0095-1137/09/$08.00⫹0 doi:10.1128/JCM.01607-08
Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Utility of Multilocus Variable-Number Tandem-Repeat Analysis as a
Molecular Tool for Phylogenetic Analysis of
Shigella sonnei
䌤
Chien-Shun Chiou,
1,2* Haruo Watanabe,
3You-Wun Wang,
1Wan-Ling Wang,
1Jun Terajima,
3Kwai-Lin Thong,
4Dac Cam Phung,
5and Sheng Kai Tung
1Central Region Laboratory, Center for Research and Diagnostics, Centers for Disease Control, Taichung City 40855, Taiwan1; Institute of Medicine, Chung Shan Medical University, Taichung City 40201, Taiwan2; National Institute of
Infectious Diseases, Tokyo, Japan3; University of Malaya, 50603 Kuala Lumpur, Malaysia4; and Division of Enteric Infections, National Institute of Hygiene and Epidemiology, Vietnam5
Received 19 August 2008/Returned for modification 5 December 2008/Accepted 11 February 2009
A panel of 916 isolates, including 703 closely related IST1 isolates, were characterized by inter-IS1spacer typing (IST), pulsed-field gel electrophoresis (PFGE), and multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) to evaluate the utility of MLVA as a molecular tool for the phylogenetic analysis ofShigella sonnei. The global phylogenetic patterns determined by IST, PFGE, and MLVA were concordant. MLVA was carried out using 26 VNTR loci with a range of degrees of variability. MLVA data for the 703 IST1 isolates revealed that diversification among the closely related isolates was attributed mainly to four highly variable loci. The phylogenetic pattern for the closely related isolates determined using MLVA profiles of 8 highly variable loci was in agreement with that determined using the 26-locus profiles. A clustering analysis using the profiles of 18 loci with limited variability established clear phylogenetic relationships among IST clonal groups. Accordingly, MLVA is a useful tool for the phylogenetic analysis ofS. sonnei. Combined VNTR loci with higher variability are useful markers for resolving closely related isolates, whereas combined loci with lower vari-ability are suitable for establishing clear phylogenetic relationships between strains or clones that have evolved over a longer timescale.
Shigella sonneiis one of the causative agents of shigellosis. Unlike the other threeShigellaspecies,S. dysenteriae,S. flex-neri, andS. boydii, which are prevalent in developing countries,
S. sonneiis predominant in industrialized countries (6) and is one of the major causes of travel-associated diarrheal disease (3). Transmission ofS. sonneistrains between countries occurs frequently via international travel (7, 14).
The analysis of bacterial isolates by various genotyping meth-ods provides useful information for establishing the genetic relat-edness among isolates for the purposes of epidemiological in-vestigation and phylogenetic study. Among these genotyping methods, pulsed-field gel electrophoresis (PFGE) has been proven to be a powerful tool for discriminatingShigellastrains and has become a standardized method for an international mo-lecular-subtyping network for food-borne-disease surveillance (13). However, PFGE is, at times, too discriminatory for investi-gating clonal relationships among Shigella strains that have evolved over years or decades. In order to study anS. sonnei
epidemic, we previously developed an inter-IS1 spacer typing (IST) method for subtyping ofS. sonnei strains (1). IST is less discriminative than PFGE forS. sonnei, but it is more suitable than PFGE for investigating the clonal relationships amongS. sonneistrains in circulation over a short timescale (15).
Multilocus sequence typing (MLST) is a sequence-based typing method widely adopted for the phylogenetic study of a number of bacterial pathogens, as listed on the website http:
//www.mlst.net/. The MLST method has been used for phylogenetic analysis of Shigella spp. with the protocol developed forEscherichia coli (16). This method reveals the closest phylogenetic relationship between Shigella and the enteroinvasiveE. colistrains. However, MLST is not sufficiently discriminative;Shigellastrains with different serotypes, or even strains from different Shigella species, may share common sequence types (ST) (2, 16). To date, there is no appropriate molecular tool that is sufficiently discriminative to resolve closely relatedS. sonneistrains and that can also provide useful information for establishing phylogenetic relationships among strains over different evolutionary scales. In a previous study, we developed a multilocus variable-number tandem-repeat (VNTR) analysis (MLVA) method for fine typing ofS. sonnei
isolates. A total of 26 VNTR loci were identified, and MLVA typing based on the 8 most variable loci exhibited a higher discriminatory power than PFGE in distinguishing closely related isolates for outbreak investigation (8). Since the VNTR loci have a range of degrees of variability, they may also be applicable to establishing phylogenetic relationships among strains that have evolved over different timescales. In this study, we characterized 916S. sonnei isolates, including 703 closely related IST1 isolates, by using IST, PFGE, and MLVA methods to evaluate the utility of MLVA for establishing phylogenetic relationships among S. sonneistrains that have evolved over different timescales.
