DESIGN DEVELOPMENT AND IN-VITRO EVALUATION
CONTROLLED RELEASE TABLETS
Divya Sahu*, Dr. Arun Patel, Prof. Shailendra Patel and Prof. Neelesh Dwivedi
Department of Pharmaceutics, Shri Ram Group of Institutions Jabalpur (M.P.).
ABSTRACT
The present investigation was aimed at designing controlled release
matrix tablets of isradipine, (C19H21N3O5) using various natural
polymers like guar gum, karaya gum, xanthan gum and veegum,
individually and in combinations of various concentrations, to produce
controlled release of isradipine in order to improve the efficacy and
patient compliance and to compare the drug release profiles of
optimized formulation and commercial formulation by similarity and
difference factors. The drug release from the formulation was found to
be zero order. Using Higuchi's model and the Korsmeyer equation, the
drug release mechanism from the controlled release tablets was found
to be Anomalous (non-Fickian) diffusion. The values of difference factor, f1 and similarity
factor, f2 was found to be 1.74 and 63.38 indicating similarity between drug release profiles
of optimized formulation and reference product.
KEYWORDS: Buccal delivery, Isradipine, Mucoadhesion, First pass metabolism, Permeation Enhancers.
1. INTRODUCTION
For many decades treatment of an acute disease or a chronic illness has been mostly
accomplished by delivery of drugs to patients using various pharmaceutical dosage forms,
including tablets, capsules, pills, suppositories, creams, ointments, liquids, aerosols and
injectables as drug carriers. High patient compliance and flexibility in developing dosage
forms made the oral drug delivery systems the most convenient mode of drug administration
compared to other dosage forms. 1.
Volume 8, Issue 13, 1057-1062. Research Article ISSN 2277– 7105
*Corresponding Author
Divya Sahu
Department of
Pharmaceutics, Shri Ram
Group of Institutions
Jabalpur (M.P.).
Article Received on 13 Oct. 2019,
Revised on 03 Nov. 2019, Accepted on 24 Nov. 2019
2. FORMULATION OF ISRADIPINE TABLET
There are various methods for preparing matrix tablets, namely Direct compression, Wet
granulation, Melt granulation, Response surface methodology etc.
2.1. Wet Granulation
Required quantities of drug and polymer were mixed thoroughly, and a sufficient volume of
granulating agent is added slowly. After enough cohesiveness was obtained, the mass is
sieved through 22/44 mesh.
2.2 Direct Compression
Direct compression is regarded as a relatively quick process where the powdered materials
are compressed directly without changing the physical and chemical properties of the drug.
3. MATRIX DEVICES
One of the least complicated approaches to the manufacture of sustained release dosage
forms involves the direct compression of the blends of drug, retardant material and additives
[image:2.595.138.450.414.536.2]to form a tablet in which the drug is embedded in a matrix core of the retardant.
Figure 6: Schematic Diagram of Matrix Systems.
In this model, drug in outside layer exposed to the bathing solution is dissolved first and
diffused out of the matrix. This process continues with the interface between bathing solution
and the solid drug moving controls, the rate of dissolution of drug particles within the matrix
must be faster that the diffusion rate of dissolved drug leaving matrix.
4. Preformulation Studies
Preformulation study may be described as a phase of the research and development process
where the formulation scientist characterizes the physical, chemical and mechanical
properties of new drug substances, in order to develop stable, safe and effective dosage
www.ajptr.com evaluation, possible interaction with various inert ingredients intended for
use in final dosage form was also considered.
Drug-Excipients Compatability Study by FT-IR Spectroscopy
FT-IR Spectroscopy of pure drug (Isradipine) and its formulations were carried out on Bruker
FTIR16000 model to investigate any possible interaction between the drug and the utilized
polymers (PEG 4000, PVP K30, Lycoat RS720). The samples were finely grounded with
KBr to prepare the pellets under a hydraulic pressure of 600 psi and a spectrum was scanned
in the wavelength range of 400 and 4000 cm-1 using Bruker FT‐IR spectrophotometer. The
compatibility of drug in the formulation was confirmed by comparing FTIR spectra of pure
drug with FTIR of its formulation.
CONSTRUCTION OF CALIBRATION Preparation of Standard Stock Solution
10 mg of Isradipine was accurately weighed and dissolved in 100ml volumetric flask
containing phosphate buffer of pH 6.8 and subjected to sonication. The volume is made up to
100ml with pH 6.8 phosphate buffer to produce a concentration of 100μg/ml, which is a stock
solution.
Determination of λmax Above solution was scanned between the range of 200-400nm by
Shimadzu 1700 model UV spectrophotometer. From the scan it was concluded that the λmax
of Isradipine was 285nm.
Calibration curve of Isradipine in phosphate buffer of pH 6.8.
From the standard stock solution aliquots 1ml, 2ml, 3ml, 4ml and 5ml were pipette out into
concentration of 10, 20, 30, 40 and 50μg/ml respectively. The absorbance of each
concentration was measured at λmax 285nm using UV Visible spectrophotometer against
blank (phosphate buffer of pH 6.8).
Melting method (fusion method)
Solid dispersion of Isradipine in PEG4000 or polyvinyl pyrollidone (PVP K30) containing
three different ratios (1:1, 1:1∙5 and 1:2 w/w) as seen in Table(1) were prepared by fusion
method. Required amount of drug and polymer were mixed in china dish, the mixture was
then heated using water bath at 70°C till it was completely melted.
