Ames Laboratory Publications
Ames Laboratory
10-2015
Dynamic Nuclear Polarization Solid-State NMR in
Heterogeneous Catalysis Research
Takeshi Kobayashi
Iowa State University, [email protected]
Frédéric A. Perras
Iowa State University, [email protected]
Igor I. Slowing
Iowa State University, [email protected]
Aaron D. Sadow
Iowa State University, [email protected]
Marek Pruski
Iowa State University, [email protected]
Follow this and additional works at:
http://lib.dr.iastate.edu/ameslab_pubs
Part of the
Chemistry Commons
The complete bibliographic information for this item can be found at
http://lib.dr.iastate.edu/
ameslab_pubs/299
. For information on how to cite this item, please visit
http://lib.dr.iastate.edu/
howtocite.html
.
Dynamic Nuclear Polarization Solid-State NMR in Heterogeneous
Catalysis Research
Abstract
A revolution in solid-state nuclear magnetic resonance (SSNMR) spectroscopy is taking place, attributable to the rapid development of high-field dynamic nuclear polarization (DNP), a technique yielding sensitivity improvements of2–3 orders of magnitude. This higher sensitivity in SSNMR has already impacted materials research, and the implications of new methods on catalytic sciences are expected to be profound.
Disciplines
Chemistry
Comments
Reprinted (adapted) with permission fromACS Catalysis5 (2015): 7055, doi:10.1021/acscatal.5b02039. Copyright 2015 American Chemical Society.
Dynamic Nuclear Polarization Solid-State NMR in Heterogeneous
Catalysis Research
Takeshi Kobayashi,
†Fre
de
́
ric A. Perras,
́
†Igor I. Slowing,
†,‡Aaron D. Sadow,
†,‡and Marek Pruski
*
,†,‡†U.S. DOE Ames Laboratory, Ames, Iowa 50011-3020, United States
‡Department of Chemistry, Iowa State University, Ames, Iowa 50011-3020, United States
1. INTRODUCTION
A revolution in solid-state nuclear magnetic resonance (SSNMR) spectroscopy is taking place, attributable to the rapid development of high-field dynamic nuclear polarization (DNP), a technique yielding sensitivity improvements of2−3 orders of magnitude. This higher sensitivity in SSNMR has already impacted materials research, and the implications of new methods on catalytic sciences are expected to be profound. With their unique sensitivity to the local electronic environment, the nuclear spins can play the role of perfect reporters in the quest for a fundamental understanding of the catalytic processes at the atomic-scale. Indeed, during the last several decades, SSNMR spectroscopy has evolved to become one of the premier analytical methods for structural character-ization of heterogeneous catalytic systems, providing in-depth knowledge about catalyst supports, active sites, reacting molecules, and their interactions.1−3 Noteworthy is also NMR’s ability to investigate a wide range of dynamic processes at solid−liquid and solid−gas interfaces under catalytically relevant pressures and temperatures. The development of sophisticated SSNMR instrumentation, methodology, and advances in theory have endowed the researchers with an ever increasing ability not only to identify and quantify individual chemical sites but also to determine the three-dimensional (3D) catalytic structures, which are often non-periodic and disordered. Of importance are also the active site distribution and the interactions between these sites and the reacting molecules. This area of multidimensional correlation NMR spectroscopy can open new frontiers for the definite characterization of increasingly complex catalytic materials, provided thatthe issue of low sensitivity can be overcome.
The intrinsically low sensitivity of NMR spectroscopy, which operates at the lowest-energy end of the electromagnetic spectrum, poses the fundamental challenge for catalytic applications, where fast detection of small quantities of nuclei is essential. The traditional remedies for NMR’s sensitivity woes include advances in magnet and probe technologies (e.g., cryoprobes, probes capable of ultrafast magic angle spinning, MAS, low-temperature (LT)MAS probes, or probes for in situ studies), isotope enrichment, the use of large sample sizes, the development of new pulse sequences, as well as improvements in data acquisition and processing protocols.
As briefly mentioned, a newly accessible strategy for increasing the polarization of nuclear spins, DNP, can offer unparalleled enhancements of SSNMR signals, and even greater
savings in experimental time of 4−5 orders of magnitude. By enabling the detection of insensitive nuclei, short-lived intermediates, or minute concentrations of molecules and the examination of much smaller surfaces than possible using
conventional CP MAS methods, DNP SSNMR is poised to significantly impact materials research. In the following, we wish to briefly highlight some of the new opportunities that DNP SSNMR brings to thefield of catalysis.
2. DNP SSNMR
A common strategy used in NMR for signal enhancement involves the polarization of the observed nuclei via more sensitive spins possessing larger gyromagnetic ratios (γ). The resulting signal enhancement is determined by the relative sizes of the Zeeman interaction for the excited and observed spins, i.e., by a factor ofε= γexc/γobs. In conventional SSNMR, this
strategy is commonly applied to enhance the polarization of lower-γnuclei (such as13C or15N) via cross-polarization under MAS conditions (CPMAS), resulting in signal-to-noise ratio (S/N) enhancements per scan ofε=γ1H/γ13C≈4 orγ1H/γ15N
≈ 10.4,5 Clearly, given the electron’s much larger γ, considerably higher ε values can be achieved by applying a similar principle to the electron−nucleus spin pairs; this is the basic idea behind DNP. Such hyperpolarization of nuclear spins can potentially elicit NMR signal enhancements ofε=γel/γ1H
≈660 orγel/γ15N≈ 6500, which translates to time savings of (660)2 and (6500)2, respectively. In practice, the technique relies upon saturation of the electron paramagnetic resonance (EPR) line of unpaired electrons by microwave (MW) irradiation and subsequent transfer of polarization to the material’s nuclei of interest. Of key importance is the improvement in S/N per unit of experimental time, given by
εtime = (γ
exc/γobs)(T1,obs/T1,exc)1/2 = ε(T1,obs/T1,exc)1/2, where
T1,obs and T1,exc denote longitudinal relaxation times of the observed and excited nuclei, which often, but not always, increase at lower temperatures.
