 proteins, for reactiva-tion from viral latency. In addireactiva-tion, ICP0 appears to play a role in promoting reactivation from neuronal latency in vivo, al-though it is not absolutely required (36a). The reactivation efficiencies of various ICP0 mutants correspond to the mu-tants’ transactivation efficiency in transient transfection assays (8).
Everett et al. (21) have suggested that ICP0 may act directly on the latent virus genome, displacing chromatin proteins and permitting access of the RNAP II transcription machinery to latent viral DNA. The latent genome, unlike the viral DNA of lytic infections, is assembled into nucleosomal structures re-sembling cellular chromatin (17, 39). This hypothesis would explain the transcriptional promiscuity of ICP0 and the lack of necessity for ICP0 in high-MOI lytic infections in which VP16 and ICP4 may be abundant enough to stimulate transcription of nucleosome-free viral genomes in the absence of ICP0.
It is possible that inhibition of DNA-PK activity is an im-portant mechanism for increasing the levels of RNAP II tran-scription on viral DNA or on transfected DNA. DNA-PK is known to inhibit transcription by RNAP I (33, 34) and by RNAP II (26). It appears to inhibit RNAP I transcription in vitro by phosphorylating a component of the RNAP I basal transcription complex. Although the mechanism by which DNA-PK represses RNAP II transcription in vivo on the mouse mammary tumor virus promoter is unknown, it involves sequence-specific binding of DNA-PK to a negative regulatory element and may involve phosphorylation of the glucocorticoid receptor and Oct-1. We suggest that DNA-PK may interfere with the basal or activated levels of RNAP II transcription of viral genes, both when viral DNA first enters the nucleus and when viral DNA enters a latent state in neurons. DNA-PK may accomplish this repression by phosphorylating (and inactivat-ing) transcription factors such as Oct-1 that enhance viral gene expression. ICP0 may counteract the repressive activities of DNA-PK by binding to lytic or latent viral genomes and dis-placing DNA-PK and other chromatin proteins from the DNA. Once p350/DNA-PKcs is displaced from DNA (and dissoci-ated from Ku), it may be vulnerable to rapid degradation. Alternatively, ICP0 could directly contribute to the loss of p350/DNA-PKcs through a specific effect on its proteolysis. Therefore, ICP0, by directly or indirectly targeting the p350/ DNA-PKcs subunit of DNA-PK, may facilitate enhanced levels of RNAP II transcription from the viral genome.
The depletion of DNA-PK during infection could also alter the recombination and repair mechanisms of the host cell in such a way as to favor viral DNA replication and/or packaging. However, as yet, ICP0 has not been shown to affect viral DNA replication or packaging of viral DNA.
Interestingly, DNA-PK is also inactivated, and p350/DNA-PKcs is specifically cleaved, during apoptosis (56), perhaps facilitating the DNA double-strand cleavage that accompanies programmed cell death. However, the mechanisms by which
p350/DNA-PKcs is degraded during HSV-1 infection are prob-ably different from those operating during apoptosis, as the specific cleavage products seen during apoptosis are not de-tected following infection with HSV-1 (56). Our observation that three different antibodies to p350/DNA-PKcs detect no breakdown products on immunoblots suggests that p350/DNA-PKcs is degraded to small fragments rapidly following infec-tion.
Finally, it is unlikely that depletion of DNA-PK activity is solely responsible for the aberrant phosphorylation of the large subunit of RNAP II that follows HSV-1 infection. RNAP II is modified similarly in cells infected with the ICP0 mutant virus and in cells infected with wild-type virus (47); therefore, mod-ifications to RNAP II occur in the presence or absence of wild-type levels of DNA-PK. Hence, alterations to other ki-nases and/or phosphatases must account for virus-induced modifications to RNAP II phosphorylation.
ACKNOWLEDGMENTS
We thank Neal DeLuca, Sandy Weller, and Priscilla Schaffer for providing HSV-1 mutants. The plasmid expressing amino acids 2018 to 2135 of p350/DNA-PKcs was a gift from C. W. Anderson and S. P. Jackson. S.P.L.-M. thanks Lauri G. Lintott for technical assistance and D. W. Chan for purified DNA-PK.
This research was supported by Medical Research Council of Can-ada (C.A.S.), Alberta Heritage Foundation for Medical Research (C.A.S., S.A.R., M.C.L., and S.P.L.-M.), National Science and Engi-neering Research Council of Canada (S.P.L.-M. and M.C.L.), and the National Cancer Institute of Canada (S.A.R.).
REFERENCES
1.Ajmani, A. K., M. Satoh, E. Reap, P. L. Cohen, and W. H. Reeves.1995. Absence of autoantigen Ku in mature human neutrophils and human pro-myelocytic leukemia (HL-60) cells and lymphocytes undergoing apoptosis. J. Exp. Med.181:2049.
