organic papers
o3636
Udaya lakshmiet al. C9H7NO4 doi:10.1107/S1600536805031879 Acta Cryst.(2005). E61, o3636–o3638 Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
3-Nitrocinnamic acid
K. Udaya Lakshmi,a*
S. Thamotharan,bM. Srinivasan,c K. Ramamurthiaand
B. Varghesec
aSchool of Physics, Bharathidasan University, Tiruchirappalli 620 024, India,bMolecular Biophysics Unit, Indian Institute of Science, Bangalore 560 012, India, andcSophisticated Analytical Instrument Facility, Indian Institute of Technology Madras, Chennai 600 036, India
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 293 K
Mean(C–C) = 0.003 A˚ Rfactor = 0.046 wRfactor = 0.146
Data-to-parameter ratio = 11.5
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
The title compound, C9H7NO4, forms centrosymmetric dimers
through intermolecular O—H O hydrogen bonds in the crystal structure. The nitro group deviates slightly from coplanarity with the benzene ring. The benzene ring and the carboxylic acid group are in an E configuration about the ethylenic double bond.
Comment
Various cinnamic acid derivatives form substrate inter-mediates with the enzyme papain (Huber, 1985). m -Nitro-cinnamic acid crystallizes in two modifications and the unit-cell dimensions of these polymorphs have been reported previously (Schmidt, 1964). In this paper, we report the crystal structure of thepolymorph ofm-nitrocinnamic acid, (I).
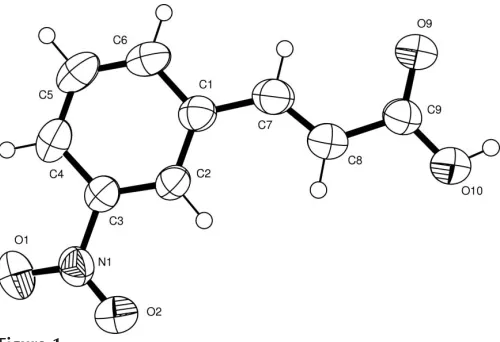
A perspective view of (I), with the atomic numbering scheme, is shown in Fig. 1. The bond lengths and angles agree well with literature values (Allenet al., 1987). The C1—C7— C8—C9 torsion angle of 179.5 (2)indicates that the benzene
ring and the carboxylic acid group are in an Econfiguration about the C7 C8 bond and the propenoic acid moiety exists in an extended conformation. The alkenecarbonyl
[image:1.610.208.458.545.716.2]conforma-Received 30 September 2005 Accepted 6 October 2005 Online 12 October 2005
Figure 1
tion [C7—C8—C9—O9 =2.5 (4)] is synperiplanar, which is
the most common conformation for trans-cinnamic acids (Leiserowitz, 1976).
The dihedral angle between the 3-nitro group and the benzene ring is 8.9 (9). In a related structure, viz. p
-nitro-cinnamic acid (Kageyama et al., 1993), the nitro group is coplanar (2.2) with the benzene ring. With respect to the plane of the benzene ring, the 3-nitro group is oriented at an angle of 45.3 in 4-dimethylamino-3-nitrocinnamic acid
(Huber, 1985), 3.6in 3,5-dinitrocinnamic acid and 2.3in the
3,5-dinitrocinnamic acid 2,5-dimethoxycinnamic acid complex (Desiraju & Sharma, 1991), 3.0 in the 3,5-dinitrocinnamic
acid 4-(N,N-dimethylamino)benzoic acid complex and 6.1in
the 3,5-dinitrocinnamic acid 4-(N,N-dimethylamino)cinnamic acid complex (Sharmaet al., 1993).
