STATE-OF-THE-ART REVIEW ARTICLE
New Approaches to Progeria
Mark W. Kieran, MD, PhDa,b, Leslie Gordon, MD, PhDc, Monica Kleinman, MDd
aDepartment of Pediatric Oncology, Dana-Farber Cancer Institute, Boston, Massachusetts; Departments ofbPediatric Oncology anddCritical Care, Children’s Hospital Boston, Boston, Massachusetts;cDepartment of Pediatrics, Warren Alpert Medical School, Brown University, Providence, Rhode Island
The authors have indicated they have no financial relationships relevant to this article to disclose.
ABSTRACT
Progeria (Hutchinson-Gilford progeria syndrome) is a rare genetic disorder that offers considerable insight into the biology of premature aging. This review sum-marizes the clinical characteristics of this disease and the underlying mutation in the lamin A (LMNA) gene that results in this phenotype. Modifications in the processing of prelamin A through alterations in farnesylation are detailed, because this pathway offers a possible drug target. Finally, discussion of an ongoing clinical trial for these children, including possible parameters for evaluation, are discussed. In the span of less than a decade, this disease has progressed from an interesting phenotype to one in which the gene defect has been identified, animal models have been created and tested with drugs that target the primary disease pathway, and significant clinical baseline data for the support of a clinical trial have been obtained.
www.pediatrics.org/cgi/doi/10.1542/ peds.2007-1356
doi:10.1542/peds.2007-1356
Key Words
progeria, premature aging, review article, farnesyltransferase inhibitor, FTI, lamin A
Abbreviation
FTI—farnesyltransferase inhibitor
Accepted for publication May 9, 2007
H
UTCHINSON-GILFORDprogeria syndrome (or proge-ria) is a rare sporadic autosomal-dominant disorder that is characterized by the premature appearance of signs of aging. Although fewer than 50 patients world-wide are currently known to be alive with the disease, progeria has generated enormous interest as we attempt to understand the biological basis of growing old. In this review we summarize the clinical manifestations of progeria, chronicle recent advances that led to the mo-lecular identification of the genetic defect responsible for it, and describe the development of animal models that have been designed to test possible treatments. On the basis of these efforts, we are now ready to embark on the first-ever clinical trial of a biological agent that may prevent, ameliorate, or reverse the clinical manifesta-tions of this disease. A number of recent reviews have been published on different aspects of progeria, and readers are referred to them for more in-depth informa-tion.1–7CHARACTERISTICS OF THE DISEASE
The characteristic phenotype of progeria was first recog-nized more than a century ago.8,9 Progeria has an
inci-dence of ⬃1 in 4 000 000 live births10 and is one of a
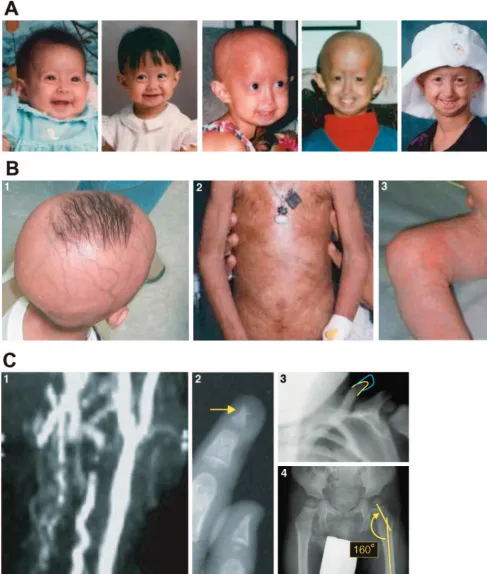
number of “segmental aging syndromes,” so named be-cause patients lack certain typical features of aging such as neurocognitive decline or increased cancer rates. Al-though some variability in the onset and severity of the physical attributions of progeria occur, most patients with the classic gene mutation are born with a normal appearance and progressively develop pathognomonic early signs and symptoms beginning at 9 to 12 months of age. The phenotype is typified by alopecia (loss of hair including scalp and eyebrows), prominent scalp veins and forehead, classical facial features including micro-gnathia (small jaw), prominent eyes and a convex nasal profile (beak-like nose), and circumoral cyanosis. De-layed and abnormal dentition is common, and most of these patients tend to have high-pitched voices. Patients with progeria have significant loss of subcutaneous fat (which results in thin, often tight skin) and short stature. Skeletal manifestations include frequent osteolysis, lim-ited joint mobility (contractures), coxa valga, and short-ened clavicles, which result in a horse-riding stance and narrowed shoulders. Together with a final height of only 3 to 3 1/2 feet, these features give the overwhelming impression of a diminutive, wasted-looking older per-son.10Clinical features of the disease are presented in Fig
1.