2.Allalunis-Turner, J. M., L. G. Lintott, G. M. Barron, R. S. Day III, and S. P. Lees-Miller.1995. Lack of correlation between DNA-dependent protein kinase activity and tumor cell radiosensitivity. Cancer Res.55:5200–5202. 3.Anderson, C. W.1994. Protein kinases and the response to DNA damage.
Semin. Cell Biol.5:427–436.
4.Anderson, C. W., M. A. Connelly, S. P. Lees-Miller, L. G. Lintott, H. Zhang, J. A. Sipley, K. Sakaguchi, and E. Appella.1995. The human DNA-activated protein kinase, DNA-PK: substrate specificity, p. 395–406.InM. Z. Atassi and E. Appella (ed.), Methods in protein structure analysis. Plenum Press, New York.
5.Anderson, C. W., and S. P. Lees-Miller.1992. The human DNA-activated protein kinase, DNA-PK. Crit. Rev. Eukaryotic Gene Expression2:283–314. 6.Blunt, T., N. J. Finnie, G. E. Taccioli, G. C. M. Smith, J. Demengeot, T. M. Gottlieb, R. Mizuta, A. J. Varghese, F. W. Alt, P. A. Jeggo, and S. P. Jackson. 1995. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine SCID mutation. Cell80:813–823.
7.Boubnov, N. V., K. T. Hall, Z. Wills, S. E. Lee, D. M. He, D. M. Benjamin, C. R. Pulaski, H. Band, W. Reeves, E. A. Hendrickson, and D. T. Weaver. 1995. Complementation of the ionizing radiation sensitivity, DNA end bind-ing and V(D)J recombination defects of double-strand break repair mutants by the p86 Ku autoantigen. Proc. Natl. Acad. Sci. USA92:890–894. 8.Cai, W., T. L. Astor, L. M. Liptak, C. Cho, D. M. Coen, and P. A. Schaffer.
1993. The herpes simplex virus type 1 regulatory protein ICP0 enhances virus replication during acute infection and reactivation from latency. J. Virol. 67:7501–7512.
9.Cai, W., and P. A. Schaffer.1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol.63:4579–4589.
10. Cai, W., and P. A. Schaffer.1991. A cellular function can enhance gene expression and plating efficiency of a mutant defective in the gene for ICP0, a transactivating protein of herpes simplex virus type 1. J. Virol.65:4078– 4090.
11. Cai, W., and P. A. Schaffer.1992. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol.66:2904–2915.
12. Carter, T., I. Vancurova, I. Sun, W. Lou, and S. DeLeon.1990. A DNA-activated protein kinase from HeLa cell nuclei. Mol. Cell. Biol.10:6360– 6471.
13. Chan, D. W., and S. P. Lees-Miller.1996. Autophosphorylation of the
on November 9, 2019 by guest
http://jvi.asm.org/
activated protein kinase, DNA-PK, correlates with inactivation and dissoci-ation of the catalytic subunit, DNA-PKcs. J. Biol. Chem.271:8936–8942. 14. Chan, D. W., C. H. Mody, S. Y. Ting, and S. P. Lees-Miller.1996. Purification
and characterization of the double-stranded DNA-activated protein kinase, DNA-PK, from human placenta. Biochem. Cell Biol.74:67–73.
15. Chen, J., and S. Silverstein.1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol.66:2916– 2927.
16. Desai, P., R. Ramakrishnan, Z. W. Lin, B. Osak, J. C. Glorioso, and M. Levine.1993. The RR1 gene of herpes simplex virus type 1 is uniquelytrans
activated by ICP0 during infection. J. Virol.67:6125–6135.
17. Deshmane, S. L., and N. W. Fraser.1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol.63:943–947.
18. Dvir, A., S. R. Peterson, M. W. Knuth, H. Lu, and W. S. Dynan.1992. Ku autoantigen is the regulatory component of a template-associated protein kinase that phosphorylates RNA polymerase II. Proc. Natl. Acad. Sci. USA 89:11920–11924.
19. Everett, R. D., P. O’Hare, D. O’Rourke, P. Barlow, and A. Orr.1995. Point mutations in the herpes simplex virus type 1 Vmw110 RING finger helix affect activation of gene expression, viral growth, and interactions with PML-containing nuclear structures. J. Virol.69:7339–7344.
20. Everett, R. D., A. Orr, and M. Elliott.1991. High level expression and purification of herpes simplex virus type 1 immediate early polypeptide Vmw110. Nucleic Acids Res.19:6155–6161.
