0095-1137/07/$08.00⫹0 doi:10.1128/JCM.02078-06
Copyright © 2007, American Society for Microbiology. All Rights Reserved.
Identification of Variable-Number Tandem-Repeat (VNTR) Sequences
in
Legionella pneumophila
and Development of an Optimized
Multiple-Locus VNTR Analysis Typing Scheme
䌤
Christine Pourcel,
1* Paolo Visca,
2,3Baharak Afshar,
4Silvia D’Arezzo,
2Gilles Vergnaud,
1,5and Norman K. Fry
4Univ Paris-Sud, Institut de Ge´ne´tique et Microbiologie, CNRS, Orsay F-91405, France1; Istituto Nazionale per le Malattie Infettive
Lazzaro Spallanzani I.R.C.C.S.2and Dipartimento di Biologia, Universita` Roma Tre,3Rome, Italy; Health Protection Agency,
Respiratory and Systemic Infection Laboratory, Centre for Infections, London, United Kingdom4; and
Division de Microbiologie Analytique, Centre d’Etudes du Bouchet, Vert le Petit, France5
Received 9 October 2006/Returned for modification 29 November 2006/Accepted 17 January 2007
The utility of a genotypic typing assay forLegionella pneumophilawas investigated. A multiple-locus variable number of tandem repeats (VNTR) analysis (MLVA) scheme using PCR and agarose gel electrophoresis is proposed based on eight minisatellite markers. Panels of well-characterized strains were examined in a multicenter analysis to validate the assay and to compare its performance to that of other genotyping assays. Excellent typeability, reproducibility, stability, and epidemiological concordance were observed. The MLVA type or profile is composed of a string of allele numbers, corresponding to the number of repeats at each VNTR locus, separated by commas, in a predetermined order. A database containing information from 99 L. pneumophilaserogroup 1 strains and four strains of other serogroups and their MLVA profiles, which can be queried online, is available from http://bacterial-genotyping.igmors.u-psud.fr/.
The genus Legionella currently comprises 50 validly pub-lished species, and many more await full characterization (12). Legionellae are widely distributed in the environment, where they can multiply inside amoebae and ciliates. More than one-third of characterized Legionella species have been isolated from patients, butLegionella pneumophilais found in the ma-jority of culture-confirmed cases of Legionnaires’ disease. Three subspecies have been defined:L. pneumophila subsp.
pneumophila, L. pneumophila subsp.fraseri, and L.
pneumo-philasubsp. pascullei(1).
L. pneumophilacan be differentiated into at least 15
sero-groups (sg) (5).L. pneumophilasg 1 isolates account for more than 90% of clinical isolates (13), with the remaining 14 sg being differently associated with human infection. A high genetic diversity is observed withinL. pneumophilaand even within sg 1 isolates, as shown by other studies (6, 21, 22). Sequencing of the genome of three sg 1 strains associated with large outbreaks, the original Philadelphia-1 strain, the Paris strain, and the Lens strain have confirmed the considerable intraspecific genetic divergence, with up to 10% of DNA se-quences unique to one of the three strains (2, 3).
Many phenotypic and genotypic methods for epidemiologi-cal typing ofL. pneumophilahave been developed, some being costly and time-consuming and not always enabling interlabo-ratory comparison (7). Amplified fragment length polymor-phism (AFLP) analysis was adopted as a standard by the Eu-ropean Working Group for Legionella Infections (EWGLI) (6, 8, 9) and was widely used to allocate L. pneumophila sg 1
strains into coded types by interrogation of a reference data-base through an internet-data-based protocol. However, this tech-nique relies on the analysis of a multiband pattern, and too many variables exist which complicate unambiguous type as-signation. The nucleic acid sequence typing approach, com-monly called multilocus sequence typing (18), is now regarded as the method of choice to generate truly portable data. In addition, it is easily codable, and data can be stored into da-tabases. In a typical multilocus sequence typing assay, data derived from parts of seven housekeeping genes are analyzed. A simpler sequence-based typing (SBT) scheme forL.
pneu-mophilasg 1 was described, initially based on the investigation
of three genes (11) and more recently extended to six genes, namelyflaA,pilE,asd,mip,mompS, andproA(10). SBT was applied to the typing of 105 predominantly clinical isolates mostly belonging to sg 1 (10, 11). In a recent comparison of three different genotyping methods, SBT was reported to be the most rapid and the easiest to perform in an outbreak setting and provided unambiguous results (23). The major problem with SBT is the high cost which makes problematic its routine use by laboratories which are not equipped with local sequencing facilities. It is presumable that this method will be restricted to a subset of most relevant isolates. Hence, addi-tional screening methods combining low cost with good typing performance are still needed.
Polymorphic tandem repeats have been successfully used for epidemiological typing studies of several bacterial species (17, 26). The so-called multiple-locus variable number of tandem repeats (VNTR) assays (MLVA) are based on the analysis of short to long tandemly repeated sequences (also called micro-satellites, up to 9 bp, and minimicro-satellites, more than 9 bp in length). An assay is defined by a set of loci spread throughout the bacterial genome (16). Previous studies of the
polymor-* Corresponding author. Mailing address: Institut de Ge´ne´tique et Microbiologie, Baˆt 400, Universite´ Paris-Sud, 91405 Orsay cedex, France. Phone: 33 1 69 15 30 01. Fax: 33 1 69 15 66 78. E-mail: [email protected].
䌤Published ahead of print on 24 January 2007.
1190
on May 16, 2020 by guest
http://jcm.asm.org/
phism of tandem repeats in L. pneumophila suggested that VNTRs could be used for genotyping (21) in spite of the fact that this species is much more genetically heterogeneous than other species for which MLVA can be regarded as a reference method, such asBacillus anthracis,Mycobacterium tuberculosis,
andYersinia pestis(15, 16, 20). Seven VNTRs have so far been
described and used to type reference strains ofL. pneumophila sg 1 to sg 14 plus 27L. pneumophilastrains isolated from the environment or from patients. The number of repeat units was assessed by measuring the size of PCR products encompassing the VNTR. This initial report was based on the single genome sequence available at that time (L. pneumophilasg 1 type strain Philadelphia-1), with the drawback that a number of primer sets would amplify only a subset of the isolates examined. The release of two additionalL. pneumophilasg 1 genomes (Lens and Paris) (2) provided the basis for identification of additional markers using the methods described by Denoeud and Verg-naud (4; see also the MLVA web service, http://minisatellites .u-psud.fr). As a result, we propose here an extended MLVA comprising up to 10 VNTR loci ofL. pneumophila.
The aims of the present study were to set up a standard MLVA protocol forL. pneumophila sg 1 including standard PCR and gel-based analysis, evaluate the overall performances of the standard MLVA typing system, i.e., typeability (T), re-producibility (R), epidemiological concordance (E), stability (S), and discriminatory power, and establish the first MLVA database forL. pneumophilawhich can be directly queried via the internet.
MATERIALS AND METHODS
Participants.Participants from three institutes representing three European countries took part in the study: Genotyping Public MLVA Site, Institut de Ge´ne´tique et Microbiologie, Universite´ Paris-Sud 11, Orsay, France (center I); National Institute for Infectious Diseases Lazzaro Spallanzani, Rome, Italy (cen-ter II); and the Health Protection Agency Centre for Infections, Respiratory and Systemic Infection Laboratory, London, United Kingdom (center III). Two of these have extensive experience of participation in multicenter studies on Le-gionellatyping (centers II and III), and one was the originating laboratory of an MLVA forLegionella(center I) (21). The study was coordinated by center III.
Guidelines for appropriate use and evaluation of microbial epidemiologic typing systems were followed (25).