The DNP effect was predicted by Overhauser6 and confirmed experimentally by Slichter7in the early 1950s. The mechanism at play in those studies is known as the Overhauser effect; however, several other DNP mechanisms were subsequently demonstrated, including the solid effect,8,9 thermal mixing,10,11and the cross-effect.11−15Still, for several decades, the technique remained in a“dormant state”, hindered by the lack of suitable technology compatible with high-field NMR instruments.16The turnaround is mainly attributable to Griffin and his co-workers who, during the past decade, have brought high-field DNP SSNMR to prominence by the combined development and implementation of advanced gyrotron technology,17,18 low-temperature MAS probes,19,20
Received: September 14, 2015
Viewpoint
pubs.acs.org/acscatalysis
© XXXX American Chemical Society 7055 DOI: 10.1021/acscatal.5b02039
and improved polarizing agents.21−24 The progress in DNP instrumentation and methodology has been scrutinized in several excellent reviews.16,25−27In short, current state-of-the-art DNP in solids requires the use of exogenous biradical polarizing agents for which the EPR frequencies of the two unpaired electrons differ roughly by the nuclear Larmor frequency. This EPR frequency separation facilitates DNP by matching the condition for an efficient three-spin polarization exchange involving two unpaired electrons and a nucleus, known as the cross-effect. Measurements involving the detection of low-γ nuclei can be carried out using a single polarization step: electron → low-γ nucleus (referred to as direct DNP) or, more commonly, via protons with the use of a CP transfer: electron→1H→low-γnucleus (indirect DNP). Direct DNP typically leads to lower sensitivity enhancements due to the increased destructive interference between positive and negative DNP enhancement conditions and the more difficult to satisfy cross-effect condition in lower-γnuclei. Direct DNP does, however, offer theoretically higher enhancements and simplifies the experimental methodology, particularly for quadrupolar nuclei. Promising developments have been made in recent years toward efficient direct DNP using trityl radicals.28,29It should be noted that the DNP transfer step is most efficient at low temperatures, and therefore, most experiments are presently carried out at temperatures not exceeding 110 K.
Whereas challenges remain in developing instrumentation and optimal sample formulations to approach the theoretical limits for ε in various classes of materials, the capabilities of DNP SSNMR have already proved revolutionary in several areas of research, with initial efforts being focused on biological systems. Importantly for catalysis, Emsley and others have demonstrated the utility of DNP in the studies of mesoporous silica nanoparticles (MSNs) and metal−organic frameworks
[image:4.625.157.468.64.342.2](MOFs), using the approach termed DNP surface-enhanced NMR spectroscopy (DNP-SENS).30−35Examples are shown in
Figure 1, which depicts DNP-SENS 13C{1H} and 15N{1H} CPMAS spectra of several functional groups deposited at natural isotopic abundance on the surface of mesoporous silica.34The spectra were acquired in 10 min for13C and 3.5 h
for 15N using the bCTbK biradical dissolved in a 1,1,2,2-tetrachloroethane solution, which yieldedεvalues on the order of 80. The best currently known polarizing agents for DNP-SENS are the bulky nitroxide biradicals referred to as TEKPol and AMUPol,23,24which under favorable conditions can afford
ε values of over 200. At present, these conditions include a moderate strength of the magneticfield (e.g., 9.4 T) and a low temperature (≤110 K), and apply to samples in which the radicals can be administered sufficiently close to the observed nuclei (within tens or hundreds of nm). The indirect DNP experiments require that the spin system comprises a sufficiently dense network of dipolar coupled1H spins (either intrinsic in the sample and/or in the solvent) to enable efficient transport of enhanced 1H polarization to the nuclei of
interest.36,37 The samples of catalytic material are typically prepared by wetting the surfaces with a solution of the biradicals in a glass-forming medium to prevent agglomeration of polarizing agents. For TEKPol, which is currently the top performing radical, this involves the use of a 16 mM solution in 1,1,2,2-tetrachloroethane. In general, however, a solvent in which the sample is insoluble should be used and solvents possessing methyl groups should be avoided.38 The global sensitivity enhancements offered by DNP, which are determined by the above-mentioned changes in T1relaxation,
quenching of NMR response by paramagnetic effects (typically on the order of tens of %), the presence of frozen solvent, changes in the efficiency of cross-polarization, and other experimental factors, strongly vary between different samples. A
Figure 1.DNP-enhanced 13C{1H} (B) and 15N{1H} (C) CPMAS spectra of hybrid organic−inorganic silica materials decorated with several catalytic precursors (A) under natural13C and15N abundance. Adapted with permission from ref34.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062
systematic analysis of these contributions for functionalized mesoporous silica material can be found in ref35.