Although these manifestations describe the physical findings in progeria, it is the global, accelerated cardio-vascular and cerebrocardio-vascular diseases that result in pre-mature death between the ages of 7 and 20 years. The vasculopathy associated with progeria results in myocar-dial ischemia and infarction as well as stroke. Major cardiac or neurologic events may be preceded by angina,
chronic congestive heart failure, or transient ischemic attacks. Despite the dramatic effects of classic progeria on growth and the cardiovascular system, it is important to recognize that multiple other organs seem to be unaf-fected by this disease, including the liver, kidney, lung, gastrointestinal tract, bone marrow, and brain.
A significant portion of the cellular defects in progeria result from the accumulation of a mutant protein, pro-gerin, within the nuclear membrane. Progerin is present in significant concentration in the cells of patients with progeria, which results in distortion of the nuclear mem-brane and decreased cell life span. The predilection of the cardiovascular and cerebrovascular manifestations of the disease may be related to differential accumulation of progerin in vascular endothelial and smooth muscle cells.11 It is unclear why other organs that possess the
abnormal protein are not affected adversely; for in-stance, the teeth are severely impacted, whereas the brain is relatively spared.12,13
MUTATION IN LAMIN A CAUSES PROGERIA
Originally thought to be an autosomal-recessive disor-der,14,15more recent evidence has identified the genetic
basis for progeria to be a single nucleotide mutation with autosomal-dominant expression.16–18 The identification
of the gene defect resulted from a multiinstitutional collaboration within the Progeria Research Foundation Genetics Consortium.18 Using material from a few
pa-tients with the disease, including 3 with chromosomal modifications rather than a single base-pair mutation, the location of interest was refined to a small segment of chromosome 1q. With identification of the disease locus for progeria on a limited region of 1q, candidate genes within this area were sequenced, and one,LMNA (pro-nounced lamin A), was noted by Collins’ group at the National Human Genome Research Institute to possess a mutation in patients with progeria when compared with parental DNA.18 LMNA normally codes for a protein
called lamin A, and in progeria the gene produces some normal lamin A and some mutated lamin A (progerin). Lamin A is one member of a group of lamin proteins. Lamins are filamentous structures that are critical com-ponents of the nuclear membrane and function to reg-ulate chromatin and nuclear integrity as well as nuclear shape.19 The lamin gene is made up of 12 exons.