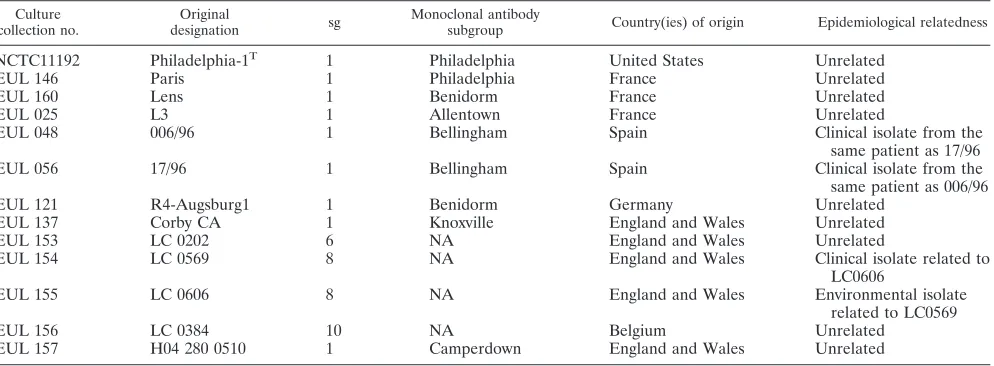
Bacterial strains.The type strain ofL. pneumophila, Philadelphia-1 (NCTC 11192), was obtained from the National Collection of Type Cultures, London, United Kingdom, and the Lens and Paris reference strains were kindly provided by Jerome Etienne, Centre National de Re´fe´rence des Le´gionelles, Lyon, France. All other isolates were obtained from the EWGLILegionellaculture collection, which was established by EWGLI members to facilitate epidemiolog-ical typing studies. All isolates from this collection have a unique number (EUL number; see http://www.ewgli.org) and have previously been characterized by multiple phenotypic and genotypic methods (7–11) (Tables 1 and 2).
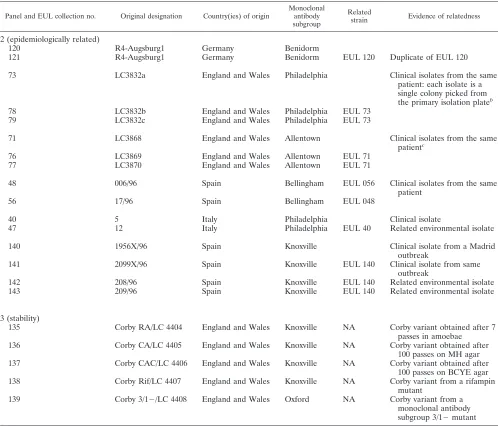
A total of 103 clinical and environmental isolates ofL. pneumophilawere analyzed, including 99L. pneumophilasg 1 strains and 4L. pneumophilastrains from other serogroups. The proficiency panel comprised 10 coded strains ofL. pneumophilafrom the EUL strain collection plus the three reference strains (Table 1). Ninety-five of theL. pneumophilasg 1 isolates, from nine European countries, were selected to produce one epidemiologically unrelated panel of 79 clinical isolates (panel 1), one epidemiologically related panel of 16 isolates (panel 2), and one stability panel of 5 isolates (panel 3). Panel 2 comprises five sets of epidemiologically related isolates and two replicates of the same isolate (EUL 120 and EUL 121), while panel 3 comprises five variants of the same strain (Table 2). Details of strains included in panels 1 to 3 have been published elsewhere (11).
Strategy.For training purposes, the proficiency panel was analyzed by the three centers to establish the reproducibility (R) of the method. The laboratory protocols for MLVA were agreed upon by the three centers prior to the study and are made available to the public at http://bacterial-genotyping.igmors.u-psud .fr/. This pilot panel comprised two pairs of epidemiologically related strains (one pair belonging to sg 1 and one pair belonging to sg 8), and six other epidemio-logically unrelated strains (four belonging to sg 1, one belonging to sg 6, and one belonging to sg 10). The results for these 13 strains were analyzed locally by each center and then sent to center I for interlaboratory comparative analysis. Fol-lowing agreement of optimal analytical settings, the second phase of the study was initiated. This was for all three centers to analyze the 16 strains of panel 2. These data were also analyzed to determine the epidemiological concordance (E). Discriminatory power and stability (S) were determined using panel 1 and panel 3 strains, respectively. Typeability (T) was assessed by the analysis of 99 unique isolates from all panels (i.e., the proficiency panel plus panels 1 and 2, with the exclusion of panel 3).
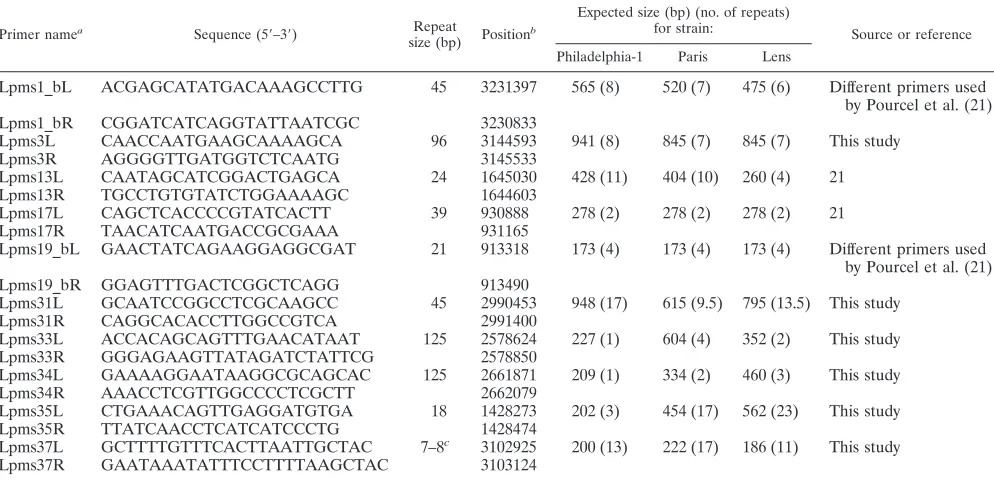
[image:2.585.46.543.81.264.2]VNTR markers.Polymorphic tandem repeats were identified in the sequenced genomes ofL. pneumophilasg 1 Philadelphia-1, Lens, and Paris reference strains using the strain comparison tool developed by Denoeud and Vergnaud (4), available at http://minisatellites.u-psud.fr/. Ten VNTR markers were used for the present study, including four from the previously described set of nine VNTR markers (21). The primers used to amplify the markers are designated Lpms1_b, Lpms3, Lpms13, Lpms17, Lpms19_b, Lpms31, Lpms33, Lpms34, Lpms35, and
TABLE 1. L. pneumophilaproficiency panel and three reference strainsa
Culture collection no.
Original
designation sg
Monoclonal antibody
subgroup Country(ies) of origin Epidemiological relatedness
NCTC11192 Philadelphia-1T 1 Philadelphia United States Unrelated
EUL 146 Paris 1 Philadelphia France Unrelated
EUL 160 Lens 1 Benidorm France Unrelated
EUL 025 L3 1 Allentown France Unrelated
EUL 048 006/96 1 Bellingham Spain Clinical isolate from the
same patient as 17/96
EUL 056 17/96 1 Bellingham Spain Clinical isolate from the
same patient as 006/96
EUL 121 R4-Augsburg1 1 Benidorm Germany Unrelated
EUL 137 Corby CA 1 Knoxville England and Wales Unrelated
EUL 153 LC 0202 6 NA England and Wales Unrelated
EUL 154 LC 0569 8 NA England and Wales Clinical isolate related to
LC0606
EUL 155 LC 0606 8 NA England and Wales Environmental isolate
related to LC0569
EUL 156 LC 0384 10 NA Belgium Unrelated
EUL 157 H04 280 0510 1 Camperdown England and Wales Unrelated
a
NCTC, National Collection of Type Cultures; EUL, European UnionLegionella;T
, type strain; NA, not applicable.
on May 16, 2020 by guest
http://jcm.asm.org/
Lpms37, as shown in Table 3. Lpms1_b and Lpms19_b correspond to new sets of primers designed for the Lpms1 and Lpms19 VNTR loci described in ref-erence 21.