Disintegration time
The disintegration time was measured using modified disintegration method. For this purpose
a petri dish was filled with 10 ml of water. The film was carefully put in the centre of petri
dish. The time for the film to completely disintegrate in to fine particles was noted in Table
3.26.
7. RESULTS AND DISCUSSIONS Particle Size
The Particle size was determined using mechanical sieve shaker as per the procedure. since
95% of the drug is retained on sieve # 50, the particle size of the drug lies between #50 and
#18 i.e. 300 um an 1.00 mm. The results are shown in the table 16.
Table 1: Particle Size Anlysis.
Sieve No Microns Wt of drug + sieve (g)
Wt of the drug retained (g)
% of drug retained
Cumulative % of drug retained (µ)
#18 1000 381.4 0.4 1.9 1.9
# 50 297 374 20 95.24 97.14
#70 210 335.6 0.6 2.86 100
#120 125 329 0 0 0
#140 105 323 0 0 0
#170 88 321 0 0 0
#200 74 322 0 0 0
#200 pass 502 0 0 0
21 100
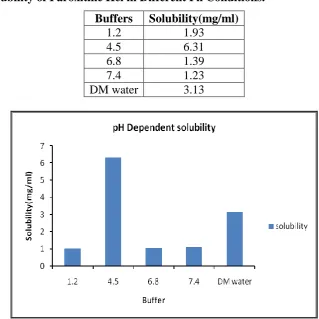
Solubility
The solubility of Paroxetine Hcl was carried out in different buffers as per the procedure and
Table 2: Solubility of Paroxitine Hcl in Different Ph Conditions. Buffers Solubility(mg/ml)
1.2 1.93
4.5 6.31
6.8 1.39
7.4 1.23
DM water 3.13
Solubility of Paroxitine Hcl in different pH conditions
7. SUMMARY AND CONCLUSION
The present study was under taken to formulate and evaluate the controlled release tablets of
Paroxetine hydrochloride by using direct compression. The study involved preformulation of
drug and excipients, formulation, evaluation and stability studies.
Matrix tablets of Paroxetine hydrochloride (F1-F6) were prepared by using hydrophilic and
hydrophobic polymers like HPMCK4M, HPMCK100M and Ethyl Cellulose. Formulation F5
with polymeric combination of HPMC and ethyl cellulose showed a better drug release at the
end of 6th hr.
CONCLUSION
It may be concluded from the present study that slow, controlled release of Paroxetine over a
period of 6 h was obtained from matrix tablets. It is evident from the results that Hydrophilic
matrix of HPMC could not control the Paroxetine release effectively where as a combination
of hydrophilic and hydrophobic matrix prepared by HPMCK4M and ethyl cellulose is a
REFERENCES
1. British Pharmacopoeia, Published by The stationary office under the license from the controller of Her Majesty’s Stationary Office for the Health Dept., 2007; 2058-2059.
2. Dr. B.K. Sharma, Instrumental Methods of Chemical Analysis, Goel Publishing House,
24th edn., 2005; 3-20: 286-385.
3. D. Fillic, M. Dumic, B. Klepic, A. Danilovski, M. Tudja, Polymorph V of Torasemide,
2007.
4. J.M. Rollinger, E.M. Gstrein, A. Burger, Crystal forms of torasemide: New Insights, Eur.
J. Pharm. Biopharm, 2002; 53: 75-86.
5. Lindsay. C, DeVane. et al., 31 Tim Cabelka, Shawn Mitchell, Karen Balwinski. et al.,
(2006)30 Tina Dasbach, Pushpa Inbasekaran, and Karen Balwinski. et al., 2003; (2002)2.
6. D.A. Skoog, D.M. West, F.J. Hollar, S.R. Crouch, Fundamental of Analytical Chemistry,
Cengage Learning India Private Limited, 8th edn., 2008; 815-995.
7. D.A. Williams, T.L. Lemke, Foye’s Principles of Medicinal Chemistry, Lippincott
Williams and Wilkins, 6th edn., 2007; 732.
8. D. Fillic, M. Dumic, B. Klepic, A. Danilovski, M. Tudja, Polymorph V of Torasemide,
2007.
9. G.D. Christian, Analytical Chemistry, John Wiley and Sons, 21st edn., 1994; 14-64,
505-514, 537-548.
10. H.H. Willard, L.L. Merrit, J.A. Dean, F.A. Settle Jr., Instrumental Methods of Analysis,
CBS Publisher and Distributors, 7th edn., 1986; 513-651.
11. Meineke, S. Engelhardt, J. Brockmoller, Improved solid-phase extraction and HPLC
measurement of torasemide and its important metabolites, J. Chromatogr. B., 2006; 831:
31-35.
12. J. Aronhime, D. Leonov, M. Kordova, A. Schwartz, B. Dolitzky, Torsemide polymorphs,
2002.
13. J.H. Block and J.M. Beale, Wilson and Gisvold’s Textbook of Organic Medicinal and
Pharmaceutical Chemistry, 11th edn., 1999; 613-614.
14. J.M. Rollinger, E.M. Gstrein, A. Burger, Crystal forms of torasemide: New Insights, Eur.
J. Pharm. Biopharm, 2002; 53: 75-86.
15. Antihypertensive+Agents at the US National Library of Medicine Medical Subject