M E T H O D
Open Access
High-throughput RNA interference screening
using pooled shRNA libraries and next generation
sequencing
David Sims, Ana M Mendes-Pereira, Jessica Frankum, Darren Burgess, Maria-Antonietta Cerone,
Cristina Lombardelli, Costas Mitsopoulos, Jarle Hakas, Nirupa Murugaesu, Clare M Isacke, Kerry Fenwick,
Ioannis Assiotis, Iwanka Kozarewa, Marketa Zvelebil, Alan Ashworth
*and Christopher J Lord
*Abstract
RNA interference (RNAi) screening is a state-of-the-art technology that enables the dissection of biological
processes and disease-related phenotypes. The commercial availability of genome-wide, short hairpin RNA (shRNA) libraries has fueled interest in this area but the generation and analysis of these complex data remain a challenge. Here, we describe novel open source computational methodologies, shALIGN and shRNAseq, that allow RNAi screens to be rapidly deconvoluted using next generation sequencing. Our computational pipeline offers efficient screen analysis and the flexibility and scalability to quickly incorporate future developments in shRNA library technology.
Background
RNA interference (RNAi) facilitates the assessment of gene function by silencing gene expression using syn-thetic anti-sense oligonucleotides or plasmids. It exploits a physiological mechanism that represses gene expres-sion, primarily by causing the degradation of mRNA transcripts. In mammalian cells, physiological RNAi is primarily mediated by non-protein-coding RNA tran-scripts, known as microRNAs (miRNAs). miRNAs are produced in a similar manner to mRNAs, but miRNAs are processed into shorter RNA species containing a hair-pin structure, known as short-hairhair-pin RNAs (shRNAs). shRNAs are in turn processed into short double-stranded pieces of RNA known as short interfering RNAs (siR-NAs). Within the multi-protein RNA-induced silencing complex (RISC), one strand of a siRNA duplex binds a protein-coding mRNA transcript that bears a comple-mentary nucleotide sequence. This interaction allows a nuclease in the RISC to cleave and destroy the protein-coding mRNA, therefore silencing the expression of the gene in a relatively sequence-specific manner.
The experimental use of synthetic siRNAs and shRNA-expressing plasmids has profoundly changed the way in which loss of function experiments can be performed. Pre-viously, techniques that were either more time consuming (gene targeting), or capricious (antisense RNA), were used. Now libraries of RNAi reagents can be purchased and used to silence almost any gene at will. While siRNAs are typically used in multiwell plate-based screening, shRNAs are commonly used for pooled competitive screening approaches, often called barcode screening.
Barcode screening offers improvements in speed and scale compared to plate-based screening. In barcode screening, a large population of cells is infected or trans-fected with a pool of different shRNA vectors. Cells are then split into two groups and one group is treated dif-ferently from the other - for example, with a drug. After this selective pressure is applied, cells are harvested from both populations and integrated hairpins extracted from the genomic DNA of each population by PCR. The rela-tive quantity of each hairpin in the two populations is then compared, to identify those genes that modulate the response to the perturbation in question. For example, in the case of drug screens, hairpins that are over- or under-represented in the drug treated sample compared to the control sample could be considered as targeting
* Correspondence: [email protected]; [email protected]
The Breakthrough Breast Cancer Research Centre, The Institute of Cancer Research, 237 Fulham Road, London, SW3 6JB, UK
genes that modulate sensitivity or resistance to the drug, respectively.
Traditionally, Sanger sequencing has been used as a readout for positive selection screens. However, this approach is costly, time consuming and in general not scalable. In the case of negative selection screens, micro-array hybridization is frequently used as a readout [1,2]. This approach requires the production of custom micro-array chips for each library, has a limited dynamic range and is restricted by the varying effectiveness of individual probes. Next generation sequencing (NGS) technologies have recently emerged as a cost-effective means of gener-ating large quantities of sequence data in a short time. Using massively parallel sequencing in place of Sanger sequencing or microarray-based approaches offers several potential advantages in terms of flexibility of input library, scalability and dynamic range.
Already, a small number of laboratories have used shRNA screens coupled to NGS [1,3-5]. One critical issue that limits the wider exploitation of this technology is the absence of a freely available and simple package for the analysis of shRNA NGS data. With this in mind, we describe here detailed protocols for pooled shRNA screening coupled to NGS screen deconvolution. As part of our optimization of this technology, we have also developed a computational pipeline to analyze NGS data from shRNA screens and describe two open source ana-lysis packages, shALIGN and shRNAseq, designed to simplify barcode screen analysis. Using shRNA pools with engineered depletion, we also assess the sensitivity and reproducibility of this method. As the cost of both shRNA libraries and NGS is rapidly decreasing, these methods and analytical tools may aid the wider adoption of this powerful technology.
Results and discussion
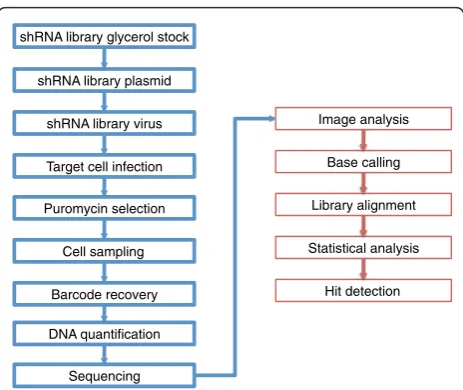
shRNA barcode screening is a lengthy procedure that required considerable optimization. Here we describe how methods were selected from principles and proce-dures established by McManus [3], Hannon, Elledge and Lowe [2,4,5] and optimized for the entire shRNA barcode screening workflow from library production to statistical analysis (Figure 1).