Importantly, Bruker has recently developed commercial DNP SSNMR instruments operated at 9.4, 14.1, and 18.8 T, with the corresponding gyrotron accessories at 263, 395, and 527 GHz, respectively. One of thefirst such spectrometers dedicated to materials research has been installed in 2014 at the Ames Laboratory (Figure 2A). The conventional (“MW off”) and DNP-enhanced (“MW on”) 13C{1H} CPMAS spectra of a standard reference sample (proline, Figure 2B) exemplify the highest enhancement (ε= 260) that can be achieved at 100 K, at least currently, by using AMUPol as a polarizing agent. The instrument uses the lowest of thefields listed above (9.4 T), which at present appears to be the field of choice for DNP applications in catalysis. This point is illustrated in Figure 3, where we compare the 13C{1H} DNP CPMAS spectra taken
back-to-back on 9.4 and 14.1 T instruments for the same sample (MSN material functionalized with 3-(N -phenylureido)-propyl groups, denoted as PUP-MSN) under identical conditions: MAS rateνR= 10 kHz, T = 100 K, and acquisition
time = 11 min per spectrum. We make several key observations: (1) The spectra exhibit field-independent resolution due to inhomogeneous line broadening. Inhomoge-neous broadening is typically encountered in noncrystalline solids due to local disorder, and it lowers the magneticfield dependence of the S/N ratio from (B0)3/2 to B
0. (2) In the
high-field spectrum (Figure 3B), the intensities are distributed within the manifolds of intense MAS sidebands (denoted as
“*”), which interfere with other isotropic resonances. The lower-field spectrum displays minimal interference. (3) Most importantly, the enhancement factorεat 9.4 T is twice as high than at 14.1 T. This is expected: since the line width of the EPR spectrum increases withB0, the degeneracy condition between
the three-spin states involved in the cross-effect becomes more difficult to satisfy at higher magneticfields.25(4) Lastly, the 9.4 T instrument is considerably less expensive. We should also note that the development of improved polarizing agents, MAS capabilities, and optimization schemes will undoubtedly result in higher sensitivity gains at both low and highfields; consider, for example, that theεvalues reported inFigure 3have already been far surpassed by the improved polarizing agents: bCTbK, TEKPol, and AMUPol.
3. DNP SSNMR OF CHALLENGING NUCLEI
One of the major avenues that DNP enables is the study of unreceptive nuclei, such as15N,17O,35Cl,43Ca,89Y, and119Sn. Due to the difficulties in studying these nuclides, which may stem from low natural abundance, low value of γ, broad lineshapes, or any combination of these factors, they are often referred to as“exotic”; although, with DNP, this terminology may soon become obsolete.
For example, one of the most ubiquitous elements in chemistry and materials science, yet one of the most challenging elements for spectroscopic investigation by NMR, is oxygen.39 More than 99.96% of oxygen nuclei are NMR silent, and the scarce NMR active isotope (17O) has a low gyromagnetic ratio (γ1H/γ17O ≈ 7.4), a spin of 5/2 and a moderate quadrupole moment (Q =−25.58 mb). The
second-Figure 2.(A) Gyrotron (left), microwave guide, and NMR magnet (right) of the newly installed 9.4 T DNP SSNMR spectrometer at the Ames Laboratory. Gyrotron control cabinet, cryo-MAS control cabinet, and NMR console are not shown. (B) Comparison between conventional (“MW off”) and DNP-enhanced1H{13C} CPMAS spectra of proline dissolved in a 10 mM AMUPol glycerol-d
[image:5.625.152.475.69.241.2]8/D2O/H2O solution at∼100 K.
Figure 3. Comparison of 13C{1H} DNP CPMAS spectra of PUP-MSNs impregnated with an aqueous solution of TOTAPOL, recorded at 9.4 T (A) and 14.1 T (B) under identical conditions. Asterisks denote the MAS sidebands. “CTAB” refers to cetyltrimethylammo-nium bromide, CH3(CH2)15N(CH3)3Br, typically used as a surfactant
during the synthesis of the MSN materials.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062
[image:5.625.357.564.286.516.2]order quadrupolar interaction prominently affects most 17O NMR spectra, because the bonding of oxygen atoms in many solids produces strong electric field gradients at the nuclei, thereby broadening the NMR lines and further reducing the sensitivity. These properties make the use of DNP particularly attractive.29,40 Indeed, in spite of all of these shortcomings, DNP-enhanced NMR spectroscopy has enabled the measure-ment of 17O NMR spectra at natural isotopic abundance.41,42 For example, DNP-enhanced, natural abundance, 17O{1H}
MAS spectra of inorganic hydroxides were recently obtained in our laboratory, enabling the swift measurements of O−H distances and heteronuclear correlation (HETCOR) spectra.42 Remarkably, DNP has also enabled us to measure thefirst17O spectra of surface hydroxyl groups in natural-abundance silica (Figure 4A).42 Further improvements in sensitivity will undoubtedly enable the measurement of multidimensional
17O NMR data of surfaces and allow the site-specific
characterization of supported catalysts43 and host−guest interactions at natural abundance.
Nitrogen, which is also ubiquitous in materials, including catalysts, is similarly indisposed to NMR studies. 14N NMR spectra suffer from low sensitivity (γ1H/γ14N≈14) and severe quadrupolar line broadening, due to the nuclide’s spin of 1 yielding no sharp central transition. The other NMR-active
isotope of nitrogen,15N, also incurs poor sensitivity, in this case due to the low gyromagnetic ratio (γ1H/γ15N ≈10) and low natural abundance (0.37%). Although a > 10-fold boost in sensitivity has been afforded by an indirect detection of low-γ
15N nuclei through high-γ1H nuclei,44,45
such improvement is often insufficient for the acquisition of15N spectra of catalytic
surfaces or bulk materials with low nitrogen content. The use of DNP SSNMR enabled the measurement of 1D as well as 2D
15N spectra of naturally abundant catalytic species within a
reasonable experimental time. For example,15N DNP SSNMR
measurements made it possible to monitor the chemical reactions on functionalized mesoporous silica surface.34 In another study, DNP-enhanced 1H−15N HETCOR spectra of
Pt2+-loaded metal−organic frameworks (UiO-66-NH2)
pro-vided direct evidence of host−guest interactions between Pt2+
and−NH2groups in UiO-66-NH2(Figure 4D).46Within this
virtual special issue, we also report that commonly-used DNP conditions are compatible with the studies of surface-grafted organotransition metal complexes, such as catalytically active zirconium dimethylamide groups.47 With the improved sensitivity gain owing to the newest polarizing agents, 15N is
indeed no longer an“exotic”nucleus.
[image:6.625.113.510.71.436.2]Most recently, spectra of other traditionally challenging nuclei were recorded using DNP, including59Co MAS spectra
Figure 4. DNP-enhanced NMR experiments on challenging nuclei at natural abundance. (A) A DNP-SENS17O{1H} MAS spectrum of the
mesoporous silica SBA-15 used as catalyst support. (B)119Sn{1H} CPMAS spectra of hydrated and dehydrated Sn-Beta zeolite (top); a conventional
NMR experiment run for over 10 days of acquisition is also shown (bottom). (C)1H−89Y HETCOR spectrum acquired on yttrium doped barium
zirconate. (D)1H−15N HETCOR spectrum of Pt@UiO-66-NH
2MOF. Adapted with permission from refs42,50,52, and46.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062
in a paramagnetically doped inorganic crystalline lattice,48
119Sn{1H} CPMAS and 2D 1H−119Sn HETCOR spectra of
silica-bound surface complexes and Sn-Beta zeolites (Figure 4B),49,50as well as Sn/SnO nanoparticles,5189Y{1H} CPMAS and 2D 1H−89Y HETCOR study of hydrated yttrium doped
barium zirconate (Figure 4C),52 and wide-line, still unpub-lished, 195Pt spectra of Pt2+-loaded MOFs in our laboratory.
The range of nuclei amenable to DNP SSNMR will undoubtedly expand in the near future through further advances in instrumentation and pulse sequences, and the development of tailored sample formulations.
4. CHALLENGING SSNMR EXPERIMENTS
Aside from enabling the studies of rare or unreceptive nuclei, DNP also greatly facilitates the implementation of challenging experiments. The acquisition of conventional 2D homo- or heteronuclear correlation spectra is often unfeasible on catalytic systems without isotope enrichment. However, signal enhance-ments of over 100 approach the benefits obtained from moderate isotopic enrichment when performing 13C−13C
homonuclear correlation experiments, while avoiding the complicated, time-consuming, and expensive syntheses asso-ciated with labeling. Several reports effectively show that
13C−13C through-space or through-bond 2D spectra can be
acquired in a few minutes to hours.53−56An added benefit of natural abundance measurements is that the 3-spin recoupling
effects and dipolar truncation are essentially absent, and carbon−carbon distances may be straightforwardly measured by assuming isolated spin pairs.57 Similarly, 29Si−29Si correlation experiments of the surfaces of functionalized silica nanoparticles have been performed at natural isotopic abundance using DNP.58These experiments will enable studies of the spatial distributions of species on catalyst’s surface. The sensitivity improvements afforded by DNP will also undoubt-edly lead to the detection of 13C−15N correlations at natural
isotopic abundance in the near future (corresponding to only 0.004% of carbon−nitrogen bonding partners). Note that with an enhancement factor of 260, which can already be obtained using the AMUPol biradical, see Figure 2B,24 a natural abundance 13C−15N correlation experiment would have
comparable sensitivity to a conventional 13C{1H} CPMAS
experiment.
Correlation experiments involving quadrupolar nuclei, which have low sensitivity due to the quadrupolar spins’ fastidious response to rf pulses, can also be significantly aided using DNP. For example, homonuclear 27Al−27Al,59,60
[image:7.625.163.464.70.401.2]as well as hetero-nuclear13C−27Al61and29Si−27Al62correlation experiments can be performed in only a few hours. Furthermore, carbon− nitrogen correlation experiments can be performed at natural isotopic abundance in under an hour to a few hours on amino acids by detecting the 13C’s correlation to the overtone transition of 14N.63 Further improvements are expected with
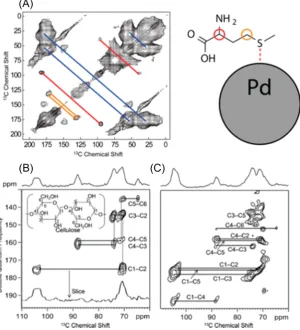
Figure 5.Examples of DNP-enhanced 2D13C−13C correlation SSNMR spectroscopy. (A) Single-quantum spin diffusion spectrum of methionine
and the low-coverage products of its oxidative breakdown on Al2O3-supported Pd nanoparticle catalyst; the cross-peaks are identified on the scheme
to the right where circles identify the points of oxidation and/or breakdown responsible for a given cross-peak (blue in (A) corresponds to unreacted methionine). (B), (C) Double-quantum single-quantum MAS spectra of natural abundance microcrystalline cellulose obtained using 1 ms (B) and 3.5 ms (C) of Post-C7 recoupling. Figures (B) and (C) are reproduced with permission from ref53.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062
the development of ultrahigh field DNP, offering additional exciting prospects for studying the quadrupolar nuclei in catalytic systems.
5. NOVEL APPLICATIONS OFFERED BY DNP SSNMR
The emergence of modern DNP redefines the sensitivity limits of SSNMR spectroscopy and opensfirst-time opportunities for the structural studies of bulk materials, molecular structures, and reactions on exceedingly small catalytic surfaces.
The previously mentioned DNP-SENS approach is now being used to examine materials with progressively smaller surface areas that are beyond the current capabilities of conventional NMR methods. For example, DNP-enhanced
13C{1H} CPMAS experiments enabled the detection of signals
from 13C-enriched sucrose adsorbed on low-surface-area (∼1 m2/g) hydrated tricalcium silicates.64
In our laboratory, we recently used DNP to study molecules reacting on the surface of alumina-supported Pd nanoparticle catalyst. Here, DNP offered >2500-fold time-savings, enabling the detection of 2D
13C−13C spin diffusion (Figure 5A) and INADEQUATE
spectra of low-coverage products from oxidative degradation of13C-enriched methionine adsorbed on the Pd surface.65
To the best of our knowledge, this is thefirst DNP SSNMR study involving catalytic chemistry at the surface of noble metals, which should inspire further similar investigations in this broad area of catalysis. In another study, the DNP-enhanced29Si{1H}
CPMAS spectra of a trace amount of Si catalytic species deposited onγ-Al2O3(∼60 m2/g) were obtained in less than 1
h;66even a week of signal accumulation using a conventional CPMAS experiment was not sufficient to obtain a spectrum with a reasonable S/N ratio. Most recent, still unpublished results from our laboratory indicate that DNP 27Al{1H}
SSNMR signals can be detected from the surface of AlxOy
thinfilms having a surfaceas small as 1 cm2contained in a MAS sample rotor.
DNP-MAS NMR is not only applicable to the studies of surfaces, where a strong contact with the radicals can be obtained, but can also be applied to bulk materials. 1H spin diffusion has been shown, both numerically and experimentally, to lead to the efficient hyperpolarization of the bulk nuclei within microcrystals with diameters on the order of tens of microns.36,37,54This is particularly noteworthy for studying the feedstocks and byproducts of catalytic transformations. See, for example,Figure 5B,C, showing the natural abundance13C−13C double-quantum correlation spectra of cellulose, in which all expected one-bond (spectrum (B), acquired in only 20 min) and one- and two-bond (spectrum (C), acquired in 2 h) correlation peaks are well resolved.53
■
SUMMARY AND OUTLOOKThe hyperpolarization of nuclear spins by DNP forces us to rethink what is possible with SSNMR; note that in many of the studies shown above the DNP spectra acquired within 1 h would require∼2 years of experimental time on a conventional spectrometer. This unprecedented shift in SSNMR’s capa-bilities offers new opportunities for applications in heteroge-neous catalytic systems, which include studies of small surfaces (i.e., materials with surface areas of <1 m2/g), “unreceptive” spin-1/2 and quadrupolar nuclei (e.g., 15N or 17O in
organometallic catalytic groups under natural abundance), elusive intermediates, low-coverage species, and so forth.
In spite of these achievements, DNP is still an emerging technology with a large amount of untapped potential. New frontiers in sensitivity will undoubtedly be opened through further, foreseeable or not imagined, advances in DNP probe capabilities (especially fast MAS at low temperatures, LTMAS67), dedicated pulse sequences, new types of polarizing agents, and improved sample formulations yielding higher ε values and minimizing the intrusion of solvent signals. For example, ultra-LTMAS probes, using helium gas, have been shown to enable the achievement ofε values surpassing 600 with little or no relaxation penalty.68 The LTMAS probes capable of achieving spinning frequencies above 40 kHz will enable further improvements in sensitivity by the use of indirect detection. Pulsed schemes allowing a deeper penetration of hyperpolarized magnetization within microcrystalline com-pounds may greatly expand the scope of DNP. Notably, some of the most recent observations suggest that the Overhauser effect can become a useful mechanism for DNP, with great potential for applications at high magneticfields.69 Similarly, very recently, a promising new class of biradical polarizing agents, known as TEMTriPols, yielded an unprecedented enhancement of ε = 65 at 800 MHz, which also extends efficient DNP to the ultrahighfield regime.70On another front, it has been shown that large molecular weight nitroxide biradicals can offer respectable ε values (≥10) at a temperature of 200 K, suggesting that DNP measurements at even higher temperatures may very well be around the corner.23 The current pace of evolution in thisfield is tremendously fast such that recent breakthroughs in tools and methods rapidly become obsolete and at present the possibilities for new capabilities and impact to applications appear to be endless.
■
AUTHOR INFORMATIONCorresponding Author
*E-mail: [email protected].
Notes
The authors declare no competingfinancial interest.
■
ACKNOWLEDGMENTSWe wish to thank Dr. A.J. Rossini for helpful comments and to Drs. M. Rosay and M.A. Caporini for assistance with acquisition of the spectra inFigure 3. This research is supported by the U.S. Department of Energy (DOE), Office of Science, Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences. Support for F.P. is through a Spedding Fellowship funded by the LDRD program. Ames Laboratory is operated for the DOE by Iowa State University under Contract No. DE-AC02-07CH11358.
■
REFERENCES(1)NMR techniques in catalysis, Chemical Industries Series 55; Bell, A. T.; Pines, A., Eds.; Marcel Dekker: New York, 1994; pp 1−432.
(2) Fernandez, C.; Pruski, M.Top. Curr. Chem.2012,306, 119−188. (3) Koller, H.; Weiß, M.Top. Curr. Chem.2012,306, 189−228. (4) Pines, A.; Gibby, M. G.; Waugh, J. S.J. Chem. Phys.1972,56, 1776−1777.
(5) Stejskal, E. O.; Schaefer, J.; Waugh, J. S.J. Magn. Reson.1977,28, 105−112.
(6) Overhauser, A. W.Phys. Rev.1953,92, 411−415.
(7) Carver, T. R.; Slichter, C. P.Phys. Rev.1953,92, 212−213. (8) Abragam, A.Principles of Nuclear Magnetism; Oxford University Press: New York, 1961; pp 399−401.
(9) Afeworki, M.; Schaefer, J.Macromolecules1992,25, 4092−4096.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062
(10) Wind, R. A.; Duijvestijn, M. J.; van der Lugt, C.; Manenschijn, A.; Vriend, J.Prog. Nucl. Magn. Reson. Spectrosc.1985,17, 33−67.
(11) Hall, D. A.; Maus, D. C.; Gerfen, G. J.; Inati, S. J.; Becerra, L. R.; Dahlquist, F. W.; Griffin, R. G.Science1997,276, 930−932.
(12) Kessenikh, A. V.; Lushchikov, V. I.; Manenkov, A. A.; Taran, Y. V.Soviet Physics-Solid State1963,5, 321−329.
(13) Hwang, C. F.; Hill, D. A.Phys. Rev. Lett.1967,18, 110−112. (14) Wollan, D. S.Phys. Rev. B1976,13, 3671−3685.
(15) Farrar, C. T.; Hall, D. A.; Gerfen, G. J.; Inati, S. J.; Griffin, R. G. J. Chem. Phys.2001,114, 4922−4933.
(16) Griffin, R. G.; Prisner, T. F.Phys. Chem. Chem. Phys.2010,12, 5737−5740.
(17) Becerra, L. R.; Gerfen, G. J.; Bellew, B. F.; Bryant, J. A.; Hall, D. A.; Inati, S. J.; Weber, R. T.; Un, S.; Prisner, T. F.; McDermott, A. E.; Fishbein, K. W.; Kreischer, K. E.; Temkin, R. J.; Singel, D. J.; Griffin, R. G.J. Magn. Reson., Ser. A1995,117, 28−40.
(18) Joye, C. D.; Griffin, R. G.; Hornstein, M. K.; Hu, K.-N.; Kreischer, K. E.; Rosay, M.; Shapiro, M. A.; Sirigiri, J. R.; Temkin, R. J.; Woskov, P. P.IEEE Trans. Plasma Sci.2006,34, 518−523.
(19) Rosay, M.; Lansing, J. C.; Haddad, K. C.; Bachovchin, W. W.; Herzfeld, J.; Temkin, R. J.; Griffin, R. G.J. Am. Chem. Soc.2003,125, 13626−13627.
(20) Barnes, A. B.; Mak-Jurkauskas, M. L.; Matsuki, Y.; Bajaj, V. S.; van der Wel, P. C. A.; DeRocher, R.; Bryant, J.; Sirigiri, J. R.; Temkin, R. J.; Lugtenburg, J.; Herzfeld, J.; Griffin, R. G.J. Magn. Reson.2009, 198, 261−270.
(21) Hu, K.-N.; Yu, H.-h.; Swager, T. M.; Griffin, R. G.J. Am. Chem. Soc.2004,126, 10844−10845.
(22) Song, C.; Hu, K.-N.; Joo, C.-G.; Swager, T. M.; Griffin, R. G.J. Am. Chem. Soc.2006,128, 11385−11390.
(23) Zagdoun, A.; Casano, G.; Ouari, O.; Schwarzwälder, M.; Rossini, A. J.; Aussenac, F.; Yulikov, M.; Jeschke, G.; Coperet, C.; Lesage, A.;́ Tordo, P.; Emsley, L.J. Am. Chem. Soc.2013,135, 12790−12797.
(24) Sauvée, C.; Rosay, M.; Casano, G.; Aussenac, F.; Weber, R. T.; Ouari, O.; Tordo, P.Angew. Chem., Int. Ed.2013,52, 10858−10861. (25) Maly, T.; Debelouchina, G. T.; Bajaj, V. S.; Hu, K.-N.; Joo, C.-G.; Mak-Jurkauskas, M. L.; Sirigiri, J. R.; van der Wel, P. C. A.; Herzfeld, J.; Temkin, R. J.; Griffin, R. G.J. Chem. Phys. 2008,128, 052211.
(26) Rossini, A. J.; Zagdoun, A.; Lelli, M.; Lesage, A.; Coperet, C.;́ Emsley, L.Acc. Chem. Res.2013,46, 1942−1951.
(27) Lee, D.; Hediger, S.; De Paëpe, G.Solid State Nucl. Magn. Reson. 2015,66−67, 6−20.
(28) Michaelis, V. K.; Smith, A. A.; Corzilius, B.; Haze, O.; Swager, T. M.; Griffin, R. G.J. Am. Chem. Soc.2013,135, 2935−2938.
(29) Michaelis, V. K.; Corzilius, B.; Smith, A. A.; Griffin, R. G.J. Phys. Chem. B2013,117, 14894−14906.
(30) Lesage, A.; Lelli, M.; Gajan, D.; Caporini, M. A.; Vitzthum, V.; Miéville, P.; Alauzun, J.; Roussey, A.; Thieuleux, C.; Mehdi, A.; Bodenhausen, G.; Coperet, C.; Emsley, L.́ J. Am. Chem. Soc.2010,132, 15459−15461.
(31) Lelli, M.; Gajan, D.; Lesage, A.; Caporini, M. A.; Vitzthum, V.; Miéville, P.; Heroguel, F.; Rascó ́n, F.; Roussey, A.; Thieuleux, C.; Boualleg, M.; Veyre, L.; Bodenhausen, G.; Coperet, C.; Emsley, L.́ J. Am. Chem. Soc.2011,133, 2104−2107.
(32) Lafon, O.; Rosay, M.; Aussenac, F.; Lu, X.; Trebosc, J.; Cristini,́ O.; Kinowski, C.; Touati, N.; Vezin, H.; Amoureux, J.-P.Angew. Chem., Int. Ed.2011,50, 8367−8370.
(33) Rossini, A. J.; Zagdoun, A.; Lelli, M.; Canivet, J.; Aguado, S.; Ouari, O.; Tordo, P.; Rosay, M.; Maas, W. E.; Coperet, C.; Farrusseng,́ D.; Emsley, L.; Lesage, A.Angew. Chem., Int. Ed.2012,51, 123−127. (34) Zagdoun, A.; Casano, G.; Ouari, O.; Lapadula, G.; Rossini, A. J.; Lelli, M.; Baffert, M.; Gajan, D.; Veyre, L.; Maas, W. E.; Rosay, M.; Weber, R. T.; Thieuleux, C.; Coperet, C.; Lesage, A.; Tordo, P.;́ Emsley, L.J. Am. Chem. Soc.2012,134, 2284−2291.
(35) Kobayashi, T.; Lafon, O.; Thankamony, A. S. L.; Slowing, I. I.; Kandel, K.; Carnevale, D.; Vitzthum, V.; Vezin, H.; Amoureux, J.-P.; Bodenhausen, G.; Pruski, M.Phys. Chem. Chem. Phys.2013,15, 5553− 5562.
(36) van der Wel, P. C. A.; Hu, K.-N.; Lewandowski, J.; Griffin, R. G. J. Am. Chem. Soc.2006,128, 10840−10846.
(37) Lafon, O.; Thankamony, A. S. L.; Kobayashi, T.; Carnevale, D.; Vitzthum, V.; Slowing, I. I.; Kandel, K.; Vezin, H.; Amoureux, J.-P.; Bodenhausen, G.; Pruski, M.J. Phys. Chem. C2013,117, 1375−1382. (38) Zagdoun, A.; Rossini, A. J.; Gajan, D.; Bourdolle, A.; Ouari, O.; Rosay, M.; Maas, W. E.; Tordo, P.; Lelli, M.; Emsley, L.; Lesage, A.; Copéret, C.Chem. Commun.2012,48, 654−656.
(39) Ashbrook, S. E.; Smith, M. E.Chem. Soc. Rev.2006,35, 718− 735.
(40) Michaelis, V. K.; Markhasin, E.; Daviso, E.; Herzfeld, J.; Griffin, R. G.J. Phys. Chem. Lett.2012,3, 2030−2034.
(41) Blanc, F.; Sperrin, L.; Jefferson, D. A.; Pawsey, S.; Rosay, M.; Grey, C. P.J. Am. Chem. Soc.2013,135, 2975−2978.
(42) Perras, F. A.; Kobayashi, T.; Pruski, M.J. Am. Chem. Soc.2015, 137, 8336−8339.
(43) Merle, N.; Trebosc, J.; Baudouin, A.; Del Rosal, I.; Maron, L.;́ Szeto, K.; Genelot, M.; Mortreux, A.; Taoufik, M.; Delevoye, L.; Gauvin, R. M.J. Am. Chem. Soc.2012,134, 9263−9275.
(44) Ishii, Y.; Tycko, R.J. Magn. Reson.2000,142, 199−204. (45) Wiench, J. W.; Bronnimann, C. E.; Lin, V. S.-Y.; Pruski, M.J. Am. Chem. Soc.2007,129, 12076−12077.
(46) Guo, Z.; Kobayashi, T.; Wang, L.-L.; Goh, T. W.; Xiao, C.; Caporini, M. A.; Rosay, M.; Johnson, D. D.; Pruski, M.; Huang, W. Chem. - Eur. J.2014,20, 16308−16313.
(47) Eedugurala, N.; Wang, Z.; Chaundhary, U.; Nelson, N.; Kandel, K.; Kobayashi, T.; Slowing, I. I.; Pruski, M.; SadowA. D.ACS Catal. 2015 submitted, under review.
(48) Corzilius, B.; Michaelis, V. K.; Penzel, S. A.; Ravera, E.; Smith, A. A.; Luchinat, C.; Griffin, R. G.J. Am. Chem. Soc.2014,136, 11716− 11727.
(49) Conley, M. P.; Rossini, A. J.; Comas-Vives, A.; Valla, M.; Casano, G.; Ouari, O.; Tordo, P.; Lesage, A.; Emsley, L.; Coperet, C.́ Phys. Chem. Chem. Phys.2014,16, 17822−17827.
(50) Gunther, W. R.; Michaelis, V. K.; Caporini, M. A.; Griffin, R. G.; Roman-Leshkov, Y.́ J. Am. Chem. Soc.2014,136, 6219−6222.
(51) Protesescu, L.; Rossini, A. J.; Kriegner, D.; Valla, M.; de Kergommeaux, A.; Walter, M.; Kravchyk, K. V.; Nachtegaal, M.; Stangl, J.; Malaman, B.; Reiss, P.; Lesage, A.; Emsley, L.; Copéret, C.; Kovalenko, M. V.ACS Nano2014,8, 2639−2648.
(52) Blanc, F.; Sperrin, L.; Lee, D.; Dervisoģ ̆lu, R.; Yamazaki, Y.; Haile, S. M.; De Paëpe, G.; Grey, C. P.J. Phys. Chem. Lett.2014,5, 2431−2436.
(53) Takahashi, H.; Lee, D.; Dubois, L.; Bardet, M.; Hediger, S.; De Paëpe, G.Angew. Chem., Int. Ed.2012,51, 11766−11769.
(54) Rossini, A. J.; Zagdoun, A.; Hegner, F.; Schwarzwalder, M.;̈ Gajan, D.; Copéret, C.; Lesage, A.; Emsley, L.J. Am. Chem. Soc.2012, 134, 16899−16908.
(55) Takahashi, H.; Hediger, S.; De Paëpe, G.Chem. Commun.2013, 49, 9479−9481.
(56) Rossini, A. J.; Widdifield, C. M.; Zagdoun, A.; Lelli, M.; Schwarzwälder, M.; Copéret, C.; Lesage, A.; Emsley, L.J. Am. Chem. Soc.2014,136, 2324−2334.
(57) Mollica, G.; Dekhil, M.; Ziarelli, F.; Thureau, P.; Viel, S.Angew. Chem., Int. Ed.2015,54, 6028−6031.
(58) Lee, D.; Monin, G.; Duong, N. T.; Lopez, I. Z.; Bardet, M.; Mareau, V.; Gonon, L.; De Paëpe, G. J. Am. Chem. Soc. 2014,136, 13781−13788.
(59) Lee, D.; Takahashi, H.; Thankamony, A. S. L.; Dacquin, J.-P.; Bardet, M.; Lafon, O.; Paëpe, G.J. Am. Chem. Soc.2012,134, 18491− 18494.
(60) Lee, D.; Duong, N. T.; Lafon, O.; De Paëpe, G.J. Phys. Chem. C 2014,118, 25065−25076.
(61) Pourpoint, F.; Thankamony, A. S. L.; Volkringer, C.; Loiseau, T.; Trebosc, J.; Aussenac, F.; Carnevale, D.; Bodenhausen, G.; Vezin,́ H.; Lafon, O.; Amoureux, J.-P.Chem. Commun.2014,50, 933−935.
(62) Valla, M.; Rossini, A. J.; Caillot, M.; Chizallet, C.; Raybaud, P.; Digne, M.; Chaumonnot, A.; Lesage, A.; Emsley, L.; van Bokhoven, J. A.; Coperet, C.́ J. Am. Chem. Soc.2015,137, 10710−10719.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062
(63) Rossini, A. J.; Emsley, L.; O’Dell, L. A.Phys. Chem. Chem. Phys. 2014,16, 12890−12899.
(64) Sangodkar, R. P.; Smith, B. J.; Gajan, D.; Rossini, A. J.; Roberts, L. R.; Funkhouser, G. P.; Lesage, A.; Emsley, L.; Chmelka, B. F.J. Am. Chem. Soc.2015,137, 8096−8112.
(65) Johnson, R. L.; Perras, F. A.; Kobayashi, T.; Schwartz, T. J.; Dumesic, J. A.; Shanks, B.; Pruski, M. Chem. Commun. 2015, submitted.
(66) Mouat, A. R.; George, C.; Kobayashi, T.; Pruski, M.; R.P, v. D.; Marks, T. J.; Stair, P. C. Angew. Chem., Int. Ed. 2015, in press. DOI:10.1002/anie.201505452.
(67) Tycko, R.Acc. Chem. Res.2013,46, 1923−1932.
(68) Bouleau, E.; Saint-Bonnet, P.; Mentink-Vigier, F.; Takahashi, H.; Jacquot, J. F.; Bardet, M.; Aussenac, F.; Purea, A.; Engelke, F.; Hediger, S.; Lee, D.; De Paëpe, G.Chem. Sci. 2015, in press. DOI:10.1039/ c5sc02819a.
(69) Can, T. V.; Caporini, M. A.; Mentink-Vigier, F.; Corzilius, B.; Walish, J. J.; Rosay, M.; Maas, W. E.; Baldus, M.; Vega, S.; Swager, T. M.; Griffin, R. G.J. Chem. Phys.2014,141, 064202.
(70) Mathies, G.; Caporini, M. A.; Michaelis, V. K.; Liu, Y.; Hu, K.-N.; Mance, D.; Zweier, J. L.; Rosay, M.; Baldus, M.; Griffin, R. G. Angew. Chem., Int. Ed.2015,54, 11770−11774.
ACS Catalysis Viewpoint
DOI: 10.1021/acscatal.5b02039
ACS Catal.2015, 5, 7055−7062