DNA extraction and PCR amplification.Genomic DNA from all EUL strains and the reference strains was extracted by the coordinating laboratory as de-scribed previously (11) and distributed by courier. Oligonucleotide primers tar-geting the 5⬘and 3⬘flanking regions of VNTR loci were used for amplification. The following conditions were used. PCRs were performed with 15-l volumes containing 1 to 5 ng of DNA, 1⫻reaction buffer, 1.5 mM MgCl2, 1 U ofTaq DNA polymerase (Roche, Promega, or Invitrogen), 200M concentrations of each deoxynucleoside triphosphate (dNTP), 0.3M concentrations of each flanking primer (Eurogentec or MWG Biotech). Amplification was locally per-formed with different thermocyclers (including PTC 200 or PTC 225 DNA Engine [MJ Research] and MyCycler [Bio-Rad]) using the following conditions: initial denaturation cycle for 5 min at 94°C, 35 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 60°C, and elongation for 45 s at 72°C, plus a final elongation step for 10 min at 72°C.
Unless otherwise stated, 3l of PCR products was separated in a 2% agarose (UltraPure electrophoresis grade; Invitrogen) gel or in a 4% agarose gel
com-prising 2% Metaphor (FMC Bioproducts-Cambrex) plus 2% regular agarose (as above) for Lpms37. Electrophoresis was performed on 20-cm-wide gels made in 0.5⫻Tris-borate-EDTA buffer (Sigma), run at 8 V/cm. For each PCR run, a reference strain (L. pneumophilaPhiladelphia-1, Paris, or Lens) was included. DNA size markers, including the 100-bp ladder (routinely) or 20-bp ladder (only for Lpms37), were from Bio-Rad, MBI Fermentas, or Euromedex. The gels were stained after the run in 0.5 to 1.0g/ml ethidium bromide for 15 to 30 min, rinsed with water, and photographed under UV illumination.
[image:3.585.43.541.80.506.2]Agarose gel image analysis.The band size was determined using the software Quantity One v. 4.2.1 (Bio-Rad; available in center II) or the BioNumerics software v. 4.0 or v. 4.5 (Applied Maths; available in centers I and III). First, the position of the cursor relative to the DNA band was adjusted to achieve optimum size matching with theL. pneumophilaPhiladelphia-1, Lens, or Paris reference strain used as an internal control; then the cursor was similarly positioned for all the other strains run in the same gel. Size assignments were confirmed by visual inspection of gels and comparative analysis of strains for each marker. The images produced by the three centers were inspected by one center “ex post,” and then differences or errors in data reporting on this training set were adjusted and/or corrected.
TABLE 2. Characteristics ofL. pneumophilasg1 strains used to assess epidemiological concordance and stability of MLVAa
Panel and EUL collection no. Original designation Country(ies) of origin
Monoclonal antibody subgroup
Related
strain Evidence of relatedness
2 (epidemiologically related)
120 R4-Augsburg1 Germany Benidorm
121 R4-Augsburg1 Germany Benidorm EUL 120 Duplicate of EUL 120
73 LC3832a England and Wales Philadelphia Clinical isolates from the same
patient: each isolate is a single colony picked from the primary isolation plateb
78 LC3832b England and Wales Philadelphia EUL 73
79 LC3832c England and Wales Philadelphia EUL 73
71 LC3868 England and Wales Allentown Clinical isolates from the same
patientc
76 LC3869 England and Wales Allentown EUL 71
77 LC3870 England and Wales Allentown EUL 71
48 006/96 Spain Bellingham EUL 056 Clinical isolates from the same
patient
56 17/96 Spain Bellingham EUL 048
40 5 Italy Philadelphia Clinical isolate
47 12 Italy Philadelphia EUL 40 Related environmental isolate
140 1956X/96 Spain Knoxville Clinical isolate from a Madrid
outbreak
141 2099X/96 Spain Knoxville EUL 140 Clinical isolate from same
outbreak
142 208/96 Spain Knoxville EUL 140 Related environmental isolate
143 209/96 Spain Knoxville EUL 140 Related environmental isolate
3 (stability)
135 Corby RA/LC 4404 England and Wales Knoxville NA Corby variant obtained after 7 passes in amoebae 136 Corby CA/LC 4405 England and Wales Knoxville NA Corby variant obtained after
100 passes on MH agar 137 Corby CAC/LC 4406 England and Wales Knoxville NA Corby variant obtained after
100 passes on BCYE agar 138 Corby Rif/LC 4407 England and Wales Knoxville NA Corby variant from a rifampin
mutant
139 Corby 3/1⫺/LC 4408 England and Wales Oxford NA Corby variant from a
monoclonal antibody subgroup 3/1⫺mutant
a
NA, not applicable; MH, Mueller-Hinton; BCYE, buffered charcoal yeast extract. b
EUL 073, 078, and 079 are each a single colony picked from the primary isolation plate. c
EUL 071 was isolated from sputum by direct culture, EUL 076 was isolated from sputum via amoebal culture, and EUL 077 was isolated from feces by direct culture.
on May 16, 2020 by guest
http://jcm.asm.org/
Nomenclature and description of MLVA profiles.The repeat length and num-ber of repetitions was determined in the three sequenced genomes available at Columbia Genome Center (http://genome3.cpmc.columbia.edu/⬃legion/g_info .html) or Institut Pasteur (http://genolist.pasteur.fr/LegioList/) using the Micro-bial Tandem Repeats Database (http://minisatellites.u-psud.fr) (4, 16). Amplifi-cation of DNA from these three reference strains using primers described in Table 3 produced amplicons of the expected size. The number of repeats in new alleles was estimated by subtracting the invariable flanking region from the amplicon size and then dividing by the repeat unit length, as determined for reference strain Philadelphia-1. Intermediate-sized alleles (which may result from intermediate-size repeat units or from small deletions in the flanking sequence) were reported as half-sized, when observed.
The polymorphism index of individual or combined VNTR loci was calculated using panel 1 strains and the Hunter-Gaston diversity index (HGDI) (14), an application of the Simpson’ s index of diversity (24). The allelic profile for 8 loci (MLVA-8) was defined as the number of repeats at each VNTR locus in the order Lpms1_b, Lpms3, Lpms13, Lpms17, Lpms19_b, Lpms33, Lpms34, and Lpms35. The not amplified designation was given when no amplification was repeatedly observed at a given locus. The null (0) allele designates a locus that contains both flanking sequences but no repeat unit.
DNA sequence analysis.The full-length sequences of selected PCR products were determined on both strands following DNA purification by the QIAquick PCR purification kit (QIAGEN). Sequencing reactions were performed using the BigDye terminator technology according to the manufacturer’s recommen-dation (Applied Biosystems), and products were analyzed in an ABI 3100 cap-illary electrophoresis system equipped with the POP 4 matrix (Applied Biosys-tems). Data obtained with forward and reverse sequencing primers were combined, and sequences were manually aligned. DNA sequences have been deposited in the EMBL Nucleotide Sequence Database (see “Nucleotide se-quence accession numbers” below).
Criteria for evaluation ofL. pneumophilasg 1 MLVA.Standard efficacy criteria ofL. pneumophilasg 1 MLVA, including typeability (T), reproducibility (R), stability (S), epidemiological concordance (E), and discriminatory power (ex-pressed asHGDI) were determined as reported elsewhere (25).
Nucleotide sequence accession numbers.DNA sequences have been deposited in the EMBL Nucleotide Sequence Database under accession numbers AM420643 to AM420666 as follows (the corresponding strain name is shown in parentheses): for Lpms1 sequences, AM 420643 (EUL 103), AM 420644 (EUL 031), AM 420645 (EUL 048), AM 420646 (EUL 087), and AM 420647 (ATCC
35096); for Lpms17 sequences, AM 420648 (ATCC 33154), AM 420649 (EUL 052), and AM 420650 (ATCC 33290); for Lpms31 sequences, AM 420651 (EUL 006), AM 420652 (EUL 027), AM 420653 (EUL 063), AM 420654 (EUL 135), AM 420655 (Paris), AM 420656 (Lens), AM 420657 (EUL 111), AM 420658 (Philadelphia), AM 420659 (EUL 048), and AM 420660 (EUL 070); and for Lpms37 sequences, AM 420661 (Paris), AM 420662 (Lens), AM 420663 (Phil-adelphia), AM 420664 (EUL 111), AM420665 (EUL 032), and AM 420666 (EUL 076). Sequence data are available at http://bacterial-genotyping.igmors.u-psud.fr/.