FIGURE 1
which is to allow proteins to be embedded into mem-branes (many important cellular proteins that need to be associated with the cell membrane to function, such as ras, are also farnesylated). After addition of the farnesyl group, the “aaX” motif is removed and replaced by a carboxymethyl group through the function of an en-zyme called ZMPSTE24.20Using an internal splice site in
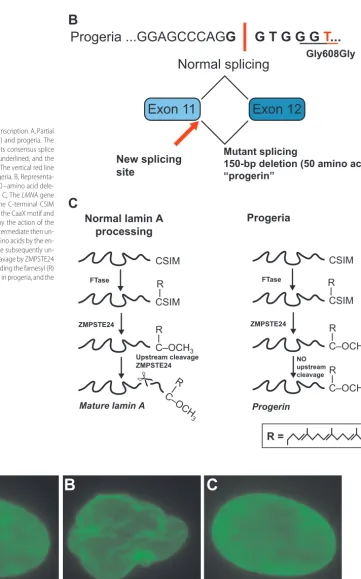
exon 11, the C-terminal end of the protein, including the farnesyl group, is then removed, which results in mature lamin A. With the loss of the farnesyl group, lamin A is no longer embedded into the cell membrane. Patients with classical progeria have a single nucleo-tide substitution in exon 11 of the prelamin gene, whereby the sequence change GGC3GGT occurs at po-sition 1824. Although this mutation does not alter the amino acid sequence of the protein (a conserved or “silent” mutation), it introduces an alternative splice site that results in the removal of a 150-nucleotide stretch of exon 11. Because exon 12 is retained, the first 3 steps of prelamin processing occur normally (farnesylation of the CaaX site, removal of the aaX, and addition of the car-boxymethyl group). Unfortunately, the missing segment of exon 11 contains the recognition site for the enzyme responsible for cleaving the C-terminal component of the molecule with its attached farnesyl group. This new molecule, with a 50 –amino acid deletion from the exon 11 protein product and preservation of the 3⬘ farnesyl group, is called “progerin”21,22(see Fig 2). A number of
related progeroid syndromes have been described that result from different mutations within the same gene or in other genes within the same processing pathway. Each of these mutations is even rarer than classical pro-geria. Readers are referred to the review by Ramı´rez et al,1which describes these related diseases in detail.
With the identification of the genetic mutation and characterization of the resulting mutant protein, efforts were directed toward understanding how this abnormal protein affects cellular function. Because the mutant prelamin A remains farnesylated, it stays embedded in the nuclear membrane. Morphologically, this results in nuclear membrane “blebbing” as a result of the accumu-lation of progerin (Fig 3).18,23Because nuclear
localiza-tion of progerin is a result of the presence of the farnesyl group, many investigators embarked on a series of stud-ies to target this molecule.
Unrelated to progeria, many researchers had been studying the inhibition of farnesylation in connection with cancer. Ras is a major regulator of cellular signaling that influences decisions about proliferation, migration, apoptosis, and angiogenesis.24Up to 50% of human
tu-mors demonstrate mutations in ras that leave it consti-tutively activated. Both normal and mutant ras require localization to the cell membrane to transmit their sig-nal, and this localization is also achieved through addi-tion of a farnesyl group to the 3⬘end of the molecule (also using a CaaX domain). Pharmaceutical companies,
therefore, have exerted significant effort to find inhibi-tors of this process. One class of drug, called farnesyl-transferase inhibitors (FTIs), are small molecular moi-eties that reversibly bind the CaaX domain and prevent a farnesyl group from being added.2,25
Investigators from several laboratories proceeded to treat progeria cells with FTIs in vitro. Remarkably, nuclei of the treated cells demonstrated resolution of the mor-phologic nuclear abnormalities within 36 hours of FTI administration at concentrations that are considered comparable to in vivo treatment levels.26–28These results
indicate that the nuclear membrane abnormalities in cells that are already affected by the accumulation of progerin can be reversed if additional progerin produc-tion is inhibited (an important aspect of treating patients who already demonstrate clinical characteristics of the disease). The results also imply that treatment likely has to be prolonged and continuous, because release of in-hibition of farnesylation allowed cells to return to their abnormal shape. It should be noted that FTIs prevent farnesylation and localization of progerin to the cell membrane but do not repair the function of the abnor-mal progerin protein within the cytoplasm, which may result in abnormalities in cell function and DNA repair that, therefore, would not be treated with these drugs.29,30
With exciting clues as to the importance of progerin in the cellular defects associated with progeria and the ability of FTIs to reverse the nuclear blebbing in progeria cells, a number of animal models have been developed to test the role of these drugs further in more clinically relevant systems.31Animals made null for lamin A and
prelamin A were phenotypically normal (which suggests that absence of lamin A is not the cause of the disease), whereas those that expressed the mutant protein pro-gerin, even in the presence of normal lamin A, were abnormal.32,33These data would suggest that the mutant