Bacterial culture
One factor that could affect screen performance is the variation of representation of individual hairpins within a screening pool. Since library production relies on the growth of thousands of bacterial cultures, it is inevitable that there will be some variation in growth in individual wells within a plate, and between plates within a screen-ing pool. Consequently, it is important to be systematic about the generation and pooling of bacterial cultures. First, all liquid handling was performed robotically to
ensure that most errors are systematic and can be easily traced. Second, growth temperatures and times were tightly controlled. Culture plates were stacked evenly to ensure even air circulation to all plates and wells. Hairpin plasmids were grown in small batches (ten plates) to facilitate quality control. Since recombination was a pro-blem in previous generations of shRNA libraries, the quality of plasmid DNA was checked by restriction enzyme digest following plasmid purification. Once screening pools had been constructed, the plasmid pool was sequenced on the Illumina Genome Analyzer IIx (GAIIx) to determine hairpin representation (Figure 2a), using a shRNA targeted sequencing procedure described by Zuber [4], Silva [5] and colleagues. Although it is somewhat difficult to normalize the representation of individual hairpins in large screening pools, it is impor-tant to minimize the variation within the population to reduce the chances that observed screen results can be attributed to issues in starting hairpin abundance. Although these issues can be partially mitigated at the statistical analysis stage (see below), careful library pre-paration and quality control can minimize variance in shRNA representation.
Lentiviral packaging
Packaging of hairpin plasmid into lentiviral vectors requires large numbers of packaging cells and high trans-fection efficiency to ensure faithful representation of the plasmid pool in the viral supernatant. We have success-fully employed two approaches to transfection of shRNA plasmids into packaging cell lines, calcium phosphate-and lipid-based transfection. Both methods were routi-nely used and returned viral supernatants of similar titer
shRNA library glycerol stock
shRNA library plasmid
shRNA library virus
Target cell infection
Barcode recovery
DNA quantification
Sequencing
Image analysis
Library alignment
Statistical analysis Puromycin selection
Cell sampling
Base calling
Hit detection e ca
alig
[image:2.595.306.538.86.281.2]cal a
(data not shown). cDNA generated from viral superna-tant was sequenced and compared to the plasmid DNA to ensure good representation of the library has been achieved (Figure 2a). Typically, cDNA from viral super-natant showed slightly greater variance in hairpin repre-sentation than the plasmid pool. Furthermore, hairpin representation at early time points post-viral integration demonstrated better correlation with plasmid representa-tion than with virus (Figure 2a). This suggested that the viral cDNA preparation step was a considerable source of noise and thus plasmid shRNA sequence most likely represents a better reference for starting hairpin repre-sentation than virus. This analysis also demonstrated a high concordance between technical replicates, where the same DNA library was sequenced on different GAIIx runs.
Typically, lentiviral stocks were transduced using a multiplicity of infection (MOI) of 0.7 to reduce the likeli-hood of multiple integrations per cell and the emergence of combinatorial phenotypes. Accurate determination of
viral titer in target cell lines allowed subsequent infection of screening cell lines at intended efficiencies. We tested a wide range of breast tumor cell line models and the majority infected at >60% using viral titers of 106to 107 TU/ml (Figure 2b). Those that did not infect at high effi-ciency were puromycin selected to give a final green fluorescent protein (GFP)-positive cell population of >90% (Figure 2c).
Viral transduction and cell sampling
Regardless of the design of a particular screen, the man-ner in which the viral transduction and subsequent cell culture are performed is crucial to the success of the screen. The maintenance of hairpin representation (the number of cells infected with each shRNA) and logarith-mic cell growth are of particular importance [3-5]. Throughout all shRNA barcode screens, we maintained an average representation of 1,000 cells per shRNA con-struct to maximize the potential for phenotypic effects from each shRNA being observed in the final analysis.
50% 98%
A549 Post-puromycin selection
GFP+ A549 pre-puromycin selection
GFP
-GFP
-GFP+
(a) (b)
[image:3.595.57.538.87.403.2](c)
Previous screens have shown similar levels of shRNA representation to be suitable for the detection of shRNA depletion and enrichment [3,4]. Since barcode screening is a competitive growth screen, ensuring cells are in log growth at all times during the screen is critical to mini-mize changes in representation caused by localized restriction of cell growth due to over-confluence. Conse-quently, we recommend ensuring that cells are never allowed to achieve more than 70% confluence.
After viral integration and puromycin selection were complete, cultures were divided into two or more sets, depending on the experimental design. For example, in a typical drug sensitivity/resistance screen, cultures are divided into reference (vehicle treated) and test (drug treated) sets. Alternatively, in a simple viability screen, a sample of cells can be taken and stored for analysis at each passage, to generate a viability time-course. One of the strengths of the lentiviral system is the stable inte-gration of hairpins; this allows the use of longer experi-mental time-courses than could generally be performed using siRNA screening. As a consequence, final screen results were typically assessed 2 to 3 weeks after divid-ing the cells into two arms. Every time the cultures were divided or sampled, aliquots were taken to assess the cell number (to construct growth curves) and the
percentage of GFP-positive cells (to assess the number of cells required to maintain hairpin representation). To minimize screen variability, we use the same passage cells for each screen replicate. We also maintain consis-tent batches of media, serum, viral supernatant and tis-sue culture plasticware for all screen replicates, again to minimize experimental variation.