The angle between the mean plane of the benzene ring and the mean plane of the propenoic acid moiety is 3.5 (7)in (I)
and 2.6 in 4-dimethylamino-3-nitrocinnamic acid (Huber,
1985). The corresponding angles in 4-chlorocinnamic acid (Gluskeret al., 1975), 4-iodocinnamic acid (Goudet al., 1993),
p-nitrocinnamic acid (Kageyama et al., 1993), 3,5-dinitro-cinnamic acid and the 3,5-dinitrocinnamic acid 2,5-dimethoxycinnamic acid complex (Desiraju & Sharma, 1991) are 14.1, 13.8, 4.7, 28.7 and 6.4, respectively. In the
3,5-dinitrocinnamic acid 4-(N,N-dimethylamino)cinnamic acid complex, the propenoic acid group is twisted by 7.6out of the
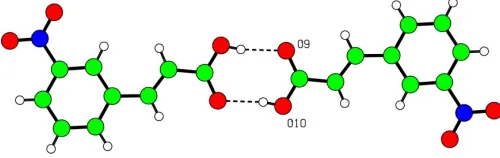
mean plane of the benzene ring (Sharmaet al., 1993). In the crystalline state, the molecules form O—H O hydrogen-bonded dimers across an inversion centre (Table 1). These dimers are stacked along the shortest cell axis and lead to anR2
2(8) motif (Fig. 2) (Bernsteinet al., 1995).
Experimental
The title compound, (I), was prepared by dissolving m -nitro-benzaldehyde (6 g, 0.04 mol) and malonic acid (8.3 g, 0.08 mol) in a mixture of 5 ml of pyridine and 0.25 ml of piperidine. The solution was allowed to reflux for 1 h, with rapid evolution of CO2. The
resulting title compound was recrystallized from ethanol.
Crystal data
C9H7NO4
Mr= 193.16
Monoclinic,P21=n a= 3.7756 (2) A˚
b= 9.4584 (13) A˚
c= 24.295 (4) A˚
= 90.875 (8)
V= 867.52 (18) A˚3
Z= 4
Dx= 1.479 Mg m
3
CuKradiation Cell parameters from 25
reflections
= 20–30
= 1.02 mm1
T= 293 (2) K Block, colourless 0.300.200.20 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
!–2scans
Absorption correction: scan (Northet al., 1968)
Tmin= 0.751,Tmax= 0.823
1733 measured reflections 1478 independent reflections 1068 reflections withI> 2(I)
Rint= 0.043
max= 67.9
h= 0!4
k= 0!11
l=29!29 3 standard reflections
frequency: 120 min intensity decay: none
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.046
wR(F2) = 0.146
S= 1.03 1478 reflections 129 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0726P)2
+ 0.3649P]
whereP= (Fo2+ 2Fc2)/3
(/)max< 0.001
max= 0.30 e A˚ 3
min=0.17 e A˚ 3
Extinction correction:SHELXL97
Extinction coefficient: 0.0054 (11)
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O10—H10 O9i
0.82 1.83 2.636 (3) 169
Symmetry code: (i)xþ1;y1;z.
All the H atoms were placed in idealized positions (C—H = 0.93 A˚ and O—H = 0.82 A˚ ) and constrained to ride on their parent atoms, withUiso(H) = 1.2Ueq(parent atom).
Data collection: CAD-4 EXPRESS (Enraf–Nonius, 1994); cell refinement:CAD-4 EXPRESS; data reduction:MolEN(Fair, 1990); program(s) used to solve structure:SIR92 (Altomare et al., 1994); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:ORTEP3 for Windows(Farrugia, 1997); software used to prepare material for publication:SHELXL97andPLATON
(Spek, 2003).
KU and KR thank Professor R. Jeyaraman and Dr K. Sarkunam, School of Chemistry, Bharathidasan University, Tiruchirappalli, India, for providing the chemicals.
References
Allen, F. H., Kennard, O., Watson, D. G., Brammer, L., Orpen, A. G. & Taylor, R. (1987).J. Chem. Soc. Perkin Trans. 2, pp. S1–19.
Altomare, A., Cascarano, G., Giacovazzo, C., Guagliardi, A., Burla, M. C., Polidori, G. & Camalli, M. (1994).J. Appl. Cryst.27, 435.
Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995).Angew. Chem. Int. Ed. Engl.34, 1555–1573.
Desiraju, G. R. & Sharma, C. V. K. (1991).J. Chem. Soc. Chem. Commun.pp. 1239–1241.
Enraf–Nonius (1994). CAD-4 EXPRESS. Version 5.1/1.2. Enraf–Nonius, Delft, The Netherlands.
Fair, C. K. (1990).MolEN. Enraf–Nonius, Delft, The Netherlands. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Glusker, J. P., Zacharias, D. E. & Carrell, H. L. (1975).J. Chem. Soc. Perkin Trans. 2, pp. 68–74.
Goud, B. S., Pathaneni, S. S. & Desiraju, G. R. (1993).Acta Cryst.C49, 1107– 1111.
Huber, C. P. (1985).Acta Cryst.C41, 1076–1079.
organic papers
[image:2.610.314.564.73.152.2]Acta Cryst.(2005). E61, o3636–o3638 Udaya lakshmiet al. C9H7NO4
o3637
Figure 2
Kageyama, Y., Iwamoto, T., Haisa, M. & Kashino, S. (1993).Acta Cryst.C49, 833–834.
Leiserowitz, L. (1976).Acta Cryst.B32, 775–802.
North, A. C. T., Phillips, D. C. & Mathews, F. S. (1968).Acta Cryst.A24, 351– 359.
Schmidt, G. M. J. (1964).J. Chem. Soc.pp. 2014–2021.
Sharma, C. V. K., Panneerselvam, K., Pilati, T. & Desiraju, G. R. (1993).J. Chem. Soc. Perkin Trans. 2, pp. 2209–2216.
Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany. Spek, A. L. (2003).J. Appl. Cryst.36, 7–13.
organic papers
supporting information
sup-1 Acta Cryst. (2005). E61, o3636–o3638
supporting information
Acta Cryst. (2005). E61, o3636–o3638 [https://doi.org/10.1107/S1600536805031879]
3-Nitrocinnamic acid
K. Udaya Lakshmi, S. Thamotharan, M. Srinivasan, K. Ramamurthi and B. Varghese
3-Nitrocinnamic acid
Crystal data
C9H7NO4
Mr = 193.16 Monoclinic, P21/n
Hall symbol: -P 2yn a = 3.7756 (2) Å b = 9.4584 (13) Å c = 24.295 (4) Å β = 90.875 (8)° V = 867.52 (18) Å3
Z = 4
F(000) = 400 Dx = 1.479 Mg m−3
Cu Kα radiation, λ = 1.54180 Å Cell parameters from 25 reflections θ = 20–30°
µ = 1.02 mm−1
T = 293 K Block, colourless 0.30 × 0.20 × 0.20 mm
Data collection
Enraf–Nonius CAD-4 diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω–2θ scans
Absorption correction: ψ scan (North et al., 1968)
Tmin = 0.751, Tmax = 0.823
1733 measured reflections
1478 independent reflections 1068 reflections with I > 2σ(I) Rint = 0.043
θmax = 67.9°, θmin = 3.6°
h = 0→4 k = 0→11 l = −29→29
3 standard reflections every 120 min intensity decay: none
Refinement
Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.046
wR(F2) = 0.146
S = 1.03 1478 reflections 129 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0726P)2 + 0.3649P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001
Δρmax = 0.30 e Å−3
Δρmin = −0.17 e Å−3
Extinction correction: SHELXL97, Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4
supporting information