protein acts in a dominant fashion in progeria cells. To date, each different in vivo animal model of progeria that has been developed mimics some aspects of the human disease. Animals in one model manifested growth retar-dation, reduction in adipose tissue, micrognathia, and osteoporosis.32In a series of very important and exciting
experiments, these genetically engineered animals were treated with FTIs, which protected them from the devel-opment of most of the clinical phenotype. Specifically, there were improvements in weight and bone structure and a decrease in spontaneous bone fractures.34 A
sec-ond mouse model was recently developed that resulted in the cardiovascular disease characteristic of progeria.33
s
s
- amino acids
Progeria New splicing
site
Progeria
FIGURE 2
Effect of the progeria mutation onLMNAtranscription. A, Partial DNA sequence for normalLMNA(top line) and progeria. The sequence in bold and italic type represents consensus splice donor sequence. The code for glycine is underlined, and the mutant transition (C to T) is shown in red. The vertical red line represents the splice point created in progeria. B, Representa-tion for mutant splicing that results in a 50 –amino acid dele-tion from lamin A, thus creating progerin. C, TheLMNAgene codes for prelamin A, which possesses the C-terminal CSIM amino acid sequence. This is referred to as the CaaX motif and accepts a farnesyl group (R) added to it by the action of the enzyme farnesyltransferase (FTase). This intermediate then un-dergoes additional cleavage of the SIM amino acids by the en-zyme ZMPSTE24, and the terminal cysteine subsequently un-dergoes carboxymethylation. A second cleavage by ZMPSTE24 removes the terminal 15 amino acids, including the farnesyl (R) group. The last step in this process is absent in progeria, and the resultant protein is called progerin.
$
%
&
FIGURE 3
DEVELOPMENT OF A HUMAN CLINIC TRIAL TO TREAT PROGERIA
We now stand at a crossroads in molecular medicine. Having attained the ability to define diseases by their molecular rather than phenotypic characterization, and with a large and increasing array of drugs that attack specific pathways, we are ready to embark on the treat-ment of children with progeria.
To undertake a human clinical trial, a number of criteria should be met:
1. Patients should have a defined disease and a target of that disease against which therapy can be directed. Progeria is the disease, farnesylation is the target, and FTIs are the therapy.
2. Ideally, laboratory and preclinical models should in-volve therapies that target the defect, with a resultant improvement in the phenotype. Improvement in in vitro cellular morphology with FTIs and improve-ment in disease status of in vivo mouse models with FTIs have been demonstrated.26 –28,34,35
3. The proposed therapy should have a toxicity profile that is justifiable depending on the severity and prog-nosis of the disease being treated. FTIs have been tested in children with cancer and neurofibromatosis type 1 and have shown limited toxicity and good tolerability.36
4. The clinical trial must have a measure that can be used to evaluate the efficacy of the therapeutic inter-vention. For many diseases, this may not be improve-ment in survival, because this end point could take more than a decade to evaluate, especially for younger patients. A number of clinical parameters have been studied in this patient population that could be used in place of survival to provide a more rapid predictor of treatment effect (see below).
Over the last 100 years, many of the phenotypic characteristics of progeria have been documented.10 A
longitudinal study of weight change in patients with progeria was undertaken at Brown University by using the largest repository of progeria patient data (see www. progeriaresearch.org). In addition, a systematic study of the natural history of progeria has been undertaken at the National Institutes of Health. When combining these data sets, a characteristic pattern of specific clinical fea-tures emerges. In particular, the data from Brown Uni-versity showed that children with progeria rapidly fall off their normal weight-and-height growth curves, which results in significant failure to thrive, usually evident by the second or third year of life. Throughout childhood, patients with progeria gain limited weight per year de-spite sufficient nutritional supplementation and, thus, have their own characteristic slope for weight gain that remains stable for the remainder of their lives (Fig 4).37
Rate of weight gain as a primary clinical parameter is
sufficiently robust to be used to gain early evidence of the effect of a new therapy for patients with progeria. A variety of secondary parameters will allow us to evaluate whether other important aspects of the disease are af-fected by FTI administration.