Barcode recovery
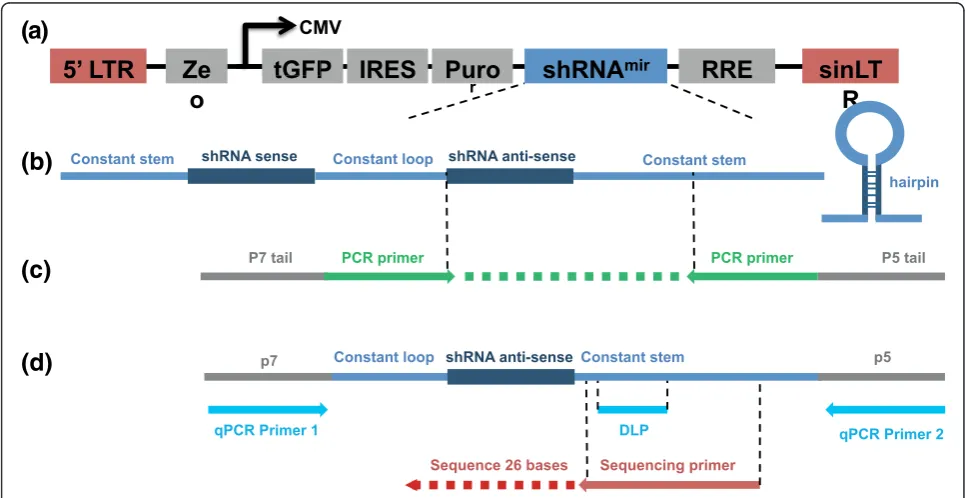
We used next generation sequencing to identify the fre-quency of each shRNA construct in screen cell popula-tions. To facilitate this we used PCR amplification of genomic DNA from screen cell populations, based upon methods previously described by McManus [3], Hannon, Elledge and Lowe [2,4,5] and Quail [6]. PCR primers complementary to constant regions found in all shRNA constructs (Figure 3) were used to amplify the shRNA target sequence that is specific to each individual shRNA construct. The PCR primers also encompassed p5 and p7 sequences that allow sequence capture and sequencing-by-synthesis on the Illumina GAIIx platform.
To enable sufficient representation of each shRNA in the screening pool, multiple PCR reactions were per-formed in parallel to generate the sequencing library from each shRNA pool. For example, to maintain a
Constant stem shRNA sense Constant loop shRNA anti-sense Constant stem
P7 tail PCR primer P5 tail
Sequencing primer Sequence 26 bases
PCR primer
5’ LTR
sinLT
R
Ze
o
tGFP IRES Puro
rtGFP
CMV
shRNA
mirRRE
(a)
(b)
(c)
(d)
p7 Constant loop shRNA anti-sense Constant stem p5DLP
hairpin
[image:4.595.56.539.404.653.2]qPCR Primer 1 qPCR Primer 2
representation of 1,000 cells per shRNA in a pool of 10,000 shRNAs at an MOI of 0.7, PCR amplification from 1 × 107 cells is required. Since a diploid human cell contains approximately 6 pg genomic DNA, we per-formed PCR amplification from 60 μg total genomic DNA using 30 parallel PCR reactions, each with 2μg of genomic DNA.
DNA quantification for next generation sequencing
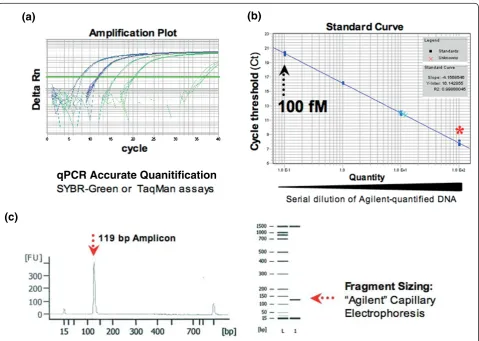
Accurate quantification of purified PCR products is required to achieve optimal cluster densities (the density of template clusters on the flowcell surface) in Illumina sequencing. Insufficient or excessive template results in poor sequencing yield with either scarce or overlapping signals. Real-time PCR has previously been used in Illu-mina sample preparation protocols to overcome the detection limits of capillary electrophoresis (typically 10 to 0.1 ng/μl) and enable standardization of cluster num-ber per tile [6]. This led us to develop two robust quan-tification assays for Illumina DNA libraries (Figure 4).
First, we utilized the complement of the Illumina adapter sequences (p5 and p7, common to all Illumina sequencing libraries) as amplification primers in an intercalating dye-based quantitative PCR (qPCR) assay (SybrGreen). This is similar to an approach applied to the quantification of 454 Roche pyro-sequencing sam-ples [7]. Second, for the quantification of DNA con-taining hairpin inserts in shRNA-derived Illumina libraries, we established a second strategy that utilized a dual-labeled probe (DLP) hydrolysis qPCR assay (Taqman). Here we designed a DLP complementary to a constant internal region in shRNA-specific Solexa PCR products. We based this approach on a reported alternative Taqman assay defined for an established Illumina sequencing application, pair-end RNA sequen-cing (RNAseq) [6]. Overall, the implementation of both of these innovative strategies allowed us to reliably quantify shRNA templates prior to massively parallel sequencing, leading to high numbers of mapped reads passing quality filters.