MATERIALS AND METHODS
Bacterial strains.Shigellaisolates were collected from hospitals or isolated in the laboratories of the Centers for Disease Control, Taiwan, from 1996 to 2004.
A total of 916S. sonneiisolates were available for IST, PFGE, and MLVA
genotyping.Salmonella enterica subsp.enterica serotype Braenderup H9812,
* Corresponding author. Mailing address: Central Region Labora-tory, Center for Research and Diagnostics, Centers for Disease Con-trol, Taichung City 40855, Taiwan. Phone: 4-24750452. Fax: 886-4-24750452. E-mail: [email protected].
䌤Published ahead of print on 18 February 2009.
1149
on May 16, 2020 by guest
http://jcm.asm.org/
kindly provided by B. Swaminathan of the Centers for Disease Control and Prevention, Atlanta, GA, was used as the PFGE size maker strain (5).
IST.IST was performed as previously described (1). The gel image was
ana-lyzed using BioNumerics, version 4.5 (Applied Maths, Kortrijik, Belgium). Only those DNA fragments with sizes within the appropriate size markers (0.1 to 3 kb) were used for comparison. A dendrogram constructed using IST patterns was generated by the unweighted-pair group method with arithmetic mean (UPGMA) using the Dice-predicted similarity value of two patterns.
PFGE.The PulseNet PFGE protocol forS. sonneiand other enterobacteria was used for PFGE analysis (11), except that 5 U of NotI was used instead of XbaI for the restriction digestion. PFGE images were analyzed using the finger-print analysis software BioNumerics, version 4.5 (Applied Maths). A PFGE pattern with one or more DNA bands different from the others was taken to be a unique PFGE pattern. A dendrogram constructed using the NotI-digested PFGE patterns was generated by the UPGMA algorithm using the Dice-pre-dicted similarity value of two patterns.
MLVA.MLVA was performed as previously described (8). Twenty-six VNTR loci were characterized for every isolate studied. The number of repeat units for each locus was saved as “character type” data in BioNumerics software, version 4.5 (Applied Maths), and then subjected to cluster analysis using a minimum
spanning tree (MST) algorithm. Nei’s diversity index was calculated as 1⫺
⌺(allele frequency)2for evaluating the allele diversity of each VNTR locus.
RESULTS
Phylogenetic patterns for the 916 isolates determined using IST.A total of 28 IST genotypes were identified among the 916
S. sonnei isolates collected from 1996 to 2004 in Taiwan.
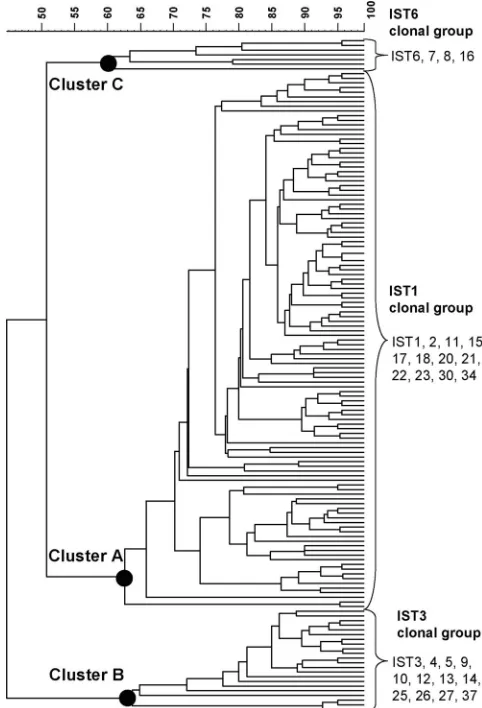
Among these isolates, 24 were recovered from travelers who had acquired infections in China, Cambodia, India, Indonesia, Thailand, and Vietnam. The IST1 genotype, identified in 710 isolates, was the most prevalent; strains with this genotype caused a number of outbreaks between 2000 and 2003 in Tai-wan. The first IST1 strain identified in Taiwan was recovered from a patient who acquired the infection in India in April 2000. A dendrogram constructed with the 28 IST patterns using the UPGMA algorithm presented three distinct clusters, designated the IST1, IST3, and IST6 clonal groups (Fig. 1). The clonal groups comprised 12, 12, and 4 IST types, respec-tively. All of the imported isolates belonged to the IST1 clonal group.