The proposed intervention, which started enrolling patients in June 2007 at Children’s Hospital Boston with the support of the Progeria Research Foundation, will include the following elements.
Eligibility
Patients with the classic (G608G) mutation, or other
LMNAmutations that result in the same phenotype as that in Hutchinson-Gilford progeria syndrome and ap-proved by the study team, are eligible. All patients also must have a minimum of 1 year of weight measure-ments that demonstrate a stable slope. They must not have severely impaired liver, kidney, gastrointestinal, or bone marrow function that would prevent them from tolerating the drug.
Exclusion Criteria
Patients who are too ill to take part or who have uncon-trolled infections are excluded.
Design
This open-label, single-arm, non–placebo-controlled trial will treat all eligible patients with an oral FTI, which will be taken twice a day by mouth (the drug is available in both pill and liquid formulations). Patients will un-dergo monthly physical examination at their local sites and will be required to come to Boston for detailed examination and testing once every 4 months for a duration of 2 years. The primary end point for analysis will be a significant increase in rate of weight gain over baseline for each patient. Multiple additional secondary measures including changes in leptin levels, glucose uti-lization, skeletal abnormalities consisting of bone min-eral density and radiograph findings, joint contractures and function, hearing loss, dental anomalies, dermato-logic changes including hair density, nutritional analysis, energy expenditure, body composition analysis by dual-energy radiograph absorptiometry scan, and cardiovas-cular function will also be evaluated. A series of bio-logical-based assays to evaluate effectiveness of farnesyltransferase inhibition will also be requested from consenting patients.
More information on this trial can be obtained from Children’s Hospital Boston (www.childrenshospital. org), the national clinic trial database (www.clinical trials.gov), or the Progeria Research Foundation (www. progeriaresearch.org).
FUTURE DIRECTIONS
this treatment in in vitro and in vivo models, and the availability of drugs that have already completed pediatric testing have generated great interest and high expecta-tions. In the field of cancer, in which innumerable targets and therapies have been identified, each with dramatic results in animal models, we still find ourselves woefully lacking in positive results in many human trials. We must combine enthusiasm with reality. It is too early to deter-mine if these drugs will have no impact or significant but transient impact or will reverse or permanently control some or all aspects of progeria. Therefore, we must con-tinue to look for additional treatment modalities at the same time that we are studying other therapies.38,39
Progeria has fascinated clinicians for a century, not just because of the unique appearance and tragic out-come for these children but also because the disease has been seen as a window into the process of aging for all of us. The recent identification of progerin accumulation in normal people with age raises interesting questions about how and why we all age40and is likely to be the
focus of a great deal of research over the coming decade.
ACKNOWLEDGMENTS
Assistance for Figs 3 and 4 was provided by Brian Capell and Jason Machan, PhD, respectively.
REFERENCES
1. Ramı´rez CL, Cadin˜anos J, Varela I, Freije JM, Lo´pez-Otı´n C. Human progeroid syndromes, aging and cancer: new genetic and epigenetic insights into old questions. Cell Mol Life Sci.
2007;64:155–170
2. Meta M, Yang SH, Bergo MO, Fong LG, Young SG. Protein
farnesyltransferase inhibitors and progeria. Trends Mol Med.
2006;12:480 – 487
3. Young SG, Meta M, Yang SH, Fong LG. Prelamin A farnesyla-tion and progeroid syndromes. J Biol Chem. 2006;281: 39741–39745
4. Rusin˜ol AE, Sinensky MS. Farnesylated lamins, progeroid syn-dromes and farnesyl transferase inhibitors.J Cell Sci.2006;119: 3265–3272
5. Navarro CL, Cau P, Le´vy N. Molecular bases of progeroid syndromes.Hum Mol Genet.2006;15:R151–R161
6. Scaffidi P, Gordon L, Misteli T. The cell nucleus and aging: tantalizing clues and hopeful promises.PLoS Biol.2005;3:e395 7. Pollex RL, Hegele RA. Hutchinson-Gilford progeria syndrome.