(a)
(b)
(c)
[image:5.595.59.539.344.685.2]qPCR Accurate Quanitification
Sequencing data analysis
Raw images from the Illumina GAIIx were processed using the GApipeline (version 1.4 or higher) and result-ing quality filtered short reads were aligned to the refer-ence shRNA library using the shALIGN script. We developed shALIGN to circumvent a number of issues associated with other open source aligners commonly used in NGS analysis. Aligners such as Bowtie [8] and BWA (Burrows-Wheeler Aligner) [9] are specifically designed to align short reads to large genome sequences, rather than short shRNA sequences. Aligning reads to the genome would cause complications in the down-stream analysis. First, using a whole genome alignment, reads could align to multiple genomic regions, which could allow the same read to be counted multiple times, or assigned at random to a particular location. Second, when using a whole genome alignment, reads with PCR or sequencing errors could align in different regions to unaltered reads, again giving a false picture of the num-ber of reads mapping to a particular target. In contrast, shALIGN aligns reads directly to the target shRNA library, aligns each read to a single library construct, and ensures that all ambiguous reads are excluded from the final analysis. Typically, >90% of short reads aligned to the reference shRNA sequence library using this method. Resulting read counts per hairpin were statistically ana-lyzed using a bespoke R-based package, shRNASeq. This analysis revealed systematic pool-specific biases in the log ratio of read counts from different screen arms at dif-ferent levels of read abundance (Figure 5a). As a
consequence, the log ratio was normalized to the average hairpin abundance using loess regression, and the nor-malized scores were re-scaled by the pool median abso-lute deviation (MAD; a robust estimator of variance) to ensure comparable distributions (Figure 5b). In some cases, we observed considerable heterogeneity in the dis-tribution of normalized scores from biological replicates from the same screen, prompting us to perform a rank normalization across replicates to ensure identical distributions.
Screen performance
We performed a number of experiments to evaluate screen performance in terms of sensitivity and reproducibility. To establish the sensitivity of the screening system, we per-formed a series of engineered depletion experiments (Addi-tional file 1). To do this, we manually altered the representation of subsets of hairpins in a single screening pool, then performed a short-term screen (long enough for viral integration, but minimizing hairpin viability effects) and examined the difference in hairpin representation between the non-manipulated reference set and the sys-tematically depleted set. These experiments demonstrated that we could detect 75% or 50% depletion in hairpin abundance with high accuracy in a single biological repli-cate (Figure 6a,b). Indeed, for the 75% depleted shRNA set, only seven depleted hairpins (0.74% of total depleted) could not be distinguished from the main population of non-manipulated hairpins at a threshold that detected no false positives (Table 1). The false negative rate was slightly
[image:6.595.61.539.469.673.2](a) (b)
increased in the 50% depleted hairpin set. Even when the shRNA constructs were depleted by 25%, half could still be distinguished from the main hairpin population with a 0% false positive rate (Table 1). The majority of false negatives in the 50% and 75% depleted hairpin groups were in hair-pins with low starting representation. Filtering of the data to remove hairpins with low representation in the reference set resulted in a reduced false negative rate associated with a 0% false positive rate (Table 1). This appears to represent a considerable improvement in sensitivity in comparison with microarray-based methods. Previous work assessing the use of custom designed microarray sets to deconvolute
(a)
(b)
(c)
(d)
0 2 4 6 8 10 12 14
20 26 33
False Positive Rate (%)
PCR Cycles
[image:7.595.59.542.89.470.2]r2=0.92
Figure 6Assessing the sensitivity and reproducibility of the screening platform. We systematically depleted subsets of hairpins by 25%, 50% or 75% within a 10k pool and compared them to a non-depleted reference set 3 days after infection of MCF7 cells (see also Additional File 1).(a)Scatter plot of log2 normalized read counts from reference and depletion sets.(b)Density plot showing the distributions of the depleted hairpin subsets: 25% depleted hairpins are plotted in red, 50% depleted in green and 75% depleted in blue. The screening methodology was capable of detecting 50% depletion in hairpin representation with high accuracy in a single experiment.(c)Scatter plot of the depletion-reference log ratio from two biological replicates, indicating a high correlation (r2 = 0.92) and thus a reproducible screening method.(d)Plot depicting the false positive rate at a fixed false negative rate of 5% in a reference depletion experiment using different numbers of PCR cycles, indicating a decrease in the false positive rate with decreased PCR cycles.
Table 1 Detection of depleted hairpins in reference depletion screens
Un-filtered Filtered Depletion Min FPR FNR Min FPR FNR
25% 0.00% 50.05% 0.00% 49.95%
50% 0.00% 4.21% 0.00% 2.89%
75% 0.00% 0.74% 0.00% 0.00%
[image:7.595.56.290.614.680.2]shRNA screens showed that even the best-performing microarrays (barcode tiling arrays) gave a mean test/refer-ence ratio of 0.78 for a 50% depletion [10], whereas our method offers a ratio of 0.54. Furthermore, barcode tiling arrays detected an 80% depletion with a test/reference ratio of 0.49 [10], whereas our NGS method gave a ratio of 0.33 for a depletion of 75%. Thus, the NGS-based approach provides a greater range of measurements and is better able to discriminate true depletions from background noise.
Although NGS performs better than microarray hybridi-zation for screen deconvolution, the barcode screening format is subject to a significant degree of stochastic noise regardless of the hairpin frequency detection method, and biological replication of screens is almost certainly required to overcome this. We performed two biological replicates of the reference depletion screen. There was a good correlation between log normalized read counts from replicate screens (reference arm r2 = 0.89). Similarly, a high correlation of depletion-reference log ratios was observed between biological replicates (r2 = 0.92; Figure 6c), suggesting that screens are highly reproducible. As expected, higher levels of noise were observed between biological replicates than between technical replicates.
Using the reference-depletion experimental frame-work, we also titrated the number of PCR cycles required to give faithful representation of the starting material. This demonstrated that PCR amplification acted as a source of noise in the experiment and that reducing the number of PCR cycles resulted in a decreased false positive rate at a set false negative rate of 5% (Figure 6d). Consequently, we opted to use 26 PCR cycles as this generated a visible band on the gel but still resulted in a relatively low error rate.