21. Everett, R. D., C. M. Preston, and N. D. Stow.1991. Functional and genetic analysis of the role of Vmw110 in herpes simplex virus replication, p. 49–76.
InE. K. Wagner (ed.), Herpesvirus transcription and its regulation. CRC Press, Inc., Boca Raton, Fla.
22. Feldman, L. T.1991. The molecular biology of herpes simplex virus latency, p. 223–244.InE. K. Wagner (ed.), Herpesvirus transcription and its regu-lation. CRC Press, Inc., Boca Raton, Fla.
23. Finnie, N. J., T. M. Gottlieb, T. Blunt, P. A. Jeggo, and S. P. Jackson.1995. DNA-dependent protein kinase activity is absent in xrs-6 cells: implications for site-specific recombination and DNA double-strand break repair. Proc. Natl. Acad. Sci. USA92:320–324.
24. Freemont, P. S., I. M. Hanson, and J. Trowsdale.1991. A novel cysteine-rich sequence motif. Cell64:483–484.
25. Getts, R. C., and T. D. Stamato.1994. Absence of Ku-like DNA end binding activity in the xrs double-strand DNA repair-deficient mutant. J. Biol. Chem. 269:15981–15984.
26. Giffin, W., H. Torrance, D. J. Rodda, G. G. Pre´fontaine, L. Pope, and R. J. G. Hache´.1996. Sequence-specific DNA binding by Ku autoantigen and its effects on transcription. Nature (London)380:265–268.
27. Gottlieb, T. M., and S. P. Jackson.1993. The DNA-dependent protein kinase: requirement for DNA ends and association with Ku antigen. Cell 72:131–142.
28. Harris, R. A., R. D. Everett, X. Zhu, S. Silverstein, and C. M. Preston.1989. Herpes simplex virus type 1 immediate-early protein Vmw110 reactivates latent herpes simplex virus type 2 in an in vitro latency system. J. Virol. 63:3513–3515.
29. Hartley, K. O., D. Gell, G. C. M. Smith, H. Zhang, N. Divecha, M. A. Connelly, A. Admon, S. P. Lees-Miller, C. W. Anderson, and S. P. Jackson. 1995. DNA-dependent protein kinase catalytic subunit: a relative of phos-phatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 82:849–856.
30. Hay, T. T., and J. Hay.1980. Properties of herpesvirus-induced “immediate-early” polypeptides. Virology104:230–234.
31. Jeggo, P. A., G. E. Taccioli, and S. P. Jackson.1995. Menage a` trois: double strand break repair, V(D)J recombination and DNA-PK. Bioessays17:949–957. 32. Kirchgessner, C. U., C. K. Patil, J. W. Evans, C. A. Cuomo, L. M. Fried, T. Carter, M. A. Oettinger, and J. M. Brown.1995. DNA-dependent kinase (p350) as a candidate gene for the murine SCID defect. Science267:1178– 1183.
33. Kuhn, A., T. M. Gottlie, S. P. Jackson, and I. Grummt.1995. DNA-depen-dent protein kinase: a potent inhibitor of transcription by RNA polymerase I. Genes Dev.9:193–203.
34. Labhart, P.1995. DNA-dependent protein kinase specifically represses pro-moter-directed transcription initiation by RNA polymerase I. Proc. Natl. Acad. Sci. USA92:2934–2938.
35. Lees-Miller, S. P., Y.-R. Chen, and C. W. Anderson.1990. Human cells contain a DNA-activated protein kinase that phosphorylates simian virus 40 T antigen, mouse p53, and the human Ku autoantigen. Mol. Cell. Biol. 10:6472–6481.
36. Lees-Miller, S. P., R. Godbout, D. W. Chan, M. W. Weinfeld, R. S. Day, G. M. Barron, and J. Allalunis-Turner.1995. Absence of the p350 subunit of DNA-activated protein kinase from a radiosensitive human cell line. Science 267:1183–1185.
36a.Leib, D. A., D. M. Coen, C. L. Bogard, K. A. Hicks, D. R. Yager, D. M. Knipe,
K. L. Tyler, and P. A. Schaffer.1989. Immediate-early regulatory gene mutants define different stages in the establishment and reactivation of herpes simplex virus latency. J. Virol.63:759–768.
36b.Maldonado, E., R. Shiekhattar, M. Sheldon, H. Cho, R. Drapkin, P. Rickert, E. Lees, C. W. Anderson, S. Linn, and D. Reinberg.1996. A human RNA polymerase II complex associated with SRB and DNA-repair proteins. Na-ture (London)381:86–89.
37. Maul, G. G., and R. D. Everett.1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol.75:1223–1233.
38. Meredith, M., A. Orr, M. Elliott, and R. Everett.1995. Separation of se-quence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology209:174–187.