Clin Genet.2004;66:375–381
8. Hutchinson J. Case of congenital absence of hair, with atrophic condition of the skin and its appendages, in a boy whose mother had been almost wholly bald from alopecia areata from the age of six.Lancet.1886;I:923
9. Gilford H. Ateleiosis and progeria: continuous youth and pre-mature old age.Br Med J.1904;2:914 –918
10. Hennekam RC. Hutchinson-Gilford progeria syndrome: review of the phenotype.Am J Med Genet A.2006;140:2603–2624 11. McClintock D, Gordon LB, Djabali K. Hutchinson-Gilford
pro-geria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody.Proc Natl Acad Sci U S A.2006;103:2154 –2159
12. Lans H, Hoeijmakers JH. Cell biology: ageing nucleus gets out of shape.Nature.2006;440:32–34
13. Csoka A, English S, Simkevich C, et al. Genome-scale expres-sion profiling of Hutchinson-Gilford progeria syndrome reveals widespread transcriptional misregulation leading to mesodermal/mesenchymal defects and accelerated atheroscle-rosis.Aging Cell.2004;3:235–243
14. Maciel AT. Evidence for autosomal recessive inheritance of pro-geria (Hutchinson Gilford).Am J Med Genet.1988;31:483– 487 15. Khalifa MM. Hutchinson-Gilford progeria syndrome: report of
, y
,
FIGURE 4
a Libyan family and evidence of autosomal recessive inheri-tance.Clin Genet.1989;35:125–132
16. Cao H, Hegele RA.LMNA is mutated in Hutchinson-Gilford progeria (MIM 176670) but not in Wiedemann-Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet. 2003;48: 271–274
17. De Sandre-Giovannoli A, Bernard R, Cau P, et al. Lamin A trun-cation in Hutchinson-Gilford progeria.Science.2003;300:2055 18. Eriksson M, Brown WT, Gordon LB, et al. Recurrent de novo
point mutations in lamin A cause Hutchinson-Gilford progeria syndrome.Nature.2003;423:293–298
19. Lammerding J, Fong LG, Ji JY, et al. Lamins A and C but not lamin B1 regulate nuclear mechanics.J Biol Chem.2006;281: 25768 –25780
20. Fong LG, Ng JK, Meta M, et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmp-ste24-deficient mice. Proc Natl Acad Sci U S A. 2004;101: 18111–18116
21. Young SG, Fong LG, Michaelis S. Prelamin A, Zmpste24, mis-shapen cell nuclei, and progeria: new evidence suggesting that protein farnesylation could be important for disease pathogen-esis.J Lipid Res.2005;46:2531–2558
22. Shackleton S, Smallwood DT, Clayton P, et al. Compound heterozygous ZMPSTE24 mutations reduce prelamin A pro-cessing and result in a severe progeroid phenotype.J Med Genet.