We also made use of the reference depletion dataset to assess the minimum amount of sequencing reads required to detect a reduction in shRNA representation. Many of the more novel sequencing platforms, such as the Illumina MiSEQ, are able to generate five million reads in a single run, offering the potential to use these cheaper platforms for shRNA screen deconvolution. We sampled reads from one reference depletion experiment to generate two additional datasets that contained either approximately 5 million reads or approximately 2.5 mil-lion reads, rather than the 10 milmil-lion reads used in the previous analysis. This analysis revealed that a 50% reduction in shRNA representation could be detected with high sensitivity and specificity even in 2.5 million reads (Table 2; Additional file 2).
To test sensitivity in a genuine screen setting using dif-ferent screening pool sizes, we performed a series of screens for viability in MCF7 cells. To assess screen per-formance, we used a number of genes where previous
observations had shown that siRNA silencing inhibited MCF7 cells as measured using a viability assay based on cellular ATP levels [11]. We validated the viability effects of a set of GIPZ library shRNAs targeting these genes using single hairpin GFP competition studies. We also validated a number of negative control non-targeting shRNAs using this method. This revealed a total of six shRNAs, which caused a >50% depletion in GFP-positive cells over 2 weeks, along with 11 non-targeting hairpins that showed no viability phenotype in the same period (Additional file 3).
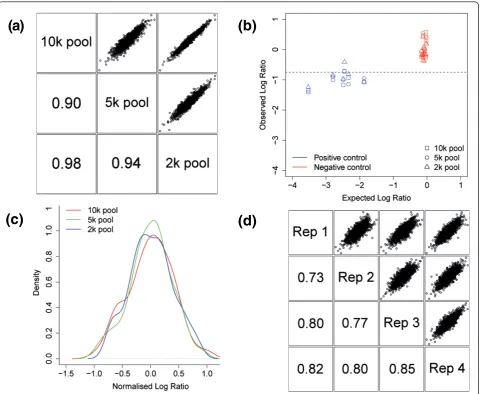
[image:8.595.305.539.112.233.2]Increased pool size leads to fewer reads per shRNA, and reduces screen sensitivity and potentially reliability. Therefore, we decided to compare the performance of pools containing 10,000, 5,000 or 2,000 shRNA con-structs in a 14 day cell viability screen. To facilitate screen comparison, the 2,000 shRNA pool was a subset of the 5,000 shRNA pool and the 5,000 shRNA pool was a subset of the 10,000 shRNA pool, and the set of vali-dated positive and negative control shRNAs described above were added to each pool. When the 2,000 shRNAs represented in all three pools were examined, there was a high correlation between different pool sizes (Figure 7a). Furthermore, five out of six positive control hairpins were identified as hits in all three screening pool sizes (Figure 7b). There was a clear distinction between vali-dated positive and negative control scores in all screens (Figure 7b). However, there was a larger than expected variation in the negative control score, with validated controls changing representation by up to 25%. This effect was also seen in the distribution of 101 non-target-ing shRNAs within the 10,000 shRNA pool (Figure 7c). Furthermore, the depletion observed in positive control hairpins was smaller than expected based on single hair-pin GFP competition assays (Figure 7b).
Table 2 Comparison of reference-depletion screens at different read depths
Total reads Depletion Min FPR FNR
10 million 25% 0.00% 49.95%
10 million 50% 0.00% 2.89%
10 million 75% 0.00% 0.00%
5 million 25% 3.41% 42.35%
5 million 50% 0.02% 1.28%
5 million 75% 0.00% 0.00%
2.5 million 25% 9.65% 49.95%
2.5 million 50% 1.70% 1.07%
2.5 million 75% 0.00% 0.00%
These results suggested that the scoring system per-forms consistently regardless of pool size and the screen is as sensitive in a 10,000 shRNA pool as in a 2,000 shRNA pool. Therefore, to maximize throughput and minimize expense, we decided to use 10,000 shRNA pools for screening. Since the genome-wide library encompasses approximately 60,000 reagents, we reasoned that a pool size of approximately 10,000 shRNAs would be an ideal choice for this library. This would enable a genome-wide screen to be run on a single eight-lane Illu-mina flow-cell along with appropriate sequencing con-trols. Furthermore, a 10,000 shRNA pool size would
require approximately 15,000,000 cells to be maintained in tissue culture over the course of the experiment, assuming an MOI of 0.7 and a representation of 1,000 cells per hairpin. This pool size enabled us to maintain the simplicity and reproducibility of the associated tissue culture work, which might become cumbersome in larger pool sizes.
To establish the reproducibility of the screening metho-dology, we repeated the screen for cell viability in MCF7 cells in a total of four biological replicates. The replicates showed high correlation despite being performed by differ-ent researchers at differdiffer-ent times, suggesting that the
(c)
(b)
(a)
[image:9.595.59.542.88.482.2](d)
screening method is robust (Figure 7d). Furthermore, >98% of library hairpins were identified in all screen repli-cates, compared to 49% of half-hairpin probes and 82% of barcode tiling probes having intensity above background in microarray-based approaches [10]. This suggests a greatly improved sensitivity for the NGS profiling approach.