RESULTS
Selection of VNTR markers and allele assignment.With the complete sequencing of threeL. pneumophilagenomes, it be-came possible to identify new polymorphic tandem repeats by the strain comparison tool available at http://minisatellites .u-psud.fr/ (4). A large proportion of tandem repeats was present in one or two strains only and, therefore, could not be used in an MLVA scheme (data not shown).
Among the nine previously described VNTR markers (21), four (Lpms1, Lpms13, Lpms17, and Lpms19) yielded amplifica-tion products of the expected size in all reference isolates and were retained for the MLVA scheme. New optimized primers were designed for Lpms1 and Lpms19 (referred to as Lpms1_b and Lpms19_b in Table 3), taking into account the published sequences for the Paris and Lens genomes. Six new VNTRs were also evaluated, namely Lpms3 (96-bp repeat unit), Lpms31 (45-bp repeat unit), Lpms33 (125-bp repeat unit), Lpms34 (125-bp re-peat unit), Lpms35 (18-bp rere-peat unit), and Lpms37 (7- to 8-bp repeat unit) (Table 3). The number of repeated units at each locus in the sequenced genomes is shown in Table 3.
[image:4.585.44.541.82.322.2]Analysis of all markers for strains included in the profi-ciency, epidemiologically related, and stability panels is shown
TABLE 3. Oligonucleotide primers used and VNTRs analyzed in this study
Primer namea
Sequence (5⬘–3⬘) Repeat
size (bp) Position b
Expected size (bp) (no. of repeats)
for strain: Source or reference
Philadelphia-1 Paris Lens
Lpms1_bL ACGAGCATATGACAAAGCCTTG 45 3231397 565 (8) 520 (7) 475 (6) Different primers used by Pourcel et al. (21)
Lpms1_bR CGGATCATCAGGTATTAATCGC 3230833
Lpms3L CAACCAATGAAGCAAAAGCA 96 3144593 941 (8) 845 (7) 845 (7) This study
Lpms3R AGGGGTTGATGGTCTCAATG 3145533
Lpms13L CAATAGCATCGGACTGAGCA 24 1645030 428 (11) 404 (10) 260 (4) 21
Lpms13R TGCCTGTGTATCTGGAAAAGC 1644603
Lpms17L CAGCTCACCCCGTATCACTT 39 930888 278 (2) 278 (2) 278 (2) 21
Lpms17R TAACATCAATGACCGCGAAA 931165
Lpms19_bL GAACTATCAGAAGGAGGCGAT 21 913318 173 (4) 173 (4) 173 (4) Different primers used by Pourcel et al. (21)
Lpms19_bR GGAGTTTGACTCGGCTCAGG 913490
Lpms31L GCAATCCGGCCTCGCAAGCC 45 2990453 948 (17) 615 (9.5) 795 (13.5) This study
Lpms31R CAGGCACACCTTGGCCGTCA 2991400
Lpms33L ACCACAGCAGTTTGAACATAAT 125 2578624 227 (1) 604 (4) 352 (2) This study
Lpms33R GGGAGAAGTTATAGATCTATTCG 2578850
Lpms34L GAAAAGGAATAAGGCGCAGCAC 125 2661871 209 (1) 334 (2) 460 (3) This study
Lpms34R AAACCTCGTTGGCCCCTCGCTT 2662079
Lpms35L CTGAAACAGTTGAGGATGTGA 18 1428273 202 (3) 454 (17) 562 (23) This study
Lpms35R TTATCAACCTCATCATCCCTG 1428474
Lpms37L GCTTTTGTTTCACTTAATTGCTAC 7–8c 3102925 200 (13) 222 (17) 186 (11) This study
Lpms37R GAATAAATATTTCCTTTTAAGCTAC 3103124
a
Lpms stands forLegionella pneumophilaminimicrosatellite (21). b
Positions (5⬘end) of all primers are according to the numbering of the reference sequence Philadelphia-1 (3). c
Existence in some strains of two different repeat units (7 or 8 bp long).
on May 16, 2020 by guest
http://jcm.asm.org/
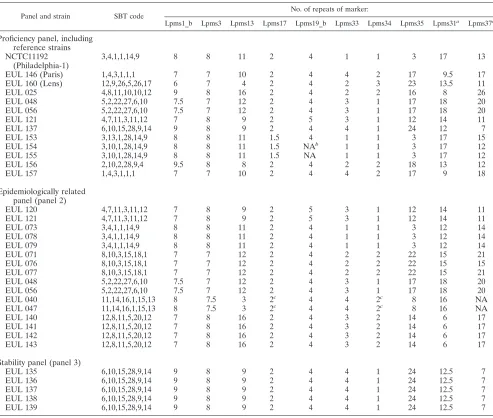
in Table 4. Half-sized alleles were observed for Lpms1_b, Lpms3, Lpms17, and Lpms31. Initially, correct assignment of the number of repeats was not straightforward for Lpms31, because of the relatively high frequency of half-size alleles, and for Lpms37, because of the existence in some strains of two different repeat units (7 bp or 8 bp long), as observed upon sequencing of selected alleles (Fig. 1). Notably, intermediate-size alleles were observed for Lpms31 also in reference strains Paris and Lens.
For all markers, except Lpms3 and Lpms31, PCR products ranged in size between 151 and 724 bp (http: //bacterial-genotyping.igmors.u-psud.fr/) and could easily be resolved by agarose gel electrophoresis. Moreover, the 96-bp repeat length of Lpms3 provided good resolution also for this marker, despite the occasional occurrence of half-sized alleles (⫾48 bp relative to the repeat unit). In the case of Lpms31
(45-bp repeat size), some of the observed alleles exceeded 1,000 bp, and extensive electrophoretic separation was re-quired for precise size estimation.
Thus, an MLVA scheme is proposed, including 8 markers, which allows unambiguous type assignment using agarose gels (MLVA-8, including Lpms1_b, Lpms3, Lpms13, Lpms17, Lpms19_b, Lpms33 Lpms34, and Lpms35). The addition of the highly informative Lpms31 and Lpms37 loci (MLVA-10) re-quires capillary electrophoresis-based fragment analysis or se-quencing for unambiguous type assignment (sequences are available at http://bacterial-genotyping.igmors.u-psud.fr/).