2005;42:e36
23. Goldman R, Shumaker DK, Erdos MR, et al. Accumulation of mutant lamin A causes progressive changes in nuclear archi-tecture in Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A.2004;101:8963– 8968
24. Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling con-trols tumour cell growth.Nature.2006;441:424 – 430 25. Adjei AA. Farnesyltransferase inhibitors.Cancer Chemother Biol
Response Modif.2005;22:123–133
26. Yang SH, Bergo MO, Toth JI, et al. Blocking protein farnesyl-transferase improves nuclear blebbing in mouse fibroblasts with a targeted Hutchinson-Gilford progeria syndrome muta-tion.Proc Natl Acad Sci U S A.2005;102:10291–10296 27. Toth JI, Yang SH, Qiao X, et al. Blocking protein
farnesyltrans-ferase improves nuclear shape in fibroblasts from humans with progeroid syndromes. Proc Natl Acad Sci U S A. 2005;102: 12873–12878
28. Capell BC, Erdos MR, Madigan JP, et al. Inhibiting farnesyla-tion of progerin prevents the characteristic nuclear blebbing of
Hutchinson-Gilford progeria syndrome. Proc Natl Acad Sci U S A.2005;102:12879 –12884
29. Liu Y, Rusinol A, Sinensky M, Wang Y, Zou Y. DNA damage responses in progeroid syndromes arise from defective matu-ration of prelamin A.J Cell Sci.2006;119:4644 – 4649 30. Shumaker DK, Dechat T, Kohlmaier A, et al. Mutant nuclear
lamin A leads to progressive alterations of epigenetic control in premature aging.Proc Natl Acad Sci U S A.2006;103:8703– 8708 31. Mounkes LC, Kozlov S, Hernandez L, Sullivan T, Stewart CL. A progeroid syndrome in mice is caused by defects in A-type lamins.Nature.2003;423:298 –301
32. Fong L, Ng J, Lammerding J, et al. Prelamin A and lamin A appear to be dispensable in the nuclear lamina.J Clin Invest.
2006;116:743–752
33. Varga R, Eriksson M, Erdos MR, et al. Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome.Proc Natl Acad Sci U S A.2006;103: 3250 –3255
34. Yang SH, Meta M, Qiao X, et al. A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J Clin Invest.2006;116: 2115–2121
35. Capell BC, Erdos MR, Eriksson M, et al. In vitro and in vivo effects of farnesyltransferase inhibitors for Hutchinson-Gilford progeria syndrome [abstract]. Presented at: American Society of Human Genetics; October 9 –13, 2006; New Orleans, LA 36. Kieran MW, Packer RJ, Onar A, et al. Phase I and
pharmaco-kinetic study of the oral farnesyltransferase inhibitor lona-farnib administered twice daily to pediatric patients with ad-vanced central nervous system tumors using a modified continuous reassessment method: a Pediatric Brain Tumor Consortium study.J Clin Oncol.2007;25:3137–3143
37. Campbell SE, Laliberte L, Wolf-Jensen N, et al. A medical and research database for Hutchinson-Gilford progeria syndrome [abstract]. Presented at: Progeria Research Foundation Inter-national Workshop on Progeria; November 3–5, 2005; Boston, MA
38. Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syn-drome.Nat Med.2005;11:440 – 445
39. Huang S, Chen L, Libina N, et al. Correction of cellular phe-notypes of Hutchinson-Gilford progeria cells by RNA interfer-ence.Hum Genet.2005;118:444 – 450
DOI: 10.1542/peds.2007-1356
2007;120;834
Pediatrics
Mark W. Kieran, Leslie Gordon and Monica Kleinman
New Approaches to Progeria
Services
Updated Information &
http://pediatrics.aappublications.org/content/120/4/834
including high resolution figures, can be found at:
References
http://pediatrics.aappublications.org/content/120/4/834#BIBL
This article cites 38 articles, 17 of which you can access for free at:
Subspecialty Collections
http://www.aappublications.org/cgi/collection/genetics_sub
Genetics
following collection(s):
This article, along with others on similar topics, appears in the
Permissions & Licensing
http://www.aappublications.org/site/misc/Permissions.xhtml
in its entirety can be found online at:
Information about reproducing this article in parts (figures, tables) or
Reprints
http://www.aappublications.org/site/misc/reprints.xhtml
DOI: 10.1542/peds.2007-1356
2007;120;834
Pediatrics
Mark W. Kieran, Leslie Gordon and Monica Kleinman
New Approaches to Progeria
http://pediatrics.aappublications.org/content/120/4/834
located on the World Wide Web at:
The online version of this article, along with updated information and services, is
by the American Academy of Pediatrics. All rights reserved. Print ISSN: 1073-0397.