[image:2.585.298.539.70.424.2]Phylogenetic patterns for the 916 isolates determined using PFGE.PFGE performed with NotI identified 144 PFGE ge-notypes among the 916 isolates. A dendrogram constructed using the 144 PFGE patterns presented three distinct large clusters, A, B, and C (Fig. 2). PFGE cluster A consisted of all of the isolates belonging to the IST1 clonal group, whereas FIG. 1. Dendrogram constructed using the IST patterns found in
the 916 isolates, and associated numbers of isolates belonging to the IST genotypes. The dendrogram was generated by UPGMA, using the Dice-predicted similarity value between two patterns. The statistics program was provided by BioNumerics, version 4.5, with settings of 1%
optimization and 0.95% tolerance. FIG. 2. Dendrogram constructed using PFGE-NotI patterns. The dendrogram was generated by UPGMA, using the Dice-predicted sim-ilarity value of two patterns. The statistics program was provided by BioNumerics, version 4.5, with settings of 1.0% optimization and 0.7% tolerance.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:2.585.48.291.74.386.2]clusters B and C consisted of all of the isolates belonging to the IST3 and IST6 clonal groups, respectively.
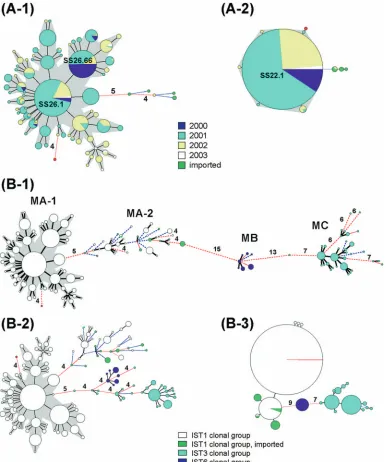
Phylogenetic patterns for the 710 IST1 isolates determined using MLVA.Of the 710 IST1 isolates, 6 were obtained from patients who acquired infections in China, India, Indonesia, and Vietnam. A total of 78 MLVA26 genotypes, which were defined by the 26-locus profiles, were identified for the IST1 isolates. A relatively polymorphic phylogenetic tree was
[image:3.585.100.488.68.530.2]con-structed with the MLVA profiles using the MST algorithm. All but one of the isolates originating in Taiwan were situated in a large, tight cluster (Fig. 3A-1). The 703 closely related isolates in the cluster were considered to belong to a newly emerged (SS26.66) clone. The first IST1 strain belonging to the SS26.66 clone emerged in October 2000 in Taiwan; since this strain was of the SS26.66 MLVA type, it was considered to be the founder of the clone. However, SS26.1, not SS26.66, was the most FIG. 3. Phylogenetic trees built with various combinations of VNTR profiles using the MST algorithm for the 710 IST1 isolates (A) and for all 916 isolates (B). (A-1) Phylogenetic tree built with 26-locus profiles. (A-2) Phylogenetic tree built with 22-locus profiles, excluding the four most variable loci, SS3, SS6, SS9, and SS11, from the 26-locus panel. (B-1) Phylogenetic tree built with 26-locus profiles. (B-2) Phylogenetic tree built with the eight most diverse locus profiles (SS1, SS3, SS6, SS9, SS10, SS11, SS12, and SS23). (B-3) Phylogenetic tree built with the 18 least diverse locus profiles, excluding the eight most diverse loci from the 26-locus panel. A distance of one locus between two MLVA types is indicated by a thick line, a distance of two loci by a thin red line, and a distance of three loci by a blue broken line. Distances of four loci or greater are marked by a red broken line, and the numbers of loci different are notated. The area of the circle is proportional to the number of isolates belonging to the MLVA type. The SS26.66 clone is marked with a gray shadow. The Indian strain is shown in red.