[image:5.585.46.539.79.497.2]Reproducibility.MLVA-8 was initially performed on a pro-ficiency panel of 10 strains from the EUL collection plus 3 reference strains (Table 1) in the three centers to assess the reproducibility of the method. The reference strains Philadel-phia-1, Paris, and Lens were analyzed together with the 10
TABLE 4. MLVA analysis of the proficiency, epidemiologically related, and stability panels
Panel and strain SBT code
No. of repeats of marker:
Lpms1_b Lpms3 Lpms13 Lpms17 Lpms19_b Lpms33 Lpms34 Lpms35 Lpms31a Lpms37a
Proficiency panel, including reference strains NCTC11192
(Philadelphia-1)
3,4,1,1,14,9 8 8 11 2 4 1 1 3 17 13
EUL 146 (Paris) 1,4,3,1,1,1 7 7 10 2 4 4 2 17 9.5 17
EUL 160 (Lens) 12,9,26,5,26,17 6 7 4 2 4 2 3 23 13.5 11
EUL 025 4,8,11,10,10,12 9 8 16 2 4 2 2 16 8 26
EUL 048 5,2,22,27,6,10 7.5 7 12 2 4 3 1 17 18 20
EUL 056 5,2,22,27,6,10 7.5 7 12 2 4 3 1 17 18 20
EUL 121 4,7,11,3,11,12 7 8 9 2 5 3 1 12 14 11
EUL 137 6,10,15,28,9,14 9 8 9 2 4 4 1 24 12 7
EUL 153 3,13,1,28,14,9 8 8 11 1.5 4 1 1 3 17 15
EUL 154 3,10,1,28,14,9 8 8 11 1.5 NAb 1 1 3 17 12
EUL 155 3,10,1,28,14,9 8 8 11 1.5 NA 1 1 3 17 12
EUL 156 2,10,2,28,9,4 9.5 8 8 2 4 2 2 18 13 12
EUL 157 1,4,3,1,1,1 7 7 10 2 4 4 2 17 9 18
Epidemiologically related panel (panel 2)
EUL 120 4,7,11,3,11,12 7 8 9 2 5 3 1 12 14 11
EUL 121 4,7,11,3,11,12 7 8 9 2 5 3 1 12 14 11
EUL 073 3,4,1,1,14,9 8 8 11 2 4 1 1 3 12 14
EUL 078 3,4,1,1,14,9 8 8 11 2 4 1 1 3 12 14
EUL 079 3,4,1,1,14,9 8 8 11 2 4 1 1 3 12 14
EUL 071 8,10,3,15,18,1 7 7 12 2 4 2 2 22 15 21
EUL 076 8,10,3,15,18,1 7 7 12 2 4 2 2 22 15 15
EUL 077 8,10,3,15,18,1 7 7 12 2 4 2 2 22 15 21
EUL 048 5,2,22,27,6,10 7.5 7 12 2 4 3 1 17 18 20
EUL 056 5,2,22,27,6,10 7.5 7 12 2 4 3 1 17 18 20
EUL 040 11,14,16,1,15,13 8 7.5 3 2c 4 4 2c 8 16 NA
EUL 047 11,14,16,1,15,13 8 7.5 3 2c 4 4 2c 8 16 NA
EUL 140 12,8,11,5,20,12 7 8 16 2 4 3 2 14 6 17
EUL 141 12,8,11,5,20,12 7 8 16 2 4 3 2 14 6 17
EUL 142 12,8,11,5,20,12 7 8 16 2 4 3 2 14 6 17
EUL 143 12,8,11,5,20,12 7 8 16 2 4 3 2 14 6 17
Stability panel (panel 3)
EUL 135 6,10,15,28,9,14 9 8 9 2 4 4 1 24 12.5 7
EUL 136 6,10,15,28,9,14 9 8 9 2 4 4 1 24 12.5 7
EUL 137 6,10,15,28,9,14 9 8 9 2 4 4 1 24 12.5 7
EUL 138 6,10,15,28,9,14 9 8 9 2 4 4 1 24 12.5 7
EUL 139 6,10,15,28,9,14 9 8 9 2 4 4 1 24 12.5 7
a
Not included in the MLVA-8 typing scheme. b
NA, no amplicon obtained. c
For these markers, weak amplification was obtained only by center I, while no amplicon was obtained by centers II and III.
on May 16, 2020 by guest
http://jcm.asm.org/
EUL isolates. For the three reference strains, the amplicon size estimates by all three centers were consistent with the predicted length of individual VNTRs, as inferred from ge-nome analysis (compare data in Tables 3 and 4). Although the electrophoresis patterns appear to be similar, some discrepan-cies were initially observed between the three centers on the sizing which was achieved by the use of locally available gel image analysis software. These discrepancies were most likely the result of gel overloading, as discussed hereafter. A second difficulty was due to the existence of alleles with unexpected size, incompatible with an exact number of repeats. This is the case, for example, for Lpms1_b in EUL 048 and EUL 056, whose allele size is intermediate between the size of a 7- and 8-repeat allele observed in EUL 157 and reference strain Phil-adelphia, respectively. We have arbitrarily scored this allele 7.5 (Table 4) to clearly reflect this fact. A similar size variability was shown also for other markers, namely Lpms3 and Lpms17. Sequencing of such alleles confirmed the existence of repeats with unexpected sizes. However, for all these markers, the half-sized alleles could easily be distinguished from the full-length counterparts by visual inspection of the gel.
All images were sent to center I, where sizing was repeated by using BioNumerics software as described above. The correct position of the cursor on a given gel was deduced from the reference strain lane, and minor adjustments were made, tak-ing into account the amount of DNA present in the band. Data from all isolates tested by the three laboratories were in com-plete agreement (R⫽1.00 for all VNTR markers).
Epidemiological concordance. Panel 2, including 16 epide-miologically related isolates that had previously been typed by different DNA-based techniques (7–9), were genotyped by MLVA in the three centers (Table 4). EUL 048, EUL 056, and EUL 121 were also retested, since they were previously in-cluded in the proficiency panel. Amplicons were obtained by the three centers for all markers except Lpms17, Lpms34, and Lpms37 in EUL 040 and EUL 047. In fact, no amplification
was obtained for these isolates by centers II and III, while a weak amplification was observed by center I using a prepara-tion of highly concentratedTaqDNA polymerase (SilverStar; Eurogentec).
Sizing was performed independently by each center, and then images were reanalyzed by center I. Table 4 shows the compiled data for 10 VNTR markers and comparison with SBT profiles. The epidemiological concordance was excellent for all VNTR markers.Evalues of 1.00 were determined for all markers except Lpms37 (E⫽0.83). This high epidemiological concordance was also seen in other sets of epidemiologically related strains, including those from outbreaks in two Paris hospitals described in the study by Pourcel et al. (21) (our unpublished data).
Stability.Analysis of five variants (EUL 135 to EUL 139) in the stability panel (panel 3 in Table 4) resulted in identical MLVA-8 allelic profiles, namely 9,8,9,2,4,4,1,24 (S⫽1.00 for all markers).
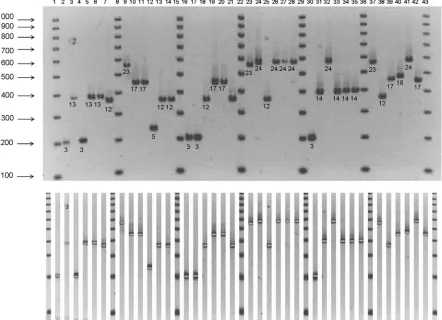
[image:6.585.117.474.71.267.2]Strain diversity and discriminatory power of MLVA. Esti-mates of individual and pooled diversity index (HGDI) values obtained with the 8 VNTRs were determined by using panel 1, which is composed of 79 unrelated isolates. Figure 2 shows the typical organization of a gel, here for marker Lpms35, on which one reference strain (Philadelphia-1 or Lens) is run next to a group of 5 isolates. On a 20-cm-wide gel, up to 30 isolates can simultaneously be analyzed for a single VNTR marker. The data for panel 1 isolates are presented in Table 5 accord-ing to the result of an MLVA clusteraccord-ing analysis performed using Hamming’s distance (the categorical coefficient) and the unweighted pair group method with arithmetic mean cluster-ing method. The SBT code is indicated for comparison. A simplified estimate of the concordance between SBT and MLVA-8 is obtained by comparing side by side the codes for each strain, as shown in Table 5. Strains clustered under a single SBT code can show differences in MLVA-8 profile (for example, EUL 081 and EUL 092), and the opposite is observed
FIG. 1. Alignment of Lpms37 repeats and flanking sequences in the three reference strains and three unrelated isolates using ClustalW. Single-nucleotide differences are present in the flanking sequences, whereas in the tandem repeats, variable numbers of 7-bp or 8-bp units are observed. The sequences have been deposited in the EMBL Nucleotide Sequence Database under the following accession numbers: Lens, AM420662; Philadelphia, AM420663; Paris, AM420661; EUL 111, AM420664; EUL 032, AM420665; EUL 076, AM420666.
on May 16, 2020 by guest
http://jcm.asm.org/
for some MLVA-8 groups (for example, EUL 029 and EUL 049). The larger group with the SBT code 1,4,3,1,1,1 is split into two groups by Lpms35, and additional differences are found when Lpms31 and Lpms37 are analyzed. On the con-trary, in the second larger group with MLVA-8 profile 7,8,9,2,5,3,1,12, SBT appears to be more discriminatory.HGDI values for each marker and for MLVA-8 are shown in Table 6. Lpms35 provided high index of discrimination (0.88). Overall, the highHGDIvalues obtained for MLVA-8 (0.93) highlights the good discriminatory power of MLVA. The number of alleles observed for the different markers and their theoretical size are provided as help file in the L. pneumophila MLVA typing site (http://bacterial-genotyping.igmors.u-psud.fr/).
