Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration
Full text
Figure
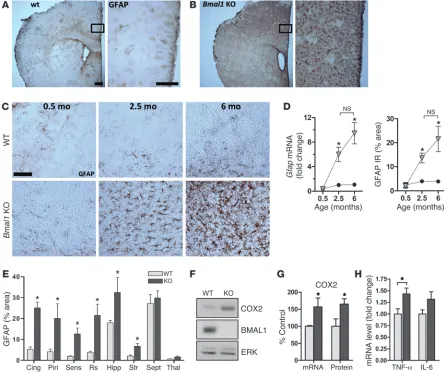
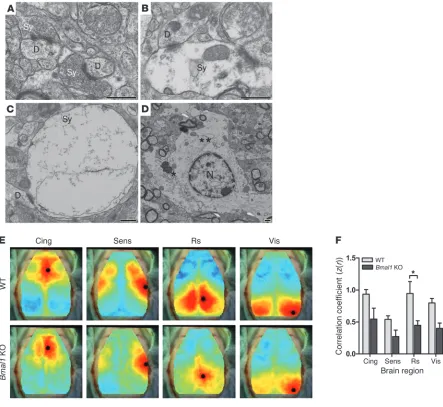
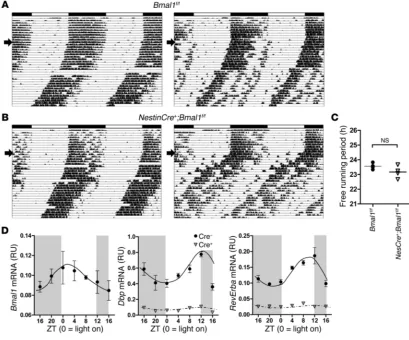
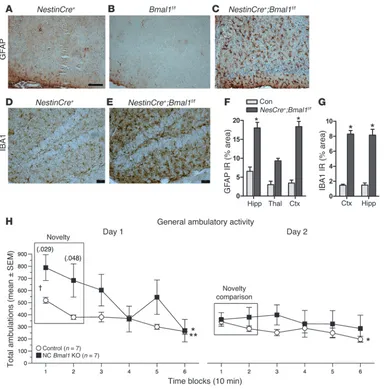
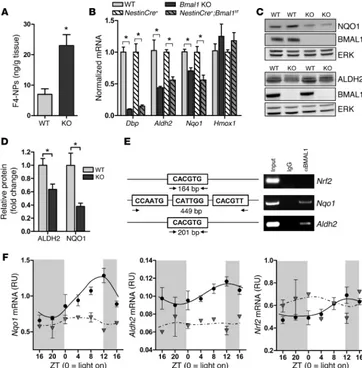
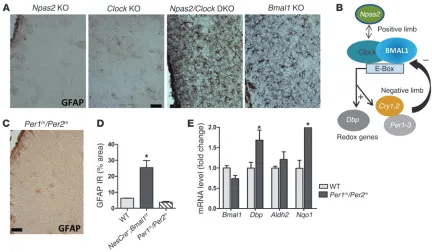
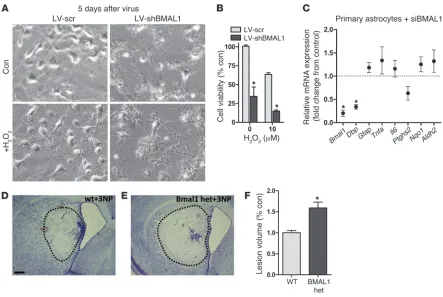
Related documents
To determine whether hyperhomocysteinaemia in humans is associated with impaired endothelium-dependent vasodilatation, studies were performed to investigate the
names and symptoms for selecting the new OTC brand product so it is necessary for pharmaceutical companies to work intensively on brand building as well as the label and
The information consumption side of the information economy must take into account the information needs of decision makers including information on the decision model used
The study aim was to determine the following: (1) sustainability of the impact of EPIQ on pain practice (assessment and management) and clinical (pain intensity) outcomes
We provide training for airline customers on product quality, airport storage and into-plane
All ventilation openings such as that providing combustion air or under floor ventilation and all flues in the cavity wall are checked. If adequate sleeving or
The highest increase in number of released vesicles observed after T cell activation could be attrib- uted to vesicles with a buoyant density of 1.14 1.17 g/ml (Fig.. Moreover,
atherothrombosis.. Laboratory and non-laboratory-based risk prediction models for secondary prevention of cardiovascular disease: the LIPID study. Eur J Cardiovasc Prev Rehabil.