on May 16, 2020 by guest
http://jcm.asm.org/
prevalent type and had more single-locus variants (SLVs) (Fig. 3A-1). The average distance from the founder (SS26.66) in-creased over time from 0.23 loci for the isolates collected in 2000 to 2.1 loci for those collected in 2003 (Table 1). The remaining seven isolates, six of which were imported, had a distance of at least five loci from the SS26.66 clone. The Indian strain had a distance of four loci from the clone (Fig. 3A-1) and differed from the founder at six loci. The Indian strain was the first IST1 strain that emerged in Taiwan that shared indistin-guishable PFGE-NotI and PFGE-XbaI patterns with the ma-jority of the isolates within the SS26.66 clone.
Since the 703 IST1 isolates belonged to a very new clone,
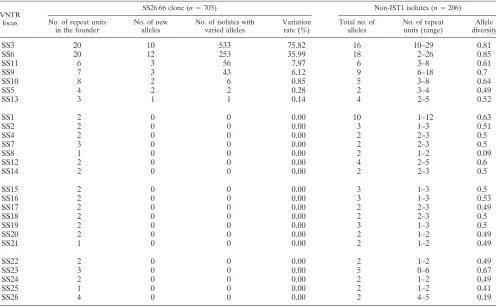
they were an excellent collection of isolates to use for evalu-ating the variability of the 26 VNTR loci over a short evolu-tionary timescale (3.25 years). Variation among the isolates was observed at seven loci. The loci SS3, SS6, SS11, and SS9 were highly variable, with variation rates among the isolates of 75.82%, 35.99%, 7.97%, and 6.12%, respectively (Table 2).
SS3, SS6, SS9, and SS11 were the four most variable loci in the SS26.66 clone. When these four loci were excluded from the 26-locus panel, the phylogenetic pattern for the SS26.66 clone was relatively monomorphic (Fig. 3A-2). The four highly variable loci were the major contributors to the diversification among the closely related isolates. Among the MLVA geno-types defined by the 22-locus profiles, SS22.1 was the predom-inant genotype, as it accounted for 97% of the isolates. All of the isolates collected in the year 2000 belonged to the SS22.1 genotype, which had five SLVs. All of the SLVs of SS22.1 emerged in 2001 and 2002. The Indian strain was a double-locus variant of SS22.1. The rest of the imported IST1 strains had a distance of at least three loci from SS22.1.
[image:4.585.44.284.90.180.2]Phylogenetic patterns for the 916 isolates determined by MLVA.A total of 146 MLVA26 genotypes were identified for the 916 isolates, which consisted of 710 IST1 and 206 non-IST1 isolates. The phylogenetic tree constructed using the MST algorithm with the 26-locus profiles presented three distinct clusters, MA, MB, and MC (Fig. 3B-1). Each cluster included genotypes sharing 19 or more of the 26 loci. The global phy-logenetic pattern for the isolates established with the MLVA data was concordant with that determined by IST and that TABLE 1. Differences from the founder genotype for isolates of
the SS26.66 clone, distributed by year recovered
No. of loci different
No. of isolates recovered in yr:
2000 2001 2002 2003
0 52 42 11
1 11 272 58 5
2 2 126 82
3 9 27 4
4 1 1
Total 65 449 179 10
aThe average numbers of loci different from the founder genotype were as
follows: for isolates recovered in 2000, 0.23; for isolates recovered in 2001, 1.23; for isolates recovered in 2002, 1.72; and for isolates recovered in 2003, 2.1. These
values were calculated as follows: distance⫽ ⌺关(no. of loci different)⫻(no. of
isolates)兴/total no. of isolates recovered in the year.