Typeability. Analysis of 99 unique isolates from all panels showed amplification and full typeability (T⫽1.00) for 5 of the 8 VNTR loci, namely Lpms1_b, Lpms3, Lpms13, Lpms33, and Lpms35. In EUL 040 and EUL 047, weak amplification with Lpms17 and Lpms34 was observed by center I, and no ampli-fication was found by the other two centers. Moreover, no amplification was observed in all three centers for Lpms19_b in EUL 154 and EUL 155 (Table 4). All of these results were
scored as failures. Therefore, aTvalue of 0.98 was determined for Lpms17, Lpms19_b, and Lpms34.
DISCUSSION
The primary aim of this study was to evaluate the utility of MLVA as a genotyping method forL. pneumophila, comple-mentary or alternative to SBT and AFLP.
In an attempt to increase the repertoire of previously estab-lished VNTR markers (21), we have compared the three
avail-ableL. pneumophilagenome sequences and have identified 10
common markers for which conserved primers could be unam-biguously derived. Amplification was obtained with almost all of the tested strains. No amplification was observed for marker Lpms19_b in EUL 154 and EUL 155 from the proficiency panel, while Lpms17, Lpms34, and Lpms37 could not consis-tently be amplified in EUL 040 and EUL 047. By comparison with MLVA profiles of previously analyzed strains (21), EUL 040 and EUL 047 would belong to L. pneumophila subsp.
fraseri. Thus, one possible explanation for the failure of
ampli-fication with specific markers may be flanking sequence
varia-FIG. 2. Analysis of the Lpms35 marker in 30L. pneumophilaisolates. Philadelphia-1 (lanes 2, 16, and 30) and Lens (lanes 9, 23, and 37) reference strains were alternately used as internal controls. Lanes and test strains are, respectively: 3, EUL 101; 4, EUL 099; 5, EUL 102; 6, EUL 100; 7, EUL 103; 10, EUL 110; 11, EUL 104; 12, EUL 111; 13, EUL 105; 14, EUL 112; 17, EUL 118; 18, EUL 116; 19, EUL 119; 20, EUL 117; 21, EUL 120; 24, EUL 136; 25, EUL 121; 26, EUL 137; 27, EUL 135; 28, EUL 138; 31, EUL 141; 32, EUL 139; 33, EUL 142; 34, EUL 140; 35, EUL 143; 38, EUL 121; 39, EUL 157; 40, EUL 156; 41, EUL 137; 42, EUL 048. The 100-bp DNA size marker is shown in lanes 1, 8, 15, 22, 29, 36, and 43. The repeat unit size of Lpms35 is 18 bp. The expected sizes for Philadelphia-1 and Lens strains are, respectively, 202 bp (3 repeats, allele 3) and 562 bp (23 repeats, allele 23) (Table 3). The number of repeats is indicated under each band.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:7.585.72.514.71.391.2]tion in these strains compared to the majority of theL.
pneu-mophilasubsp.pneumophilasg 1 strains tested.
In theory, the optimal MLVA scheme for epidemiological studies should comprise a set of markers with variable indices of diversity. Markers with a moderate diversity and small num-ber of alleles (presumably reflecting a low mutation rate) are important to define families, whereas markers with more rapid evolution provide variability inside clonal lineages.
[image:8.585.51.541.81.478.2]There is no fixed rule regarding the number of markers to be used in an MLVA, since the association of individual markers (with variable index of diversity) offers the opportunity of mod-ulating the discriminatory power of MLVA. In its simpler version, MLVA of L. pneumophila sg 1 could consider the association of only five easily manageable markers, namely Lpms13, Lpms19_b, Lpms33, Lpms34, and Lpms35, and ex-clude Lpms_1b, Lpms3, and Lpms17 because of the occur-rence of half-sized alleles. By combining these five VNTR markers, sufficiently high discrimination can be achieved (HGDI⫽0.92, 34 genotypes). However, the addition of three
TABLE 6. HGDIfor individual or combined VNTR loci (MLVA-8) calculated from 79 unrelated isolates ofL. pneumophila
VNTR marker
No. of alleles
or genotypes HGDI
Lpms1_b 6 0.65
Lpms3 3 0.52
Lpms13 10 0.78
Lpms17 2 0.03
Lpms19_b 2 0.30
Lpms33 5 0.70
Lpms34 3 0.64
Lpms35 18 0.88
MLVA-8a 36b 0.93
Lpms31c 11 0.86
Lpms37c 23 0.92
aNumbers of alleles andHGDI were calculated for a combination of 8 (MLVA-8) VNTR markers.
bNumber of genotypes.
[image:8.585.43.283.564.691.2]cNot included in the MLVA-8 typing scheme.
TABLE 5. Allelic profiles of 79 unrelatedL. pneumophilaisolates (panel 1)
Isolate
Allelic profilea
Isolate
Allelic profilea
SBT MLVA-8 SBT MLVA-8
EUL 002 6,10,19,3,19,4 8,8,9,2,4,2,2,21 EUL 070 5,10,22,15,6,2 7.5,7,12,2,4,4,1,16
EUL 072 1,10,19,1,9,4 8,8,9,2,4,2,2,17 EUL 031 5,10,22,15,6,2 7.5,7,12,2,4,4,1,16
EUL 087 2,4,3,10,9,4 9,8,8,2,4,2,2,18 EUL 068 5,1,22,5,6,10 7.5,7,12,2,4,4,1,16
EUL 081 6,10,21,12,9,4 9,8,8,2,4,3,3,24 EUL 086 5,1,22,5,6,10 7.5,7,12,2,4,4,1,16
EUL 092 6,10,21,12,9,4 9,8,8,2,4,3,3,25 EUL 103 5,1,22,26,6,10 7.5,7,7,2,4,4,1,12 EUL 083 6,10,15,24,17,14 9,8,8,2,4,2,3,26 EUL 048 5,2,22,27,6,10 7.5,7,12,2,4,3,1,17 EUL 025 4,8,11,10,10,12 9,8,16,2,4,2,2,16 EUL 111 2,6,17,15,12,8 8,8,10,2,4,3,1,5
EUL 093 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 018 2,6,21,12,12,8 8,8,6,2,4,3,1,6
EUL 082 1,4,3,1,1,1 7,7,10,2,4,4,2,18 EUL 073 3,4,1,1,14,9 8,8,11,2,4,1,1,3
EUL 084 1,4,3,1,1,1 7,7,10,2,4,4,2,18 EUL 069 3,4,1,1,14,9 8,8,11,2,4,1,1,3
EUL 085 1,4,3,1,1,1 7,7,10,2,4,4,2,18 EUL 033 3,6,1,14,14,9 8,8,11,2,4,1,1,3
EUL 088 1,4,3,1,1,1 7,7,10,2,4,4,2,18 EUL 030 3,4,1,14,14,9 8,8,11,2,4,1,1,3
EUL 067 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 020 3,10,1,3,14,9 8,8,11,2,4,1,1,3
EUL 119 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 118 3,4,1,1,14,9 8,8,11,2,4,1,1,3
EUL 017 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 074 1,4,1,1,14,9 8,8,10,2,4,1,1,3
EUL 038 1,10,3,1,1,1 7,7,10,2,4,4,2,17 EUL 098 3,10,1,3,14,9 8,8,12,2,4,1,1,3
EUL 042 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 097 3,10,1,3,14,9 8,8,12,2,4,1,1,3
EUL 043 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 019 3,10,1,3,14,9 8,8,12,2,4,1,1,3
EUL 110 1,6,3,1,1,1 7,7,10,2,4,4,2,17 EUL 052 3,10,3,1,14,9 8,8,11,1.5,4,1,1,3
EUL 055 6,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 099 3,13,1,25,14,9 8,8,11,2,4,1,1,3
EUL 117 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 101 7,6,17,3,13,11 8,8,11,2,5,4,1,13
EUL 060 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 100 7,6,17,3,13,11 7,8,11,2,5,4,1,13
EUL 013 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 102 7,6,17,3,13,11 8,8,10,2,5,4,1,13
EUL 014 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 116 4,7,11,3,11,12 7,8,9,2,5,3,1,12
EUL 016 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 112 1,4,3,1,1,1 7,8,9,2,5,3,1,12
EUL 037 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 105 4,7,11,3,11,12 7,8,9,2,5,3,1,12
EUL 104 1,6,3,1,1,1 7,7,10,2,4,4,2,17 EUL 006 4,7,9,3,11,12 7,8,9,2,5,3,1,12
EUL 001 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 039 4,7,11,3,11,12 7,8,9,2,5,3,1,12
EUL 003 1,4,3,1,1,1 7,7,10,2,4,4,2,17 EUL 050 4,6,11,3,11,12 7,8,9,2,5,3,1,12
EUL 053 1,6,3,1,14,1 7,7,10,2,4,4,2,16 EUL 027 4,7,11,3,11,12 7,8,9,2,5,3,1,12
EUL 032 2,10,18,10,2,1 7,7,10,2,4,4,3,18 EUL 075 4,7,11,3,11,12 7,8,9,2,5,3,1,12
EUL 028 2,3,9,10,2,1 8,7,11,2,4,4,3,27 EUL 120 4,7,11,3,11,12 7,8,9,2,5,3,1,12