Conclusions
Here we describe detailed and optimized methods for high-throughput shRNA screening using NGS. Using massively parallel sequencing in place of microarray hybridization for deconvolution of shRNA barcode screen results offers several advantages. Firstly, we demonstrate that NGS profiling offers greater sensitivity, enabling more hairpins to be detected above background. Secondly, we show that NGS profiling has a better dynamic range when compared to literature examples of microarray-based approaches, since sequencing cannot be easily saturated in the same way that hybridization can, thus enabling a greater separation of true effects from background noise. Thirdly, sequencing of tags is both scalable and flexible, enabling new hairpins to be incorporated into the work schema without having to print a new batch of custom microarrays. Finally, our computational pipeline offers the ability to identify sequences where bases are not called due to sequencing errors, or mutated during PCR, since short reads can be matched inexactly to the reference library of shRNA barcode sequences.
We do note that one of the major limitations of any shRNA library is the efficacy of gene silencing, an issue we have not addressed here. However, the ability to rapidly assess silencing capacity and thus develop algo-rithms that would predict effective gene knockdown is improving [12]. In addition, an increase in the redun-dancy of shRNA libraries (the number of different shRNA constructs per gene) will also improve the gen-eral effectiveness of pooled RNAi screens. Finally, improvements in sequencing technology will undoubt-edly increase read number per lane; these developments will thus enable the use of larger shRNA pool sizes that could accommodate libraries with higher levels of redundancy. Nevertheless, even with the existing com-mercial shRNA libraries and also the ever-increasing availability of cost-effective NGS, the methods we describe here should enable the wider applicability of this powerful technology.
Materials and methods shRNA library
Although the following methods are suitable for most viral shRNA libraries, the work described here used the
Thermo Scientific Open Biosystems GIPZ Lentiviral human shRNAmir library (version 2). These methods could be used for other shRNA libraries, such as the RNAi consortium library [13], with altered PCR and sequencing primers. The GIPZ Lentiviral human shRNAmir library we used encompasses 61,416 distinct hairpin constructs targeting 15,739 human protein coding genes (based upon the Ensembl 56 build). In the GIPZ vector the 19-nucleo-tide siRNA sense sequence is inserted into a human mir-30 backbone [14]. shRNA sequences are designed to have destabilized 5’ends in the antisense strand to encourage stand-specific incorporation into the RISC [14]. The vector backbone includes a GFP-coding sequence that is tran-scribed as part of a bicistronic transcript with the shRNA sequence allowing the visualization of shRNAmir expres-sing cells, and a puromycin resistance marker for selecting infected cells.
Bacterial culture
LB media (1 ml) containing 50 μg/ml ampicillin was added to 96-deep-well microplates using a multidrop (Thermo Fisher, Waltham, MA, USA), and 1μl of each bacterial inoculant (extracted from fully thawed 96-well glycerol stocks) was seeded per well using a Beckman FX robot. Culture plates were stacked evenly in shaking incubators (200 rpm) ensuring even air circulation. Following 16 hours of incubation at 37°C, cultures from a single batch of plates were pooled and plasmid DNA was isolated using a Plasmid Maxi kit (Qiagen, Crawley, UK) and normalized to a standard concentration (0.5 μg/ml). Plasmid DNA pools were combined in equal concentrations to create screening pools of differ-ent complexities.
Lentiviral packaging
Viral transduction and cell sampling
Cells were infected with viral pools using a MOI of 0.7 and media replenished after 16 hours. Seventy-two hours post-infection, when viral integration was presumed complete, cells were exposed to 1 mg/l puromycin for 2 days to select for cells with viral integration. Following puromycin selection, cultures were divided into two or more replica cultures, and continuously cultured for 2 to 3 weeks, dividing cultures regularly to ensure continued logarithmic growth and to maintain hairpin representa-tion at approximately 1,000 cells per shRNA [4,5]. Cells were harvested in suspension and either stored frozen at -20°C or processed immediately as described below.
Barcode recovery
Genomic DNA extraction and purification from cultured cells was carried out using a Gentra Puregene kit (Qiagen). shRNA sequences integrated into genomic DNA were recovered by PCR amplification using the following primers and a procedure based upon that described by Hannon, Elledge and Lowe [4,5]: p5+mir30, 5’ -AATGATACGGC-GACCACCGACTAAAGTAGCCCCTTGAATTC-3’; p7+ Loop, 5’-CAAGCAGAAGACGGCATACGATAGTGAAG CCACAGATGTA-3’. PCR was performed on PTC-225 3DNA Engine Tetrads (Bio-Rad, Hemel Hempstead, UK) using Amplitaq Gold polymerase and 20 to 33 cycles of denaturation (95°C, 30 s), annealing (52°C, 45 s) and exten-sion (72°C, 60 s). In general, 2μg genomic DNA was used per 100μl PCR reaction and 48 PCR reactions were per-formed per 10,000 shRNA pool. PCR products from multi-ple parallel reactions were subsequently pooled, concentrated and purified using a QIAquick gel extraction kit (Qiagen).
Quantitative PCR DNA quantification
For absolute DNA quantification, real-time PCR was per-formed using two alternative strategies: SybrGreen and TaqMan. Both approaches used the appropriate universal 2X PCR mix from Applied Biosystems and the following oligonucleotides at 250 nM each: P5, AATGATACGGC-GACCACCGA (20-mer); and P7, CAAGCAGAA-GACGGCATACGA (21-mer). For TaqMan only, 250 nM of a 21-mer DLP was included (CCCTTGAATTCC-GAGGCAGTA), with 5’reporter (6FAM fluorescein) and 3’quencher (tetramethylrhodamine (TAMRA)) as detec-tors, and ROX passive reference dye. Forty-cycle measure-ments of triplicate 25 μl reactions containing 10% template sample at a theoretical concentration of 10 pM (as defined by Agilent Bioanalyzer) were carried out in an ABI 7900 instrument. Solexa library concentrations were then inferred by comparing measurements to the standard curve using the Sequence Detection System (SDS) v2.2.1 software. To generate the standard curve, a 10-fold
dilution series of a standard 100 nM sample (7.7 ng/μl for 117 bp) was used (final range 0.1 to 100 pM).