TABLE 2. Characteristics of VNTR loci for the 703 IST1 isolates of the SS26.66 clone and the 206 non-IST1 isolates
VNTR locus
SS26.66 clone (n⫽703) Non-IST1 isolates (n⫽206)
No. of repeat units in the founder
No. of new alleles
No. of isolates with varied alleles
Variation rate (%)
Total no. of alleles
No. of repeat units (range)
Allele diversity
SS3 20 10 533 75.82 16 10–29 0.81
SS6 20 12 253 35.99 18 2–26 0.85
SS11 6 3 56 7.97 6 3–8 0.61
SS9 7 3 43 6.12 9 6–18 0.7
SS10 8 2 6 0.85 5 3–8 0.64
SS5 4 2 2 0.28 2 3–4 0.49
SS13 3 1 1 0.14 4 2–5 0.52
SS1 2 0 0 0.00 10 1–12 0.63
SS2 2 0 0 0.00 3 1–3 0.51
SS4 2 0 0 0.00 2 2–3 0.5
SS7 3 0 0 0.00 2 2–3 0.5
SS8 1 0 0 0.00 2 1–2 0.09
SS12 2 0 0 0.00 4 2–5 0.6
SS14 2 0 0 0.00 2 2–3 0.5
SS15 2 0 0 0.00 3 1–3 0.5
SS16 2 0 0 0.00 3 1–3 0.53
SS17 2 0 0 0.00 2 2–3 0.49
SS18 2 0 0 0.00 2 2–3 0.5
SS19 2 0 0 0.00 3 1–3 0.5
SS20 2 0 0 0.00 2 1–2 0.49
SS21 1 0 0 0.00 2 1–2 0.49
SS22 2 0 0 0.00 2 1–2 0.49
SS23 3 0 0 0.00 5 0–6 0.67
SS24 2 0 0 0.00 2 1–2 0.49
SS25 1 0 0 0.00 2 1–2 0.41
SS26 4 0 0 0.00 2 4–5 0.19
on May 16, 2020 by guest
http://jcm.asm.org/
[image:4.585.44.540.419.726.2]determined by PFGE. Clusters MA, MB, and MC, respec-tively, included all of the isolates belonging to the IST1, IST6, and IST3 clonal groups. Strains within the MA and MC clus-ters were more divergent. Cluster MA comprised two subclus-ters, MA1 and MA2, with a distance of five loci in between. MA1 included the 703 isolates of the SS26.66 clone and an Indian strain, whereas MA2 included six IST1 isolates and the non-IST1 isolates within the IST1 clonal group. All of the imported isolates, except the one from India, were situated in the MA2 subcluster. The MB cluster had a distance of 15 and 13 loci from the MA and MC clusters, respectively. Strains in the MC cluster were quite divergent; some lineages differed at seven or more loci.
Each clonal group shared a number of common alleles. The strains in the MA cluster (IST1 clonal group), the MB cluster (IST6 clonal group), and the MC cluster (IST3 clonal group), respectively, shared 14, 21, and 10 common alleles. All of the loci with common alleles for each clonal group had low diver-sity values, except that the IST3 clonal group had a highly variable locus (SS1) and the IST6 clonal group had two highly variable loci (SS1 and SS6). It is notable that the two loci harbored only one (SS1) or two (SS6) repeat units. Loci with a low number of repeat units should be less variable.
The phylogenetic relationships among the 916 isolates were evaluated using the eight most variable loci, SS1, SS3, SS6, SS9, SS10, SS11, SS12, and SS23. These loci had the highest number of alleles and the highest level of allele diversity ob-served for the 206 non-IST1 isolates (Table 2). The groupings based on the 8-locus profiles (Fig. 3B-2) were similar to those based on the 26-locus profiles (Fig. 3B-1). In the phylogenetic tree built using the eight-locus profiles, the isolates for each IST clonal group were mostly clustered more closely, but the cluster for the IST6 clonal group was not clearly separated from that for the IST3 clonal group. The phylogenetic pattern for the closely related isolates belonging to the SS26.66 clone was concordant with that determined using the 26-locus pro-files. When the 8 most variable loci were excluded from the 26-locus panel, a phylogenetic tree constructed using the re-maining 18 loci presented a clear phylogenetic pattern for the three IST clonal groups (Fig. 3B-3).