EUL 041 2,3,9,10,2,1 8,7,11,2,4,4,3,27 EUL 091 9,6,3,10,22,15 7,8,8,2,4,1,1,11
EUL 004 2,3,9,10,2,1 8,7,11,2,4,4,3,27 EUL 054 8,10,3,15,18,1 7,7,13,2,4,2,2,22
EUL 008 2,3,9,10,2,1 8,7,9,2,4,4,3,26 EUL 071 8,10,3,15,18,1 7,7,12,2,4,2,2,22
EUL 029 2,2,18,15,2,1 8,7,7,2,4,5,3,32 EUL 063 6,4,14,12,2,3 9,8,10,2,4,5,3,28
EUL 049 2,3,18,15,2,1 8,7,7,2,4,5,3,32 EUL 066 6,10,14,10,2,1 9,8,10,2,4,5,3,28
EUL 036 2,3,3,15,2,6 8,7,9,2,4,3,3,18 EUL 051 3,10,15,3,21,7 6,8,10,2,5,2,2,17
EUL 007 2,10,9,13,2,5 8,7,10,2,4,4,3,25 EUL 040 11,14,16,1,15,13 8,7.5,3,2b,4,4,2b,8
EUL 026 2,3,6,10,2,1 8,7,11,2,4,5,3,26
a
Boldface type indicates groups of strains with identical or similar SBT and identical or similar MLVA profiles. b
For these markers and isolates, weak amplification was obtained.
on May 16, 2020 by guest
http://jcm.asm.org/
more markers, namely Lpms_1b, Lpms3, and Lpms17, for which the half-sized alleles could easily be distinguished from the full-length counterparts, improves the informativeness of the assay (HGDI⫽0.93, 36 genotypes) without complicating gel analysis. Although the diversity index for Lpms17 is low, it could be increased when other series of isolates are analyzed. To combine technical simplicity with high discriminatory power, the MLVA-8 scheme proposed in this study comprises an easily manageable combination of both moderately and highly diverse markers. In addition to MLVA-8 markers, Lpms31 and Lpms37 could contribute to further discrimina-tion in light of their high diversity index (0.86 and 0.92, respec-tively) and are here proposed for an extended MLVA panel. However, their correct allele size assignment requires the use of high-precision DNA sizing equipment, such as a sequencer, and the number of repetitions cannot be deduced from the size. It is therefore recommended to type these two markers when additional informativeness is required and to use the size expressed in bp for clustering analyses. Epidemiological con-cordance was not perfect for Lpms37, which may suggest in-stability of this marker. This point, however, warrants future investigation. In a previous study, sequencing of minisatellite Lpms4 in 14 reference strains revealed the existence of alleles made up of a completely different (in size and sequence) re-peat unit (21). Such extreme markers cannot be used in a routine gel-based MLVA. More classically, internal motif vari-ations can be the mark of different evolution pathways for a similarly sized end product causing homoplasy (19). Since MLVA relies on the sizing of PCR products encompassing polymorphic tandem repeats, it does not inform on the se-quence and will not detect homoplasy. It is the analysis of multiple VNTRs which compensates for potentially high homoplasy levels at individual loci.
The index of discrimination of MLVA-8 is greater than that reported for a standardized nonfluorescent AFLP method (7) and similar to that of SBT (10), as also shown by the compar-ison of SBT and MLVA-8 profiles (Table 5). However, phylo-genetic relationships between the different clusters obtained by SBT and MLVA-8 show only 57.2% congruence calculated using the Pearson correlation coefficient in BioNumerics (data not shown). This discrepancy might be explained by horizontal gene transfer and by the different evolution rate of individual SBT and VNTR markers.
The interlaboratory reproducibility of the assay was excel-lent, although in preliminary assays, some discrepancies were occasionally observed in the sizing of amplicons on agarose gels. These were due to the way the cursor was placed on a band and band size was estimated. When placed in the middle of a fat band resulting from DNA overloading, the observed size was generally underestimated, as indicated by the sizing of reference strains. This artifactual behavior is not taken into account by gel analysis software, which automatically positions cursors at the band peak. This error was suppressed by loading similarly low amounts of individual size markers and amplicons on the gel to get thin bands. Minimum experience and quick visual inspection is sufficient to subsequently correctly position cursors, and the proficiency panel of strains gives the oppor-tunity to develop this experience. Distortion of the migration on the gel borders must also be taken into account when sizing is performed.
In conclusion, we emphasize that MLVA-8 is a rapid, repro-ducible, and epidemiologically meaningful typing tool, which can be performed at low cost using regular equipment and agarose gel electrophoresis. To further reduce the cost and improve the accuracy of the assay, especially when large num-bers of isolates need to be analyzed, it may be interesting to perform multiplex PCR using fluorescent primers and to sep-arate the products by means of a DNA sequencer. As previ-ously shown (21), VNTR amplification can be performed on thermolysates from a single L. pneumophila colony, further facilitating and lowering the cost of the procedure. For more precise information, and particularly in the case of phyloge-netic studies, the sequencing of some markers with internal variability is advisable.
In its present form, the MLVA typing scheme cannot be used to investigate otherLegionellaspecies. Indeed, no PCR products were observed when primers were tested on 16 dif-ferent non-L. pneumophilaspecies, indicating that the assay is strictly specific forL. pneumophila. However, the availability of genome sequences and access to well-characterized strain col-lections of otherLegionellaspecies would allow the potential of this technique to be assessed for the epidemiological typing of
non-L. pneumophilaspecies.
A firstLegionella pneumophilaMLVA database can be que-ried online at http://bacterial-genotyping.igmors.u-psud.fr/.
ACKNOWLEDGMENTS
We thank E. Nebuloso for technical assistance in DNA sequencing and T. Harrison for helpful comments on the manuscript. A. Henrard and G. Corbineau performed the typing in University Paris-Sud 11.
Work on the accountability of dangerous pathogens in University Paris-Sud 11 is supported by De´le´gation Ge´ne´rale pour l’Armement and is part of the European defense project CEPA13.14. This work was also supported by grants from Ministero della Salute (ricerca corrente 2005 and ricerca finalizzata 2006) and ISPESL to P.V.
REFERENCES
1.Brenner, D. J., A. G. Steigerwalt, P. Epple, W. F. Bibb, R. M. McKinney, R. W. Starnes, J. M. Colville, R. K. Selander, P. H. Edelstein, and C. W. Moss.1988.Legionella pneumophilaserogroup Lansing 3 isolated from a patient with fatal pneumonia, and descriptions ofL. pneumophilasubsp. pneumophilasubsp. nov.,L. pneumophilasubsp.fraserisubsp. nov., andL. pneumophilasubsp.pasculleisubsp. nov. J. Clin. Microbiol.26:1695–1703. 2.Cazalet, C., C. Rusniok, H. Bru¨ggemann, N. Zidane, A. Magnier, L. Ma, M.
Tichit, S. Jarraud, C. Bouchier, F. Vandenesch, F. Kunst, J. Etienne, P. Glaser, and C. Buchrieser.2004. Evidence in theLegionella pneumophila
genome for exploitation of host cell functions and high genome plasticity. Nat. Genet.36:1165–1173.