39. Muggeridge, M. I., and N. W. Fraser.1986. Chromosomal organization of the herpes simplex virus genome during acute infection of the mouse central nervous system. J. Virol.59:764–767.
40. Mullen, M.-A., S. Gerstberger, D. M. Ciufo, J. D. Mosca, and G. S. Hayward. 1995. Evaluation of colocalization interactions between the IE110, IE175, and IE63 transactivator proteins of herpes simplex virus within subcellular punctate structures. J. Virol.69:476–491.
41. Nabel, G. J., S. A. Rice, D. M. Knipe, and D. Baltimore.1988. Alternative mechanisms for activation of human immunodeficiency virus enhancer in T cells. Science239:1299–1302.
42. Peterson, S. R., S. A. Jesch, T. N. Chamberlin, A. Dvir, S. K. Rabindran, C. Wu, and W. S. Dynan.1995. Stimulation of DNA-dependent protein kinase by RNA polymerase II transcription activator proteins. J. Biol. Chem.270: 1449–1454.
43. Peterson, S. R., A. Kurimasa, M. Oshimura, W. S. Dynan, E. M. Bradbury, and D. J. Chen.1995. Loss of the catalytic subunit of the DNA-dependent protein kinase in DNA double-strand-break-repair mutant mammalian cells. Proc. Natl. Acad. Sci. USA92:3171–3174.
44. Rathmell, W. K., and G. Chu.1994. A DNA end-binding factor involved in double-strand break repair and V(D)J recombination. Mol. Cell. Biol.14: 4741–4748.
45. Rathmell, W. K., and G. Chu.1994. Involvement of the Ku autoantigen in the cellular response to DNA double-strand breaks. Proc. Natl. Acad. Sci. USA91:7623–7627.
46. Rice, S. A., and D. M. Knipe.1990. Genetic evidence for two distinct trans-activation functions of the herpes simplexaprotein ICP27. J. Virol.64:1704– 1715.
47. Rice, S. A., M. C. Long, V. Lam, P. A. Schaffer, and C. A. Spencer.1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol.69:5550–5559.
48. Rice, S. A., M. C. Long, V. Lam, and C. A. Spencer.1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compart-ments following herpes simplex virus infection. J. Virol.68:988–1001. 49. Roizman, B., and A. E. Sears.1990. Herpes simplex viruses and their
repli-cation, p. 1795–1841.InB. N. Fields and D. M. Knipe (ed.), Virology, 2nd ed. Raven Press, New York.
50. Roth, D. B., T. Lindahl, and M. Gellert.1995. How to make ends meet. Curr. Biol.5:496–499.
51. Russell, J., and C. M. Preston.1986. An in vitro latency system for herpes simplex virus type 2. J. Gen. Virol.67:397–403.
52. Russell, J., N. D. Stow, E. C. Stow, and C. M. Preston.1987. Herpes simplex virus genes involved in latency in vitro. J. Gen. Virol.68:3009–3018. 53. Sacks, W. R., and P. A. Schaffer.1987. Deletion mutants in the gene
encod-ing the herpes simplex virus type 1 immediate-early protein ICP0 exhibit impaired growth in cell culture. J. Virol.61:829–839.
54. Smider, V., W. K. Rathmell, M. R. Lieber, and G. Chu.1994. Restoration of X-ray resistance and V(D)J recombination in mutant cells by Ku cDNA. Science266:288–291.
55. Smiley, J. R., B. Panning, and C. A. Smibert.1991. Regulation of cellular genes by HSV products, p. 151–179.InE. K. Wagner (ed.), Herpesvirus transcription and its regulation. CRC Press, Inc., Boca Raton, Fla. 56. Song, Q., S. P. Lees-Miller, S. Kumar, N. Zhang, D. W. Chan, G. C. M.
Smith, S. P. Jackson, A. Alnemri, G. Litwack, and M. F. Lavin. DNA-dependent protein kinase catalytic subunit: a target for the ICE-like protease CPP32 in apoptosis. EMBO J., in press.
57. Spencer, C. A., M. A. Kilvert, M. C. Long, and S. A. Rice.1996. Unpublished data.
58. Taccioli, G. E., T. M. Gottlieb, T. Blunt, A. Priestley, J. Demongeot, R. Mizuta, A. R. Lehmann, F. W. Alt, S. P. Jackson, and P. A. Jeggo.1994. Product of the XRCC5 gene and its role in DNA repair and V(D)J recom-bination. Science265:1442–1445.
59. Yao, F., and P. A. Schaffer.1994. Physical interaction between the herpes simplex virus type 1 immediate-early regulatory proteins ICP0 and ICP4. J. Virol.68:8158–8168.