sup-2 Acta Cryst. (2005). E61, o3636–o3638
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weightedR-factorwRand goodness of fitSare based on F2, conventional R-factors R are based
on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculatingR-factors(gt)etc. and is not relevant to the choice of reflections for refinement.R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be
even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 0.3333 (6) 0.0198 (2) 0.11430 (10) 0.0508 (6) C2 0.4539 (6) 0.0178 (2) 0.16858 (9) 0.0465 (6) H2 0.5548 −0.0636 0.1835 0.056* C3 0.4216 (6) 0.1376 (2) 0.19963 (9) 0.0458 (6) C4 0.2784 (7) 0.2620 (2) 0.17952 (12) 0.0592 (7) H4 0.2600 0.3417 0.2017 0.071* C5 0.1638 (7) 0.2640 (3) 0.12542 (13) 0.0647 (7) H5 0.0688 0.3464 0.1105 0.078* C6 0.1888 (7) 0.1455 (3) 0.09359 (11) 0.0590 (7) H6 0.1078 0.1485 0.0573 0.071* N1 0.5488 (6) 0.1356 (2) 0.25715 (9) 0.0559 (6) C7 0.3562 (7) −0.1043 (3) 0.07858 (10) 0.0592 (7) H7 0.2666 −0.0932 0.0430 0.071* C8 0.4884 (7) −0.2302 (3) 0.09062 (10) 0.0617 (7) H8 0.5826 −0.2459 0.1257 0.074* C9 0.4915 (7) −0.3454 (3) 0.05071 (10) 0.0565 (6) O1 0.4837 (7) 0.2370 (2) 0.28617 (9) 0.0890 (7) O2 0.7153 (6) 0.0337 (2) 0.27319 (8) 0.0752 (6) O9 0.3566 (5) −0.3298 (2) 0.00408 (7) 0.0708 (6) O10 0.6367 (6) −0.4602 (2) 0.06723 (8) 0.0758 (6) H10 0.6165 −0.5206 0.0432 0.091*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2005). E61, o3636–o3638
O2 0.0967 (16) 0.0674 (12) 0.0611 (12) 0.0120 (11) −0.0161 (10) 0.0039 (9) O9 0.0944 (15) 0.0670 (12) 0.0504 (10) 0.0113 (10) −0.0157 (9) 0.0000 (8) O10 0.1078 (16) 0.0607 (11) 0.0582 (11) 0.0143 (11) −0.0221 (10) −0.0038 (8)
Geometric parameters (Å, º)
C1—C2 1.389 (3) C6—H6 0.9300 C1—C6 1.398 (3) N1—O2 1.211 (3) C1—C7 1.463 (3) N1—O1 1.218 (3) C2—C3 1.368 (3) C7—C8 1.323 (3) C2—H2 0.9300 C7—H7 0.9300 C3—C4 1.381 (3) C8—C9 1.459 (3) C3—N1 1.471 (3) C8—H8 0.9300 C4—C5 1.378 (4) C9—O9 1.244 (3) C4—H4 0.9300 C9—O10 1.278 (3) C5—C6 1.366 (4) O10—H10 0.8200 C5—H5 0.9300
C2—C1—C6 118.3 (2) C5—C6—H6 119.2 C2—C1—C7 122.1 (2) C1—C6—H6 119.2 C6—C1—C7 119.7 (2) O2—N1—O1 123.3 (2) C3—C2—C1 118.8 (2) O2—N1—C3 118.4 (2) C3—C2—H2 120.6 O1—N1—C3 118.3 (2) C1—C2—H2 120.6 C8—C7—C1 128.1 (2) C2—C3—C4 123.3 (2) C8—C7—H7 116.0 C2—C3—N1 118.86 (19) C1—C7—H7 116.0 C4—C3—N1 117.9 (2) C7—C8—C9 122.2 (2) C5—C4—C3 117.7 (2) C7—C8—H8 118.9 C5—C4—H4 121.2 C9—C8—H8 118.9 C3—C4—H4 121.2 O9—C9—O10 123.7 (2) C6—C5—C4 120.4 (2) O9—C9—C8 120.6 (2) C6—C5—H5 119.8 O10—C9—C8 115.7 (2) C4—C5—H5 119.8 C9—O10—H10 109.5 C5—C6—C1 121.6 (2)
supporting information
sup-4 Acta Cryst. (2005). E61, o3636–o3638
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O10—H10···O9i 0.82 1.83 2.636 (3) 169