Barcode sequencing
Following quantification, denatured shRNA-seq libraries (3 pM in NaOH) were pumped through eight-lane flowcell channels using the cluster station of an Illumina GAIIx sequencing platform. Bridge PCR was executed using the manufacturer’s protocol (Amplification-linearization-blocking and multiple primer hybridization version 3). The sequencing primer (TAGCCCCTTGAATTCCGAGG-CAGTAGGCA) was designed to sequence from two bases upstream of the 19 bp shRNA sense sequence. Twenty-six cycles of sequencing-by-synthesis (single read) were per-formed on an Illumina GAIIx according to the manufac-turer’s protocol (version 4).
Image analysis and base calling
Raw image data were analyzed using GA pipeline v1.4. PhiX was run as a control to ensure correct phasing. Base calling was performed by the Bustard package using the Chastity filter with a threshold of 0.6. The Chastity filter was applied to bases 3 to 21 of the 26 bases sequenced (the shRNA sense sequence) only. A maximum of two uncalled bases were allowed in these 19 bases. FASTQ files from this study are available at the ROCK web page [16] and at the European Nucleo-tide Archive [17].
shRNA library alignment
program outputs the total number of short reads matching to each library hairpin in each lane. We routinely load screen results into the ROCK breast cancer functional genomics database [18], where screen reagents are fully annotated to the latest genome build. ROCK provides a mechanism for the sharing and publication of raw and processed RNAi screen results. Source code and linux bin-aries for the shALIGN program are available from the ROCK web page [16].
Statistical analysis
To facilitate statistical analysis of screen results, we have developed a novel R package, shRNAseq. This package is based on the NChannelSet class in Bioconductor [19], originally designed to handle microarray gene expression data, and serves as a single user-friendly package encompassing all of the steps required. shRNAseq reads in matrices of short read counts per shRNA construct, generated by shALIGN, along with annotations of shRNA constructs and sequenced samples.
The package is designed to compare a pair of related screen conditions. For example, in a viability screen one would compare the hairpin representation in the starting population (which could be from the plasmid, virus or an early time point post-viral integration) to the hairpin representation at the end of the screen (for example, after 2 weeks). Alternatively, for a drug screen one would com-pare the drug and vehicle treated arms at the same time point. The package can analyze multiple screen replicates simultaneously. Each screen condition is loaded into the package separately, and annotated using files describing the shRNA constructs and the sample treatments. After data loading, read counts per hairpin are log2 transformed and then the ratio of the two screen conditions is calcu-lated. This log ratio is normalized using a loess fit to the log mean read count for each screening pool. The distri-bution of normalized scores per pool is then rescaled by dividing by the pool MAD. If appropriate, screen biological replicates can be quantile (rank) normalized to ensure identical distributions, and scores can be summarized across replicates using a variety of methods (for example, median, regularizedt-test). Finally, the package plots screen distributions before and after normalization and reports a table of normalized scores. The R package is accompanied by a detailed vignette describing the metho-dology and usage at the ROCK web page [16].
Hit detection was performed using three different meth-ods. In the first method, replicate scores for each hairpin were summarized using the median and a hit threshold estimated from a quantile-quantile plot, identifying hairpin scores that significantly differed from the normal distribu-tion. Second, the Gene Set Analysis R package [20] was used to look for enrichment or depletion of sets of hair-pins targeting the same gene. This approach makes use of
the hairpin redundancy within the library and works best in libraries containing multiple hairpins per gene. Finally, the RIGER algorithm [21] in the GENE-E java package [22] was also used to look for enrichment of shRNAs tar-geting the same gene. Here we used the log fold change metric and weighted average method. Source code and documentation for the shRNAseq R-package are available from the ROCK web page [16].
Supplementary methods
Full methods, including a detailed screening manual, are provided in Additional file 4.
Additional material
Additional file 1: Engineered depletion of shRNAs. To establish the sensitivity of the screening system, we performed a series of engineered depletion experiments. We manually altered the representation of constructs in a 10,000 shRNA screening pool so that approximately 1,000 hairpins were depleted by 75%, approximately 1,000 depleted by 50% and approximately 1,000 depleted by 25%.
Additional file 2: Detection of hairpin depletion at reduced read counts. Reads were sampled at random from an engineered depletion experiment involving approximately 10 million reads to give datasets of either approximately 5 million or approximately 2.5 million reads in total. shRNA depletion was estimated from these new datasets to show that depletion of 50% could be observed in datasets containing
approximately 2.5 million reads.
Additional file 3: Positive and negative controls for the MCF7 viability screen were established using a single hairpin GFP-competition assay. The bar chart indicates the proportion of GFP positive cells remaining after 2 weeks of culture. The bar represents the average from three biological replicates. The error bars indicate the standard deviation.
Additional file 4: Detailed shRNA screening protocols. This Word document describes in detail all of the steps of the shRNA screening protocol from library generation to massively parallel sequencing.
Abbreviations
DLP: dual-labeled probe; GAIIx: Genome Analyzer IIx; GFP: green fluorescent protein; MAD: median absolute deviation; miRNA: microRNA; MOI:
multiplicity of infection; NGS: next generation sequencing; qPCR: quantitative PCR; RISC: RNA-induced silencing complex; RNAi: RNA interference; RNASeq: RNA sequencing; shRNA: short hairpin RNA; shRNAmir: shRNA encoded within a miRNA; siRNA: short interfering RNA; TU: transducing unit.