3.Chien, M., I. Morozova, S. Shi, H. Sheng, J. Chen, S. M. Gomez, G. Asamani, K. Hill, J. Nuara, M. Feder, J. Rineer, J. J. Greenberg, V. Steshenko, S. H. Park, B. Zhao, E. Teplitskaya, J. R. Edwards, S. Pampou, A. Georghiou, I.-C. Chou, W. Iannuccilli, M. E. Ulz, D. H. Kim, A. Geringer-Sameth, C. Goldsberry, P. Morozov, S. G. Fischer, G. Segal, X. Qu, A. Rzhetsky, P. Zhang, E. Cayanis, P. J. De Jong, J. Ju, S. Kalachikov, H. A. Shuman, and J. J. Russo.2004. The genomic sequence of the accidental pathogen Le-gionella pneumophila. Science305:1966–1968.
4.Denoeud, F., and G. Vergnaud.2004. Identification of polymorphic tandem repeats by direct comparison of genome sequence from different bacterial strains: a Web-based resource. BMC Bioinformatics5:4.
5.Fields, B. S., R. F. Benson, and R. E. Besser.2002.Legionellaand Legion-naires’ disease: 25 years of investigation. Clin. Microbiol. Rev.15:506–526. 6.Fry, N. K., B. Afshar, P. Visca, D. Jonas, J. Duncan, E. Nebuloso, A. Underwood, and T. G. Harrison.2005. Assessment of fluorescent amplified fragment length polymorphism analysis for epidemiological genotyping of
Legionella pneumophilaserogroup 1. Clin. Microbiol. Infect.11:704–712. 7.Fry, N. K., S. Alexiou-Daniel, J. M. Bangsborg, S. Bernander, M. Castellani
Pastoris, J. Etienne, B. Forsblom, V. Gaia, J. H. Helbig, D. Lindsay, P. Christian Lu¨ck, C. Pelaz, S. A. Uldum, and T. G. Harrison.1999. A multi-center evaluation of genotypic methods for the epidemiologic typing of
Legionella pneumophilaserogroup 1: results of a pan-European study. Clin. Microbiol. Infect.5:462–477.
on May 16, 2020 by guest
http://jcm.asm.org/
8.Fry, N. K., J. M. Bangsborg, A. Bergmans, S. Bernander, J. Etienne, L. Franzin, V. Gaia, P. Hasenberger, B. Baladron Jimenez, D. Jonas, D. Lindsay, S. Mentula, A. Papoutsi, M. Struelens, S. A. Uldum, P. Visca, W. Wannet, and T. G. Harrison.2002. Designation of the European Working Group on Legionella infection (EWGLI) amplified fragment length poly-morphism types ofLegionella pneumophilaserogroup 1 and results of inter-centre proficiency testing using a standard protocol. Eur. J. Clin. Microbiol. Infect. Dis.21:722–728.
9.Fry, N. K., J. M. Bangsborg, S. Bernander, J. Etienne, B. Forsblom, V. Gaia, P. Hasenberger, D. Lindsay, A. Papoutsi, C. Pelaz, M. Struelens, S. A. Uldum, P. Visca, and T. G. Harrison.2000. Assessment of intercentre re-producibility and epidemiological concordance ofLegionella pneumophila
serogroup 1 genotyping by amplified fragment length polymorphism analysis. Eur. J. Clin. Microbiol. Infect. Dis.19:773–780.
10.Gaia, V., N. K. Fry, B. Afshar, P. C. Lu¨ck, H. Meugnier, J. Etienne, R. Peduzzi, and T. G. Harrison.2005. Consensus sequence-based scheme for epidemiological typing of clinical and environmental isolates ofLegionella pneumophila. J. Clin. Microbiol.43:2047–2052.
11.Gaia, V., N. K. Fry, T. G. Harrison, and R. Peduzzi.2003. Sequence-based typing ofLegionella pneumophilaserogroup 1 offers the potential for true portability in legionellosis outbreak investigation. J. Clin. Microbiol. 41:
2932–2939.
12.Harrison, T. G.2005. Legionella, p. 1761–1785.InS. P. Borriello, P. R. Murray, and G. Funke (ed.), Topley & Wilson’s Microbiology & Microbial Infections. Hodder Arnold, London, United Kingdom.
13.Helbig, J. H., S. Bernander, M. Castellani Pastoris, J. Etienne, V. Gaia, S. Lauwers, D. Lindsay, P. C. Lu¨ck, T. Marques, S. Mentula, M. F. Peeters, C. Pelaz, M. Struelens, S. A. Uldum, G. Wewalka, and T. G. Harrison.2002. Pan-European study on culture-proven Legionnaires’ disease: distribution of
Legionella pneumophilaserogroups and monoclonal subgroups. Eur. J. Clin. Microbiol. Infect. Dis.21:710–716.
14.Hunter, P. R., and M. A. Gaston.1988. Numerical index of the discrimina-tory ability of typing systems: an application of Simpson’s index of diversity. J. Clin. Microbiol.26:2465–2466.
15.Le Fle`che, P., M. Fabre, F. Denoeud, J. L. Koeck, and G. Vergnaud.2002. High resolution, on-line identification of strains from theMycobacterium tuberculosiscomplex based on tandem repeat typing. BMC Microbiol.2:37. 16.Le Fle`che, P., Y. Hauck, L. Onteniente, A. Prieur, F. Denoeud, V. Ramisse,
P. Sylvestre, G. Benson, F. Ramisse, and G. Vergnaud.2001. A tandem repeats database for bacterial genomes: application to the genotyping of
Yersinia pestisandBacillus anthracis. BMC Microbiol.1:2.
17.Lindstedt, B. A.2005. Multiple-locus variable number tandem repeats anal-ysis for genetic fingerprinting of pathogenic bacteria. Electrophoresis26:
2567–2582.
18.Maiden, M. C., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell, R. Urwin, Q. Zhang, J. Zhou, K. Zurth, D. A. Caugant, I. M. Feavers, M. Achtman, and B. G. Spratt.1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. USA95:3140–3145.
19.Olsen, K. M.1999. Minisatellite variation in a single-copy nuclear gene: phylogenetic assessment of repeat length homoplasy and mutational mech-anism. Mol. Biol. Evol.16:1406–1409.
20.Pourcel, C., F. Andre´-Mazeaud, H. Neubauer, F. Ramisse, and G. Vergnaud.
2004. Tandem repeats analysis for the high resolution phylogenetic analysis ofYersinia pestis. BMC Microbiol.4:22.
21.Pourcel, C., Y. Vidgop, F. Ramisse, G. Vergnaud, and C. Tram.2003. Char-acterization of a tandem repeat polymorphism inLegionella pneumophila
and its use for genotyping. J. Clin. Microbiol.41:1819–1826.
22.Samrakandi, M. M., S. L. G. Cirillo, D. A. Ridenour, L. E. Bermudez, and J. D. Cirillo.2002. Genetic and phenotypic differences betweenLegionella pneumophilastrains. J. Clin. Microbiol.40:1352–1362.
23.Scaturro, M., M. Losardo, G. De Ponte, and M. L. Ricci.2005. Comparison of three molecular methods used for subtyping ofLegionella pneumophila
strains isolated during an epidemic of legionellosis in Rome. J. Clin. Micro-biol.43:5348–5350.
24.Simpson, E. H.1949. Measurement of diversity. Nature163:688. 25.Struelens, M. J., and members of the European Study Group on
Epidemi-ological Markers (ESGEM) of the European Society for Clinical Microbi-ology and Infectious Diseases.1996. Consensus guidelines for appropriate use and evaluation of microbial epidemiologic typing systems. Clin. Micro-biol. Infect.2:2–11.
26.Vergnaud, G., and C. Pourcel.2006. Multiple locus VNTR (variable number of tandem repeat) analysis (MLVA), p. 83–104.InE. Stackebrandt (ed.), Molecular identification, systematics and population structure of pro-karyotes. Springer-Verlag, Berlin, Germany.