DISCUSSION
The first IST1 strain of the SS26.66 clone that emerged in Taiwan belonged to the SS26.66 genotype. It caused a large outbreak of shigellosis at a high school in October 2000, and thereafter it and its derivative strains circulated throughout the country for 3.25 years (15). Since these strains were derived from a line of common evolutionary descent, they were an excellent collection of isolates to identify the highly variable VNTR loci and investigate the phylogenetic pattern for the newly emerged clone by use of combinations of loci with dif-ferent variation values.
Of the seven loci that varied in the SS26.66 clone, four (SS3, SS6, SS9, and SS11) were highly variable (Table 2). When the changes resulting from the four highly variable loci were ig-nored, little diversification was observed in the clone (Fig. 3A-2). These four loci were the primary contributors to the diversification among the isolates. Therefore, loci with higher variability are suitable markers for resolving closely related
isolates. As indicated in a previous study, MLVA with four to eight highly variable loci is sufficient to discriminate isolates for investigatingS. sonneioutbreaks (8).
The global phylogenetic patterns for the 916 isolates estab-lished with the three typing methods (IST, PFGE, and MLVA) were considerably concordant; each of the phylogenetic trees consisted of three distinct clusters, and each of the clusters in a tree shared the same set of isolates with the corresponding clusters in the other two trees. Among the three methods, IST is the least discriminatory; it is not sufficient to distinguish closely related strains but is suitable to defineS. sonneiclones. PFGE has a higher discriminatory power than IST; it can further discriminate isolates with a common IST genotype, but it sometimes is not powerful enough to distinguish epidemio-logically unrelated strains (8). In this study, we found that PFGE was not sufficient to establish clear phylogenetic rela-tionships among the strains belonging to the SS26.66 clone. In the dendrogram constructed using the PFGE patterns, some IST1 strains within the SS26.66 clone were separated by strains with other IST genotypes, including IST11, IST17, IST18, and IST23 (data not shown). MLVA is the most discriminatory among the three methods in distinguishing the strains within the SS26.66 clone from others and establishing a fine phyloge-netic pattern for closely related isolates.
Discrimination of closely related strains is attributed mainly to highly variable loci. In the phylogenetic tree established by MLVA profiles based on the 8 most variable loci, the pattern for the strains in the SS26.66 clone was concordant with that in the tree established by the 26-locus profiles (Fig. 3B-1 and B-2). Although the eight most variable loci could discriminate and establish fine phylogenetic relationships between the closely related strains, they did not separate the three clonal groups distinctly (Fig. 3B-2). In contrast, the 18 loci with lower variability were not sufficient to discriminate the closely related strains, but they were able to clearly separate the three clonal groups (Fig. 3B-3). The global phylogenetic patterns deter-mined with the eight-locus profiles suggest that the loci with higher variability are useful markers for establishing the “twig and leaves” of a phylogenetic tree for theS. sonneipopulation. In contrast, the clustering analysis based on the 18 less variable loci clearly separates the three IST clonal groups (Fig. 3B-3), indicating that loci with lower variability are more suitable for establishing the “trunk” of a phylogenetic tree.
S. sonnei infection was not previously reported to occur frequently in Taiwan, but it became prevalent in the central and eastern parts of the country during 2000 to 2002. The epidemic was caused mainly by strains with an IST1 genotype. Since the first IST1 strain detected in Taiwan was recovered from a traveler who returned from India and the isolate shared indistinguishable PFGE-XbaI and PFGE-NotI patterns with the majority of IST1 isolates, we concluded in our previous report that the Indian strain was the original source responsi-ble for the epidemic (15). However, the MLVA data show that the Indian strain differed at six loci from the founder strain of the SS26.66 clone. Since a strain is very unlikely to have varied at six loci over a period of less than 1 year, the Indian strain can be ruled out as the original source of the epidemic.
The 916S. sonneiisolates collected in Taiwan over 9 years (from 1996 to 2004) fall into three distinct clonal groups. All of the imported isolates, except the one from India, belonged to
on May 16, 2020 by guest
http://jcm.asm.org/
a single MLVA subcluster, MA2 (Fig. 3B-1). Strains in this subcluster have been found in China, Cambodia, Indonesia, Taiwan, Thailand, and Vietnam, indicating that strains within this clonal group have been circulating in Asia for years.S. sonneiis one of the primary causal agents of travel-associated diarrhea (3). Transmission of the pathogen takes place easily between countries and geographical regions between which people travel frequently. The data also suggest that it is very likely that certain clones are spread more easily. To investigate this speculation, further studies of strains collected in various geographical regions should be conducted.
Combinations of VNTR loci with different degrees of vari-ability have been used to investigate different levels of genetic relatedness among bacterial strains. Pourcel et al. (10) used MLVA as a tool for the phylogenetic analysis ofYersinia pestis, a highly monomorphic pathogen; the MLVA data based on 25 VNTR markers could effectively separate panels of isolates of different biovars, and typing based on 7 highly variable loci exhibited a resolution among the isolates comparable to that achieved by typing based on 25 loci. Schouls et al. (12) suc-cessfully used four highly variable VNTR loci to differentiate closely related Neisseria meningitidis isolates and used eight VNTR loci with limited variability to classify strains, which yielded groupings similar to those obtained by MLST. In our laboratory, we concurrently developed an MLVA with 12 VNTR loci forN. meningitidis; the MLVA ofN. meningitidis
isolates also yielded groupings similar to those obtained by MLST (9). Although a high degree of congruence between MLST and MLVA methods was obtained in the two studies of
N. meningitidis, some ST groups or ST clonal complexes were not clearly separated. Therefore, a larger set of VNTR loci would be more favorable to obtain a clearer separation of genetically distinct groups.
Recently, Gorge´ et al. (4) identified 15 VNTR loci by ex-ploring five genomic sequences of four Shigella species for typingShigellaspp. Although the MLVA method could effec-tively discriminate the isolates of the fourShigellaspecies and pathogenicE. coli, the grouping based on the MLVA profiles was not able to clearly separate the fourShigellaspecies into distinct groups. Tandem-repeat loci may have variable num-bers in one species or serotype but may be invariable in others. In our recent study, we found that differentS. flexneriserotypes shared only a very limited number of common VNTR loci (unpublished data). Therefore, MLVA may not be as appro-priate a molecular tool as MLST to establish phylogenetic relationships between different bacterial species or serotypes. Nevertheless, our study indicates that MLVA is a useful tool to establish phylogenetic relationships among strains of a mono-morphic bacterial species, such asS. sonnei.
In conclusion, VNTR loci were considered too variable to serve as suitable molecular markers for the phylogenetic anal-ysis of genetically diverse microbes. In this study, we found that the VNTR loci inS. sonneihave a range of degrees of vari-ability that are useful markers for establishing accurate phylo-genetic patterns forS. sonnei strains that have evolved over different timescales. Combined loci with higher variability can be used to resolve closely related isolates, whereas combined
loci with lower variability are suitable to establish phylogenetic relationships amongS. sonneistrains that have evolved over a longer timescale. Therefore, MLVA is a useful tool for the phylogenetic analysis ofS. sonnei, a monomorphic bacterial species.
ACKNOWLEDGMENTS
This work was supported by a grant (DOH 97-DC-2012) from the Centers for Disease Control, Department of Health, Taiwan, and by a grant (H17-Shinkou-Ippan-019) from the Ministry of Health, Labor and Welfare, Japan.
REFERENCES
1.Chiou, C. S., H. L. Wei, Y. W. Wang, J. C. Liao, and C. C. Li.2006.
Usefulness of inter-IS1spacer polymorphisms for subtyping ofShigella
son-neiisolates. J. Clin. Microbiol.44:3928–3933.
2.Choi, S. Y., Y. S. Jeon, J. H. Lee, B. Choi, S. H. Moon, L. von Seidlein, J. D. Clemens, G. Dougan, J. Wain, J. Yu, J. C. Lee, S. Y. Seol, B. K. Lee, J. H. Song, M. Song, C. Czerkinsky, J. Chun, and D. W. Kim.2007. Multilocus
sequence typing analysis ofShigella flexneriisolates collected in Asian
coun-tries. J. Med. Microbiol.56:1460–1466.
3.Ekdahl, K., and Y. Andersson.2005. The epidemiology of travel-associated
shigellosis—regional risks, seasonality and serogroups. J. Infect.51:222–229.
4.Gorge´, O., S. Lopez, V. Hilaire, O. Lisanti, V. Ramisse, and G. Vergnaud.
2008. Selection and validation of a multilocus variable-number
tandem-repeat analysis panel for typingShigellaspp. J. Clin. Microbiol.46:1026–
1036.
5.Hunter, S. B., P. Vauterin, M. A. Lambert-Fair, M. S. Van Duyne, K. Kubota, L. Graves, D. Wrigley, T. Barrett, and E. Ribot.2005. Establishment of a universal size standard strain for use with the PulseNet standardized pulsed-field gel electrophoresis protocols: converting the national databases
to the new size standard. J. Clin. Microbiol.43:1045–1050.
6.Kotloff, K. L., J. P. Winickoff, B. Ivanoff, J. D. Clemens, D. L. Swerdlow, P. J. Sansonetti, G. K. Adak, and M. M. Levine.1999. Global burden ofShigella
infections: implications for vaccine development and implementation of
con-trol strategies. Bull. W. H. O.77:651–666.
7.Lee, H. C., K. L. Chen, C. L. Tsai, C. H. Chen, T. N. Yeh, C. R. Yang, Y. L. Wang, H. Y. Chiu, C. L. Lee, H. P. Su, and T. H. Lin.2004. Imported
infection ofShigella sonneimolecular epidemiological investigation of cases
of the Bali tours. Epidemiol. Bull.20:23–42.
8.Liang, S. Y., H. Watanabe, J. Terajima, C. C. Li, J. C. Liao, S. K. Tung, and C. S. Chiou.2007. Multilocus variable-number tandem repeat analysis for
molecular typing ofShigella sonnei. J. Clin. Microbiol.45:3574–3580.
9.Liao, J. C., C. C. Li, and C. S. Chiou.2006. Use of a multilocus variable-number tandem-repeat analysis method for molecular subtyping and
phylo-genetic analysis ofNeisseria meningitidisisolates. BMC Microbiol.6:44.
10.Pourcel, C., F. Andre-Mazeaud, H. Neubauer, F. Ramisse, and G. Vergnaud.
2004. Tandem repeats analysis for the high resolution phylogenetic analysis ofYersinia pestis. BMC Microbiol.4:22.
11.Ribot, E. M., M. A. Fair, R. Gautom, D. N. Cameron, S. B. Hunter, B. Swaminathan, and T. J. Barrett.2006. Standardization of pulsed-field gel
electrophoresis protocols for the subtyping of Escherichia coliO157:H7,
Salmonella, andShigellafor PulseNet. Foodborne Pathog. Dis.3:59–67. 12.Schouls, L. M., A. van der Ende, M. Damen, and I. van de Pol.2006.
Multiple-locus variable-number tandem repeat analysis ofNeisseria
menin-gitidisyields groupings similar to those obtained by multilocus sequence
typing. J. Clin. Microbiol.44:1509–1518.
13. Swaminathan, B., P. Gerner-Smidt, L. K. Ng, S. Lukinmaa, K. M. Kam, S. Rolando, E. P. Gutierrez, and N. Binsztein.2006. Building PulseNet Inter-national: an interconnected system of laboratory networks to facilitate timely public health recognition and response to foodborne disease outbreaks and
emerging foodborne diseases. Foodborne Pathog Dis.3:36–50.
14.Terajima, J., N. Tosaka, K. Ueno, K. Nakashima, P. Kitsutani, M. K. Gay-nor, S. Y. Park, and H. Watanabe.2006.Shigella sonneioutbreak among
Japanese travelers returning from Hawaii. Jpn. J. Infect. Dis.59:282–283.
15.Wei, H. L., Y. W. Wang, C. C. Li, S. K. Tung, and C. S. Chiou.2007. Epidemiology and evolution of genotype and antimicrobial resistance of an
importedShigella sonneiclone circulating in central Taiwan. Diagn.
Micro-biol. Infect. Dis.58:469–475.
16.Wirth, T., D. Falush, R. Lan, F. Colles, P. Mensa, L. H. Wieler, H. Karch, P. R. Reeves, M. C. Maiden, H. Ochman, and M. Achtman.2006. Sex and
virulence inEscherichia coli: an evolutionary perspective. Mol. Microbiol.
60:1136–1151.