Acknowledgements
This work was supported by Breakthrough Breast Cancer, American Association of Cancer Research Stand Up to Cancer, The Breast Cancer Research Foundation and Cancer Research UK. We thank Drs. Greg Hannon, Joannas Zuber and Scott Lowe for sharing methods prior to publication. We acknowledge NHS funding to the Royal Marsden Hospital National Institute for Health Research Biomedical Research Centre.
Authors’contributions
Competing interests
The authors declare that they have no competing interests.
Received: 13 June 2011 Revised: 25 September 2011 Accepted: 21 October 2011 Published: 21 October 2011
References
1. Burgess DJ, Doles J, Zender L, Xue W, Ma B, McCombie WR, Hannon GJ, Lowe SW, Hemann MT:Topoisomerase levels determine chemotherapy response in vitro and in vivo.Proc Natl Acad Sci USA2008,105:9053-9058. 2. Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, Zhao Z,
Smogorzewska A, Sowa ME, Ang XL, Westbrook TF, Liang AC, Chang K, Hackett JA, Harper JW, Hannon GJ, Elledge SJ:Cancer proliferation gene discovery through functional genomics.Science2008,319:620-624. 3. Bassik MC, Lebbink RJ, Churchman LS, Ingolia NT, Patena W, LeProust EM,
Schuldiner M, Weissman JS, McManus MT:Rapid creation and quantitative monitoring of high coverage shRNA libraries.Nat Methods2009,
6:443-445.
4. Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, Lowe SW:
Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi.Nat Biotechnol2011,29:79-83.
5. Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, Elledge SJ, Hannon GJ, Chang K:Profiling essential genes in human mammary cells by multiplex RNAi screening.Science2008,319:617-620.
6. Quail MA, Kozarewa I, Smith F, Scally A, Stephens PJ, Durbin R, Swerdlow H, Turner DJ:A large genome center’s improvements to the Illumina sequencing system.Nat Methods2008,5:1005-1010.
7. Meyer M, Briggs AW, Maricic T, Hober B, Hoffner B, Krause J, Weihmann A, Paabo S, Hofreiter M:From micrograms to picograms: quantitative PCR reduces the material demands of high-throughput sequencing.Nucleic Acids Res2008,36:e5.
8. Langmead B, Trapnell C, Pop M, Salzberg SL:Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.
Genome Biol2009,10:R25.
9. Li H, Durbin R:Fast and accurate short read alignment with Burrows-Wheeler transform.Bioinformatics2009,25:1754-1760.
10. Boettcher M, Fredebohm J, Moghaddas Gholami A, Hachmo Y, Dotan I, Canaani D, Hoheisel JD:Decoding pooled RNAi screens by means of barcode tiling arrays.BMC Genomics2010,11:7.
11. Iorns E, Turner NC, Elliott R, Syed N, Garrone O, Gasco M, Tutt AN, Crook T, Lord CJ, Ashworth A:Identification of CDK10 as an important
determinant of resistance to endocrine therapy for breast cancer.Cancer Cell2008,13:91-104.
12. Fellmann C, Zuber J, McJunkin K, Chang K, Malone CD, Dickins RA, Xu Q, Hengartner MO, Elledge SJ, Hannon GJ, Lowe SW:Functional identification of optimized RNAi triggers using a massively parallel sensor assay.Mol Cell2011,41:733-746.
13. Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM:Genome-scale loss-of-function screening with a lentiviral RNAi library.Nat Methods
2006,3:715-719.
14. Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, Siolas D, Hu G, Paddison PJ, Schlabach MR, Sheth N, Bradshaw J, Burchard J, Kulkarni A, Cavet G, Sachidanandam R, McCombie WR, Cleary MA, Elledge SJ, Hannon GJ:Second-generation shRNA libraries covering the mouse and human genomes.Nat Genet2005,37:1281-1288.
15. Klages N, Zufferey R, Trono D:A stable system for the high-titer production of multiply attenuated lentiviral vectors.Mol Ther2000,
2:170-176.
16. ROCK - Breast Cancer Functional Genomics.[http://rock.icr.ac.uk/software/ shrnaseq.jsp].
17. The European Nucleotide Archive, Accession number ERP000908.[http:// www.ebi.ac.uk/ena/data/view/ERP000908].
18. Sims D, Bursteinas B, Gao Q, Jain E, MacKay A, Mitsopoulos C, Zvelebil M:
ROCK: a breast cancer functional genomics resource.Breast Cancer Res Treat2010,124:567-572.
19. Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J:Bioconductor: open software development for computational biology and bioinformatics.Genome Biol2004,5:R80. 20. Gene Set Analysis.[http://www-stat.stanford.edu/~tibs/GSA/].
21. Luo B, Cheung HW, Subramanian A, Sharifnia T, Okamoto M, Yang X, Hinkle G, Boehm JS, Beroukhim R, Weir BA, Mermel C, Barbie DA, Awad T, Zhou X, Nguyen T, Piqani B, Li C, Golub TR, Meyerson M, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE:Highly parallel identification of essential genes in cancer cells.Proc Natl Acad Sci USA2008,
105:20380-20385.
22. GENE-E Java Package.[http://www.broadinstitute.org/cancer/software/ GENE-E/].
doi:10.1186/gb-2011-12-10-r104
Cite this article as:Simset al.:High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing.Genome Biology201112:R104.
Submit your next manuscript to BioMed Central and take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution