D
e t e c t i n g
p o s i t i v e
s e l e c t i o n
IN PROTEIN CODING GENES
M
a r i aA
n i s i m o v aDepartment of Biology
Centre for Mathematics and Physics in the Life Sciences
and Experimental Biology (CoMPLEX)
University College London
2003
Subm itted to the University of London
ProQuest Number: 10015037
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10015037
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author. All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
A
b s t r a c tD e te c tin g p o s it iv e s e le c tio n in th e p r o te in c o d in g g e n e s
Maria Anisim ova D. Phil. Thesis
University College London University of London
Selective pressure at the protein level is typically measured by the
nonsynonym o u s /synonym ous rate ratio {cu = d u /d s ), with ûj < 1, = 1, and > 1 indicating purifying selection, neutral evolution, and positive selection,
respectively. Methods that detect positive selection using this criterion are
reviewed. I focus on maximum likelihood (ML) m ethods based on codon
substitution m odels accounting for heterogeneous selective pressure across
sites. If ML estimates indicate presence of positive selection and the likelihood ratio test (LRT) is significant, Bayes prediction can be used to identify sites
under positive selection.
I examine the accuracy and power of LRTs for positive selection and
Bayes prediction of residues under positive selection. The use of for
significance testing makes the LRT conservative, especially for small samples of
closely related lineages. Nevertheless, if a large number of lineages of sufficient divergence are analyzed, the power of the LRT can be as high as 100%. Both
accuracy and power of Bayes prediction are low for data containing only few
similar sequences. But sampling a large number of lineages improves the
performance substantially. Multiple models of heterogeneous selective
pressures among sites should be applied in real data analysis.
ML m odels are phylogeny-based and do not incorporate recombination.
To evaluate the effect of recombination on the LRTs and Bayes prediction, data
are simulated using a coalescent model with recombination. The LRT is found
to be robust to low recombination rates. H owever, for higher rates, the type-I
error rate can be very high. Identification of sites under positive selection by
the Bayes method is less affected by recombination than is the LRT. Finally, the
hepatitis D antigen gene (HDAg) is tested for positive selection. Sites predicted
to evolve under positive selection are found in imm unogenic domain and in
C
o n t e n t sA B S T R A C T ... 2
LIST OF T A B L E S ...7
LIST OF F IG U R E S...9
A C K N O W L E D G E M E N T S ...12
IN T R O D U C T IO N ...13
CHAPTER 1 STA TISTIC A L M E T H O D S FOR DETECTING PO SITIV E SELECTION IN C O D IN G G E N E S ...21
1.1 Measureofthepositiveselectionpressureo napr o tein...22
1.2 Estimationofaveragesy n o n y m o u sa n d n o n sy n o n y m o u ssubstitutionrates 23 1.2.1 A pproxim ate m e th o d s... 23
1.2.2 M axim um lik elih ood estim ation of p ositive selection p r e ssu r e ...26
The concepts of likelihood and maximum likelihood estimation...26
Markov codon m odels... 28
ML estimation of the coratio for two sequences and its accuracy...32
1.3 Methodsallow ingvariationofselectivepressurea m o n gsites...34
1.3.1 M ethods based on ancestral recon stru ction ... 36
1.3.2 M odels im p lem en ted w ithin m axim um likelihood fr a m ew o rk ... 40
Codon Substitution Models for Detecting Positive Selection at Sites...41
Likelihood calculation... 44
Likelihood ratio tests for positive selection... 47
Bayesian inference...49
1.4 Maxim umlikelihoodm ethodsallow ingvariableselectivepressureovertime 51 1.4.1 M odels of variable selective pressures am on g b r a n c h e s... 52
1.4.2 ML m o d els for detecting p o sitiv e selection at in d iv id u a l c od on sites along specific lin e a g e s...55
CHAPTER 2 ACC U R A C Y A N D POWER OF THE LIKELIHOOD R A TIO TESTS IN DETECTING A D A PT IV E M OLECULAR E V O L U T IO N ...60
2.2 Theorya n dm e t h o d s... 62
2.2.1 A ccuracy of the LRT... 62
2.2.2 P ow er of the LRT...64
2.3 Resu lts... 68
2.3.1 A c cu ra cy ...68
2.3.2 P ow er A n a ly sis... 73
2.4 Dis c u s s io n... 80
2.4.1 A ccuracy o f the A p p r o x im a tio n ... 80
2.4.2 P ow er of L R T ...82
2.4.3 D ifferences b etw een the tw o L R T s...83
2.4.4 M od ifyin g LRTs for p o sitiv e selection to increase the p o w e r ... 84
Modified LRTs comparing models M7 (beta) against MS (beta&A))...84
A penalized LRT comparing models MO (one ratio) and M3 (discrete)... 86
CHAPTER 3 ACCURACY A N D POWER OF BAYESIAN PREDICTION OF AM INO ACID SITES UND ER POSITIVE SELECTION...90
3.1 Bayesinferenceofsitesu n d erpositiveselection: Fromsequencetostructure .... 91
3.2 Me t h o d s... 92
3.2.1 S im u la tio n s... 92
3.2.2 A n a ly s is ... 94
Accuracy... 95
Pow er... 96
3.3 Resu lts... 98
3.3.1 A ccuracy o f Bayes p r e d ic tio n ... 98
3.3.2 P ow er of Bayes P red ictio n ... 101
3.4 Dis c u s s io n... 103
3.4.1 Sam pling errors of ML estim ates o f param eters and their effect on accuracy of Bayes site p r ed ictio n ... 103
CHAPTER 4 EFFECT OF RECOMBINATION O N THE ACCURACY OF THE
LIKELIHOOD METHOD FOR DETECTING POSITIVE SELECTION AT AM INO ACID
SITES... 114
4.1 Effectsc a usedbyrecombinationinmoleculard a t a... 115
4.2 Me t h o d s... 116
4.2.1 Likelihood Ratio T ests... 116
4.2.2 Coalescent Simulation with Recombination...117
4.2.3 Values of Parameters Used in the Sim ulation...118
4.3 R e s u lt s... 120
4.3.1 Impact of Recombination on the LRT... 120
4.3.2 The Effect of Incorrect Phytogeny on the LRT... 126
4.3.3 Bayes Prediction of Sites under Positive Selection in Presence of Recombination 126 4.4 D is c u s s io n...129
4.4.1 Effect of Recombination on the LRT...129
4.4.2 Detecting Positive Selection in Presence of Recom bination...132
CHAPTER 5 POSITIVE SELECTION IN THE HEPATITIS DELTA V IR U S ... 135
5.1 H e p a titis d e l t a virus: b a sic f a c t s...137
5.1.1 Classification, clinical featu res... 137
5.1.2 Genome structure and replication...139
5.1.3 Defining objectives with a special reference to the previous study of HDV by Wu et al. (1999)... 140
5.2 M e t h o d s...142
5.2.1 HDV data...142
5.2.2 Phylogeny reconstruction...145
5.2.3 Testing for recombination and positive selection... 146
5.3 Results... 149
5.3.1 Testing for recom bination...149
5.3.2 Testing for positive selection ... 154
5.4 D is c u s s io n...160
5.4.1 Recombination in HDAg-S g e n e ... 160
5.4.2 Detecting residues under diversifying selection in H D A g-S ...162
Accuracy of the prediction...162
Biological significance of predicted positive selection sites... 163
Summary and p rospects... 165
C O N C LU SIO N S... 166
Li s t o f Ta b l e s
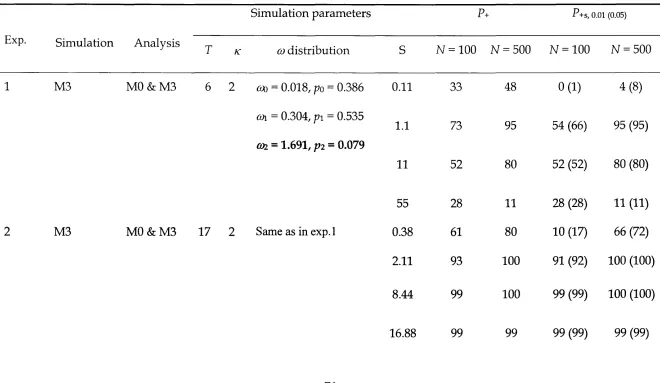
Table 1.1 - Models of œ ratio variation among sites used for simulation and
analysis
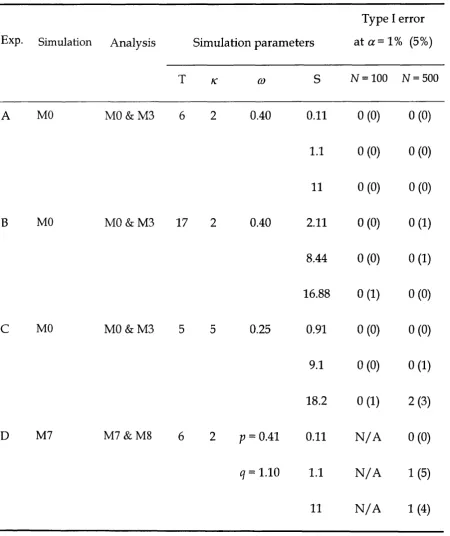
Table 2.1 - Type I error rate: number of cases out of 100 for w hich the null
hypothesis was rejected at the 1% (5%) significance levels
Table 2.2 - Power of the LRT: proportions of replicates out of 100 in which
positive selection was indicated, or detected by the LRT at the 1 %
and 5% significance levels
Table 2.3 - Comparison of the power of the standard LRT and the penalized LRT
Table 3.1 - Parameter values used in simulations
Table 3.2 - Partition of sites in a sequence used in estimation of accuracy and
power of Bayes prediction
Table 4.1 - Number of replicates (out of 100) in the likelihood analysis
comparing m odels MO (one-ratio) and M3 (discrete)
Table 4.2 - Number of replicates (out of 100) in the likelihood analysis
comparing M l (neutral) and M2 (selection)
Table 4.3 - Number of replicates (out of 100) in the likelihood analysis
comparing MO (one-tatio) and M3 (discrete)
Table 4.4 - Number of replicates (out of 100) in the likelihood analysis
comparing M7 (beta) and M8 (beta&a>)
Table 4.5 - Number of replicates (out of 100) in the likelihood analysis
comparing MO {co= 1, fixed) against MO (&> estimated)
Table 5.1 - Likelihood scores and P-values of SH test for the four inferred trees
Table 5.3 - Detecting putative recombination regions with PLATO using
HKY85 m odel with ML nucleotide frequencies and gamma rate
variation
Table 5.4 - Results of recombination tests carried out with FIST using HKY85
m odel w ith ML nucleotide frequencies and gamma rate variation
Table 5.5 - Results of LRTs and ML estimates for hepatitis delta antigen gene
Table 5.6 - Amino acids predicted to be under positive selection by Bayes
Li s t o f f i g u r e s
Figure 0.1. - Darwin's collection of finches
Figure 1.1 - Unrooted tree for two lineages
Figure 1.2 - An example of an unrooted 6-taxon tree used to explain likelihood
calculation and in simulation of Chapter 2
Figure 1.3 - Examples of nested models used in likelihood ratio tests for
detecting positive selection
Figure 2.1 - Tree topologies used to simulate the data
Figure 2.2. - Comparison of the distribution with the distribution of the 2A^
statistic in 500 simulated replicates
Figure 2.3. - Accuracy of the asymptotic theory for the LRT of Ho: û) = 1 against
HI: 1
Figure 3.1. - Accuracy of the Bayes prediction under M3 (discrete) for sequences
of 500 codons
Figure 3.2. - Accuracy and power of Bayes site prediction in data sets of 98
sequences
Figure 3.3 - Power of the Bayes prediction under M3 (discrete) for sequences of
500 codons
Figure 3.4 - Accuracy of the Bayesian prediction computed for one 6-taxon data
set of "infinitely" long (10^ codons) sequences using true parameter
values
Figure 3.5 - The effect of large sampling of extremely similar sequences on
accuracy and power of Bayesian prediction
Figure 3.6. - Bayes prediction of selected sites using different models.
Figure 4.1 - Accuracy of Bayes prediction of amino acid sites under positive
Figure 5.1 - Three RNAs detected in infected by HDV cells
Figure 5.2. - Phylogenies of 33 HDAg-S strains used to test data for positive
selection
Figure 5.3. - Posterior probabilities for sites in HDAg-S to evolve under positive
or purifying selection or under relaxed functional constraints in
discrete m odel M3
Figure 5.4. - Domains within hepatitis delta antigen gene
..if variations useful to any organic being ever do occur, assuredly individuals thus
characterized w ill have the best chance o f being preserved in the struggle for life; and
from the strong principle o f inheritance, these w ill tend to produce offspring sim ilarly
characterized. This principle o f preservation, or the su rvival o f the fittest, I have called
N atural Selection.
Charles Darwin, "The Origin of Species" (1859)
. ..when its whole significance dawns on you, you r heart sinks into a heap o f sand
within you. There is a hideous fatalism about it, a gh astly and damnable reduction o f
beauty and intelligence, o f strength and purpose, o f honor and aspiration.
G. Bernard Shaw on natural selection.
Ac k n o w l e d g e m e n t s a n d a u t h o r s h i p d e c l a r a t i o n
I would like to thank my supervisor Ziheng Yang (Z. Y.) for his patience, flexibility and
useful advice. I am also extremely grateful to Joseph P. Bielawski (J. B.) for many fruitful
discussions, the inspiration and moral support. It is thanks to these peoples' guidance,
advice and support the simulation studies presented in Chapters 2 - 4 resulted in the
following publications:
Anisimova, M., /. P. Bielawski, and Z. Yang. 2001. Accuracy and power of the likelihood ratio test
to detect adaptive molecular evolution. Mol. Biol. Evol. 18:1585-1592.
Anisimova, M., /. P. Bielawski, and Z. Yang. 2002. Accuracy and power of Bayes prediction of
amino acid sites under positive selection. Mol. Biol. Evol. 19:950-958.
Anisimova, M., R. Nielsen, and Z. Yang. 2003. Effect of recombination on the accuracy of the
likelihood method for detecting positive selection at amino acid sites. Genetics.164:1229-1236.
The methods overview presented in Chapter 1 and the empirical study presented
in Chapter 5 have been done solely by the author of this thesis. It was Z. Y.'s initiative to
examine the accuracy and power of methods developed by Z. Y. and collaborators. This
became the aim of my simulations, which I designed, analyzed and summarized myself.
The results presented in Chapters 2 and 3 were discussed with J. B. and Z. Y. in order to
find the best possible interpretation and draw up guidelines that would be useful for
experimental biologists using these methods. Z. Y. and J. B. helped me to develop and
improve my skills of writing a scientific report by suggesting changes (structural or
editorial) to my drafts. Work presented in Chapter 4 was initiated by the author of this
thesis. Rasmus Nielsen wrote a program to simulate recombinant data and together with
Z. Y. contributed to the discussion of Chapter 4. Z.Y. also made some suggestions and
editorial changes to parts of Chapter 4.
This work of three years would have not been possible without the financial
support from the Medical Research Council. I would also like to acknowledge Wa Yang
for discussions on viral data and for strong companionship. Robin Callard has been
helpful on many accounts. Finally, my special appreciation goes to my daughter Kristina
for love and understanding.
I
n t r o d u c t i o nFor many thousands of years a belief that species, in their present forms, were
created by God or that it was possible for organic matter to be spawned from
inorganic matter, predominated peoples' idea about the surrounding world.
But, as always, there were the skeptics w ho needed scientific explanation of the
amazing diversity of living creatures they observed around them. Early
evolutionary theories appeared during the classical Greek period w hen
Anaximander (ca. 611 - 546 BC) suggested that humans arose from fish, and,
later, Empedocles (ca. 492 - 432 BC) proposed that presently living creatures
evolved by chance combinations of elements and natural selection that led to
the extinction of once-living "monstrous" organisms. Nevertheless, western
philosophy was mostly influenced by the theories of Plato (347 - 427 BC) and
Aristotle (384 - 322 BC) w ho opposed the idea of evolution, and the
creationist-essentialist dogma that species were permanent and created for a specific
purpose became deeply embedded in western thought.
The first serious attempt to explain the species diversity in terms of
evolution was the theory of inheritance of acquired characteristics proposed in
1809 by Jean-Baptiste Lamarck (1744 - 1829). He pictured evolution as a
successive transformation of living forms from the very simple to more and
more sophisticated. His theory rested on two key mechanisms w hich pushed
species up this "great escalator of being": use and disuse - the loss of futile
inheritance of traits acquired by ancestors in response to changing
environment. For example, according to Lamarck the stretching by giraffes to
reach distant leaves led to the elongation of their necks and, hence, to offspring
with longer necks. This theory, however, was scientifically unfounded, and
gained little support in the scientific community.
A new chapter in the study of evolution was opened during the voyage
of HMS Beagle (1831 1836), with a young naturalist Charles Darwin (1809
-1882) on board. While observing animals and plants that inhabited Galapagos
Islands, Darwin noticed how animals, such as tortoise, mockingbird, and finch.
5-unr l-tNCM C4CTLÂ
H'fch
ISLAND PNOM LAAGE
CACTUS
P.KOH
rxt
ONCH PWCf-'
SMALL
k’WALL lAEfe RHCH
TFL:I=
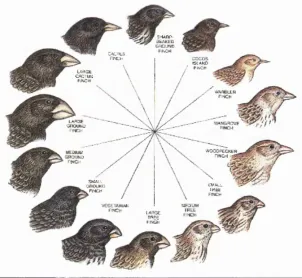
Figure 0.1. — Darwin s collection of finches: A m ong the m ost significant specim ens
collected by Darwin during his voyage on HMS Beagle w ere 14 finch species from Galapagos
Islands, w h ich developed different habits and diets d ependent on local environm ent.
Adapted from Grant (1986).
differed from island to island morphologically. His famous collection of finches
show ed that on different islands the birds developed different eating habits,
and appropriate beak, size, and scheme of organization (Figure 0.1). These
adaptations to the different environments of the islands became important
evidence used by Darwin to formulate his theory of evolution by natural
selection proposed in Origin of Species in 1859. Darwin described evolution as
a descent with modification. Species of common ancestry adapt to fit their
individual surroundings. The natural variance within a species allows some
animals to live while others die off. As the survivors breed, they pass on the
trait that allows them to live until that trait becomes a standard feature of the
new species.
Almost sim ultaneously Alfred Wallace (1823 - 1913) independently
came to amazingly similar conclusions through his ow n work on natural
selection. Throughout the second half of the nineteenth century, Darwin and
Wallace were engaged in correspondence over the evolutionary process. The
theory of common descent and evolution by natural selection sparked an
intense debate among the fellow-scientists and underwent many modifications.
Darwin's theory seem ed to be at odds with Mendelian genetics as it did not
offer a plausible explanation of heredity. After the rediscovery of MendeTs
laws, variation in populations was shown to be caused by mutation. In 1918
R.A. Fisher for the first time described the statistical m odel of quantitative
inheritance. During the 1920s and 1930s, R.A. Fisher J.B.S. Haldane and S.
Wright pioneered the developm ent of the fundamental principles of
Darwinism and M endelism were reconciled into a new theory, known as
neo-Darwinism or synthetic theory, where mutation was recognized as the ultimate
source of genetic variation, and natural selection was thought to be dominant
in shaping the genetic makeup. The discovery of the D N A structure in 1953
revealed the molecule carrying hereditary information, marking the beginning
of new era in biological research. With the advances in molecular techniques,
more and more empirical molecular data were generated and used to examine
the genetical theory; moreover, the search for evidence of adaptation and study
of adaptive mechanisms could be done at the molecular level where the species
barrier w as no longer an issue.
H aving analyzed mammalian protein sequences, Kimura (1968a) argued
that genom ic nucleotide substitution rate was too high to be compatible with
Haldane's (1957) cost of natural selection. Subsequently, the neutral theory of
molecular evolution, independently proposed by Kimura (1968b) and by King
and Jukes(1969), challenged the Darwinian concept of natural selection stating
that the main cause of the evolutionary change and variability at the molecular
level w as random fixation of selectively neutral or nearly neutral mutations,
and that the effect of natural selection was insignificant. Neutral theory had a
progressive influence on the current state of molecular evolution: it suggested
that molecular data evolved according to stochastic rather than deterministic
processes, and provided evolutionary biologists with afalsifiable null
hypothesis. Today many aspects of the neutralist school are accepted, and the
majority of scientists agree that both weak deleterious selection and occasional
positive selection are important evolutionary factors. Adaptation could no
longer be just assumed but had to be rigorously justified. Detecting selection in
the genome became an efficient strategy for finding causes of species- specific
differences or identifying genomic regions of functional, and potentially,
medical significance. This triggered the developm ent of statistical methods that
could detect various kinds of selection and hence could identify genomic
regions of functional importance. For nucleotide data, Tajima's D-test (Tajima,
1989) became one of the m ost popular tests. Tajima's D statistic is calculated as
a scaled difference between the estimates of 4N// (where N is the effective
population size, and jj, is the mutation rate per generation) based on the
number of pairwise differences and the number of segregating sites in a sample
of sequences. Under neutrality Tajima's D is expected to be 0, whereas D < 0
and D > 0 may indicate selective sw eep and balancing selection respectively.
Several other neutrality tests, based on slightly different summary statistics,
use a similar idea (e.g.. Fay and Wu, 2000; Fu and Li, 1993). Another popular
test for nucleotide data is Hudson-K reitm an-A guade or HKA test (Hudson et
al., 1987), which evaluates the neutral hypothesis through the comparison of
variability within and between species for two or more loci. Unless the locus is
under selection, levels of polymorphism (variability within species) and
divergence (variability between species) should be proportional to the
mutation rate and, thus, should show a constant ratio of polymorphism to
divergence. Tests of selective neutrality based solely on simple summary
statistics appear powerful enough to reject the strictly neutral m odel, but such
tests are very sensitive to the demographic assumptions, making it difficult to
Simonsen, 1998). The McDonald-Kreitman or MK test (McDonald and
Kreitman, 1991) for protein coding data has been more successful at detecting
selection. The basic idea behind MK test is similar to that underlying HKA test.
The MK test compares the ratio of nonsynonym ous (amino-acid altering) to
synonym ous (silent) substitutions within and between species, w hich should
be the same in absence of selection. This test is quite robust to demographic
assumptions since the effect of the demographic m odel is the same for both
nonsynonym ous and synonym ous sites (Nielsen, 2001). H owever, HK test does
not distinguish between different forms of natural selection, and thus cannot
provide explicit evidence for adaptive evolution (Nielsen, 2001; Yang and
Bielawski, 2000). Various modifications of HK test, such as those proposed by
Akashi (1995), Templeton (1996), or Akashi (1999) provide more information
about the nature of selective forces. In particular, Akashi (1999) suggested a
way to differentiate between the types of selection by examining the frequency
distribution of observed synonym ous and nonsynonym ous changes as
compared with neutral expectation. However, w hen selection is weak, or when
the fraction of adaptive mutations is small, this method becomes considerably
less powerful. Furthermore, tests based on the idea of between and within
species comparisons require population data as w ell as species sampling. This
is not always feasible in macro-evolutionary studies.
While the relative significance of deleterious, neutral and adaptive
changes remains an open issue, detecting positive Darwinian selection became
a critical aspect of understanding the mechanisms of molecular evolution.
Adaptive evolution was show n to be responsible for changes in enzymatic
functions (Irwin, 1995; Irwin and Wilson, 1990; Stewart et al., 1987), host and
parasite co-evolution (Bishop et al., 2000; Locksley, 1997; Xiao et aL, 2002),
maintenance of the specificity of reproductive proteins (Galindo et ai., 2003;
Hellberg et al., 2000; Lee et al., 1995; Swanson et al., 2001), i.e., reproductive
isolation and thus spéciation.
The m ost rigorous way of detecting positive selection in protein coding
sequences is to show that the number of nonsynonym ous substitutions per
nonsynonym ous site is significantly higher than the number of the
synonym ous substitutions per synonym ous site. Up to now , this approach
helped to obtain the m ost convincing evidence of adaptive molecular
evolution. Chapter 1 of this thesis presents the overview of statistical methods
based on the comparison of nonsynonym ous and synonym ous rates and their
successful application to real data analysis. A special em phasis is made on the
maximum likelihood based m odels that allow a variation of the selective
pressures among sites in the protein sequence. In Chapter 2 computer
simulations are used to examine the accuracy and power of the likelihood ratio
tests (LRTs) in detecting adaptive molecular evolution. These LRTs compare
nested models of variable selective pressures among sites that were described
in Chapter 1. The empirical Bayesian approach to predict amino acids under
positive selection is introduced in Chapter 3 and subsequently evaluated by
computer simulations. Methods evaluated in Chapters 2 and 3 are
phylogeny-based and do not account for the effects of recombination. Chapter 4 therefore
presents computer simulations examining the effects of recombination on the
molecular evolution. Such thorough testing of the likelihood and Bayesian
approaches helps to evaluate the feasibility of their application to data sets of
various size and divergence. Chapter 5 presents a case study where examined
m ethods were used to detect positively selected amino acid sites. I examine a
data set of the hepatitis D antigen gene that is characterized by a large sample
and sufficient divergence, ensuring the desired accuracy and a good power of
the methods.
Chapter 1
S
t a t is t ic a lM
e t h o d s f o rD
e t e c t in gP
o s it iv e1 .1 Me a s u r e o f t h e p o s i t i v e s e l e c t i o n p r e s s u r e o n a p r o t e i n
Adaptation has long been a subject of intense interest among evolutionary
molecular biologists. The most direct w ay of obtaining the evidence for
adaptive molecular evolution on a protein-coding gene is by identifying
amino acid sites where the nonsynonym ous (or replacement) substitution rate
dN exceeds the synonym ous (or silent) rate ds (first suggested by Miyata et al.,
1979; Miyata and Yasunaga, 1980). These rates are defined as numbers of
nonsynonym ous or synonym ous substitutions per nonsynonym ous or
synonym ous site respectively. Selective pressure at the protein level is
typically measured by the ratio co= d u / ds. If amino acid changes are
deleterious, purifying selection reduces their fixation rate, so that dw < ds and
6Ü < 1. If selection on the protein has no effect on fitness, nonsynonym ous
substitutions occur at the same rate as synonym ous substitutions and c o = l ,
suggesting neutral evolution. Only w hen amino acid changes offer a selective
advantage are nonsynonym ous changes fixed at a higher rate than
synonym ous changes. Thus a significantly higher nonsynonym ous
substitution rate, i.e. > 1, provides an evidence of adaptive molecular
evolution. Using this criterion, H ughes and Nei (1988) obtained unambiguous
evidence for positive selection in the major histocompatibility complex. Other
early studies utilized the same criterion and reported the evidence for positive
selection, for example, in variable-region genes of im m unoglobulins (Tanaka
and Nei, 1989); in gene regions encoding for T-cell epitopes in malaria
parasites (Hughes, 1991); in hemagglutinin gene of human influenza A (Ina
and Gojobori, 1994); in abalone sperm lysine (Lee et al., 1995); and the porB
gene of gonnococcus and meningococcus (Smith et al., 1995).
1 .2 E s t i m a t i o n o f a v e r a g e s y n o n y m o u s a n d n o n s y n o n y m o u s
S U B ST IT U T IO N RATES
1.2.1
Approximate methods
This section offers a brief overview of so-called ""approximate" m ethods for
estimating dw and ds rates. All such methods approximate rates dw and ds
using intuitive assum ptions rather than rigorous theory. Although they
greatly differ in detail, the basic procedure is the same: (i) count the numbers
of synonym ous and nonsynonym ous sites in the observed sequences; (ii)
count the numbers of synonym ous and nonsynonym ous differences by
considering all possible evolutionary pathways between the hom ologous
codons; (iii) correct for multiple substitutions at the same sites using a
standard evolutionary model.
Miyata and Yasunaga (1980) proposed one of the first m ethods for
estimating synonym ous and nonsynonym ous rates. Its simplified version (Nei
and Gojobori, 1986) gave essentially the same estimates of Jn and ds, and
subsequently became very popular. Considering the likelihood of a nucleotide to
result in a silent or replacement substitution in each of the three codon positions,
Nei-Gojobori method computes total numbers of synonym ous (S) and
comparisons the averages of S and N for the two sequences are used. The total
numbers of synonym ous (Sd) and nonsynonym ous (Nd) pairwise differences are
counted, again, by adding respective counts for all codons. When there is more
than one difference between the hom ologous codons, all possible evolutionary
pathways are considered and w eighted equally. The proportions of synonym ous
(ps) and nonsynonym ous (pw) differences can be approximated by the equations:
ps = Sd/ S and pN = Nd / N. Synonymous and nonsynonym ous rates are estimated
using Jukes and Cantor(1969) formula as a correction for multiple substitutions:
ds = - % lo g (l - 4/3ps) and dN = - % log(l - 4/3p^). Both Miyata-Yasunaga and
Nei-Gojobori methods are based on Jukes and Cantor nucleotide substitution
m odel (JC69), assume equal nucleotide frequencies, and ignore the
transition/ transversion bias. The latter is known to cause underestimation of the
number of synonym ous sites and overestimation of nonsynonym ous sites since
at the third codon positions transitions are more likely to be synonym ous than
are transversions. The problem becomes especially serious w hen sequences are
highly diverged.
Li et al. (1985) extended Miyata-Yasunaga method so 2-fold, 4-fold, and
non-degenerate nucleotide sites were treated separately (as in Perler et al., 1980),
using Kimura's (1980) 2-parameter model to correct for multiple hits. However,
JC69 m odel was still used as an underlying mutation matrix. After numerous
attempts to account for transition/ transversion rate bias (Comeron, 1995; Li,
1993; Pamilo and Bianchi, 1993) in all steps of the estimation this has been
achieved by Ina (1995) in his unweighted pathway method based on Kimura's
parameter model. Ina's (1995) simulations show ed that m ethods of
Miyata-Yasunaga, Nei-Gojobori and Li et al. caused underestimation of dN and
overestimation of ds by overestimating N and underestimating S. According to
the same study, the method of Pamilo and Bianchi and Ina's new method give
better estimates of dhi and ds) but for data with strong transition/ transversion
and nucleotide frequency biases all the above-mentioned m ethods give biased
estimates as none of them account for unequal base composition. Similar
concerns have been expressed in other studies (Bielawski et al., 2000; Moriyama
and Powell, 1997; Yang and Nielsen, 2000). Interestingly, the effect of nucleotide
bias can override the effect of transition/transversion bias. For example, in
studies of mammalian nuclear genes (Ohta, 1995; Yang and Nielsen, 1998) biased
codon frequencies led to reduced numbers of synonym ous sites; as a result,
contrary to the general belief, Nei-Gojobori method overestimated rather than
underestimated S and co.
Finally, Yang and Nielsen (2000) incorporated transition/transversion
bias and unequal base frequencies in every step of their iterative algorithm
assuming the HKY85 nucleotide substitution m odel (Hasegawa et al., 1985).
Estimates of dN and ds produced by this new approximate m ethod were
shown to be very close to the true values even for data with strong
1.2.2 Maximum likelihood estimation of positive selection pressure
The concepts of likelihood and maximum likelihood estimation
Maximum likelihood (ML) is a major statistical inference tool, w hich is widely
used in a variety of fields. The idea central to this method is to choose a
hypothesis that maximizes the likelihood of the observed data (i.e., makes the
data the m ost plausible). ML inference is based on an explicit probabilistic
model, and a hypothesis is defined as a certain set of the parameters
describing that model. Although in some cases the ML approach can be
computationally intensive, the advantages of em ploying it are apparent: (i)
ML is a classical statistical technique, it is w ell studied and has useful
statistical properties; (ii) parameters of interest can be estimated by
maximizing the likelihood of observing the data; (iii) ML enables formulation
of a variety of hypotheses as w ell as testing those hypotheses by likelihood
ratio tests (LRTs), (iv) ML can be used as a framework for Bayesian inference,
the benefits of which are predicting the most probable scenarios and
quantifying the uncertainty of such predictions in an easily interpretable
form.
More formally, if a probabilistic model is formulated in terms of
unknown parameters 6 = {Qi, ... ^»), the likelihood of the hypothesis
described by 9 is proportional to the probability of the observed data given that
hypothesis is correct. Thus, a likelihood function is defined as a joint
probability density function of all independent observations x = given
parameters 0\ l { x
|
0) =f{xi,%
%
... %
» I
0)=/(^i I ^/(%2 I 0 ./(%« I
0)-Theunknown parameters 0 are estimated by maximising the probability of
observing data x given 6 . Let è be the ML estimator (MLE) that evaluates the
parameters 9 so it maximises the likelihood
L(% |
9). The actual value of 0 iscalled an ML estimate.
As the likelihood function is a product of probabilities for all
observations it can be very close to 0. It is therefore easier to work with the
logarithm of L(% | 9). Let ^{9) = log L{9). The log-likelihood function is
maximised by solving £'(9) = 0 and t ' { 9 ) < 0. In simple cases it is possible to
solve the equation analytically. However, in the majority of cases this is done
using numerical procedures such as repeated bisection or Newton-Raphson
method.
The attraction of using ML for parameter estimation lies in the
following properties offered by the MLE 0 for regular cases (e.g., Stuart et al.,
1999; Wald, 1949):
(i) 0 is asymptotically unbiased, meaning that for large enough samples its
expectation is equal to the true value: E [ 0 ] = %
(ii) 0 is asymptotically efficient, which means that for large samples the MLE is
the estimator w ith the low est possible variance;
(iii) 0 is asymptotically normally distributed around the true value 6b with a
minimum possible variance;
(iv) 6 is a consistent estimator of 9. as the sample size increases 0 converges to
(v) à is transformation-invariant: if a single-valued twice-differentiable function
g(0) has g ' ( 0 ) ^ 0 for any 6, then g ( 0 ) is an ML estimator of
Maximum likelihood and Bayesian methods are now successfully used in
phylogenetics (Lewis, 2001; Whelan et al., 2001). The use of the ML approach in
molecular phylogenetics was first suggested by Cavalli-Sforza and Edwards
(1967). H owever, the computationally effective algorithm for calculating a
likelihood of a phylogeny was first presented by Felsenstein (1973,1981). The
ML framework, originally developed for continuous characters (Felsenstein,
1973), was later adapted to accommodate nucleotide (Felsenstein, 1981), amino
acid (Adachi and Hasegawa, 1992; Kishino et al., 1990) and codon data
(Goldman and Yang, 1994; Muse and Gaut, 1994). Since this thesis focuses on
ML m odels for coding data, the next section is devoted to stochastic models of
coding sequence evolution.
Markov codon models
Explicit probabilistic m odels of codon substitution on a phylogeny were
proposed by Goldman and Yang (1994) and Muse and Gaut (1994). These
studies demonstrated that ML was flexible in respect to a variety of
parameters that could be incorporated to reflect the transition/transversion,
codon usage biases, synonym ous and nonsynonym ous rates, and other
particulars such as chemical differences between amino acids. The m odel of
Muse and Gaut incorporated only variable nucleotide frequencies and
separate synonym ous and nonsynonym ous rates. Although incorporating
transition/transversion rate and accounting for variable codon (or amino
acid) frequencies w ould have been more realistic. Muse and Gaut (1994)
admitted this but chose a simple scenario demonstrating the technique with
minimal parameters incorporated. Goldman and Yang (1994) presented a
more realistic model, which accounted for non-uniform codon frequencies,
transition/ transversion bias and incorporated a separate parameter to
measure the gene variability. The latter parameter implicitly represented the
tendency of the gene to undergo nonsynonym ous substitutions, and was
positively correlated with the 6) ratio. In an attempt to account for selective
differences at the amino acid level Goldman and Yang (1994) used
Grantham's (1974) matrix of physiochemical distances. H ow ever, Grantham's
m odel of protein evolution was fully based on physicochemical properties
and could not directly reflect the amino acid differences enforced by
evolution. Subsequently, the m odel of Goldman and Yang (1994) w as
simplified to incorporate the 6? ratio explicitly (e.g., N ielsen and Yang, 1998).
Described versions of the codon substitution m odels are the same
conceptually, but differ in their definition of the rate matrix Q. The evolution
of codons is m odelled by a continuous-time Markov process running along a
phylogeny. Suppose p,)(f) is the probability that codon i changes into codon;
over time f. Here i and j take values from 1 to 61 representing all sense
codons. The time interval t is measured by the expected number of nucleotide
substitutions per codon (often referred to as a branch length). Transition
substitution rate from codon i to codon; is described by matrix Q = [qij]. The
m ost successful matrix incorporates codon frequencies tTj,
transition/transversion rate ratio k , and nonsynonym ous/synonym ous rate
ratio œ (simplified m odel of Goldman and Yang, 1994):
0, if / and j differ at 2 or 3 nucleotide positions
71 j , if / and j differ by 1 synonymous transversion
q.j = <1 KTTj, if i and j differ by 1 synonymous transition (1)
C07t J, if i and j differ by 1 nonsynonymous transversion
coKTTj, if i and j differ by 1 nonsynonymous transition
Substitutions are assumed to occur independently am ong the three codon
positions: only one position is allowed to change instantaneously. Note also
that diagonal values are calculated so the rows sum up to 0: qu= - ^ for
i*j
any i (e.g., Grimmet and Stirzaker, 1992, p. 241). Since time and rate of change
are confounded, the rate matrix is multiplied by a scaling factor so that the
expected number of nucleotide substitutions per codon is one:
- ^ 7T^q^. q^j = 1. All codon sites in a sequence are assum ed to evolve
i I t* j
independently according to the same Markov process (in statistical terms,
codons at different sites are independently and identically distributed).
The rate of change of the transition probability is d P { t ) / d t = P (t)x Q .
The solution of this equation with the initial conditions d P { 0 ) / d t = I (where I
is the identity matrix) is an exponential P(t)=eQ^. This can be calculated using
Taylor expansion: F(t) = ^ (Qf)”/ ^1/ where the number of terms in Taylor
n=Q
expansion is determined by a desired accuracy level. Another w ay of
calculating P{t) is through diagonalization (or spectral decomposition) of Q:
P (t) = U x diag{g^'' } x U ~ \ where U is the matrix containing eigenvectors
of Q and À i , À n are the eigenvalues of Q.
The Markov process defined by equation (1) is time reversible, i.e., in
any time t, the amount of change from codon z to j is assum ed to be the same
as the amount of change from codon; to codon z, and so mpij(t) =7Tjpji(t). This
property is convenient mathematically, and although it m ight not be justified
biologically, it is commonly assumed since the direction of codon change in
the observed data is unknown. Note also that the rate matrix is independent
of time meaning that the process of codon substitution is assumed to be the
same in different parts of the tree. This property is referred to as homogeneity;
the equilibrium distribution of a homogeneous process is also a limiting
distribution w hen time approaches infinity. Moreover, the process of codon
substitution is typically assumed to be stationary, im plying that the
evolutionary process remains at the equilibrium throughout time, i.e., codon
frequencies are approximately the same during the w hole course of the
evolution. Both hom ogeneity and stationarity are unrealistic assumptions
since the divergence rates, nucleotide composition (or other biases) may differ
in time, especially for distantly related lineages (e.g., Yang and Roberts, 1995;
Rodriguez-Trelies et al., 2001). To correct for this, non-hom ogeneous
Markov-process m odels were proposed (Yang and Roberts, 1995; Galtier and Gouy,
1998; also see Section 1.4). These models allow for different patterns of
hom ogeneity and stationarity are made for the convenience of computation,
rather than to reflect the biological reality.
ML estimation of the
coratio for two sequences and its accuracy
Once a probabilistic m odel is formulated, the likelihood function of the parameters is
constructed and maximized. Let the data contain two aligned protein-coding
sequences (Figure. 1.1), with the alignment length of N codons (sites). Then the
61
probability of observing data Xh = {i , j ) at codon site h is L(xh) = '
X r=1
where xr is the state at the root of the tree, and h and h are the lengths of the
branches connecting the root with sequences at the tips. However, the location of the
root is unknown and time-reversibility of the m odels implies that the same
probability can be calculated w hen one of the sequences is assum ed to be ancestral
to another: L(xh) = TUjPjj (f^+y or L(xh) (^1+^2)- The result remains the same
irrespective of the sequence assumed to be ancestral. That means the root of the tree
is unidentifiable, so only the sum t = h + can be estimated, but not h and ti
separately. The log-likelihood function for the w hole alignment is: k , co, )) =
N
^log{;r,p^ (r)}. Codon frequencies m are usually estimated using the observed base
h = \
Seq.l
Seq.2
Figure 1.1. - Unrooted tree for two lineages:
d u e to the reversible nature o f co d o n m od els, the
location o f root R is u n k n o w n , so on ly su m of
branch len gth s ti + tz can be estim ated.
frequencies at the three codon positions. Parameters t, k , and œ are estimated by
m axim izing the log-likelihood function. Rates dw and d s are calculated as functions
o f ML estimates of t, k , and co. By property (5) stated on p. 20, estimates of dw and d s
are also ML estimates, and therefore bear the same properties (see p. 20). In more
detail (e.g., Goldman and Yang, 1994), the proportions of synonym ous and
nonsynonym ous substitutions per codon are defined as p i = ^ Tr^qy and
'V
= \ - pI , respectively. The summation is taken for all distinct codons i and j
representing the same amino acid aui = auj. The numbers of synonym ous and
nonsynonym ous substitutions per codon are tp l and /p^, respectively. Next, the
proportions of synonym ous and nonsynonym ous sites, p \ and p \, are calculated as
pI and pI but assum ing c o = l , i.e., no natural selection at the protein level. The
numbers of synonym ous and nonsynonym ous sites per codon are respectively
?>pI and 3pjy. Synonymous and nonsynonym ous rates per site can be now calculated
as follows: = tpH{?>pl) and = % /(3 p ]^ ).
Bielawski et al. (2000) evaluated the impact of the estimation method
for 82 mammalian nuclear genes. They have demonstrated that failure of the
approximate m ethods of Nei-Gojobori (1986) and Ina (1995) to properly
account for transition/ transvertion rate bias and unequal codon usage led to
misleading conclusions about patterns of mammalian nuclear gene evolution.
Simulation study of Yang and Nielsen (2000) compared four methods
for estimating and ds for two sequences: Nei-Gojobori and Ina's methods,
contrast to ML, all three approximate methods are biased and inconsistent,
w hich was confirmed by simulation (Yang and Nielsen, 2000). H owever, for
infinite data in almost all scenarios method of Yang and N ielsen had the
smallest biases and gave similar estimates to those obtained by ML.
Furthermore, for finite data for almost all cases the ML m ethod w as show n to
be the least biased w ith lower mean squared error (MSB, defined as E [(é
-= var[^] + \ E { è \ - 6 Y ) compared to the approximate methods. This
suggested that ML method should be preferable over the ad hoc
approximations. Additionally, the probability theory underlying the ML
approach makes the method conceptually more comprehensive and flexible to
account for a variety of factors. ML accounts for all possible pathways of
codon substitution and weights them according to relative probabilities of
their occurrence; and at the same time ML naturally resolves the task of
correcting for multiple hits, which is otherwise very difficult. Finally, the ML
framework can easily accommodate comparison of multiple sequences taking
into account their phylogenetic relationship, - an advantage approximate
m ethods lack.
1 .3 M e t h o d s a l l o w i n g v a r i a t i o n o f s e l e c t i v e p r e s s u r e a m o n g s i t e s
Methods assuming the same a> ratio for all sites only detect positive selection
if the average co> 1 (Yang and Bielawski, 2000). Just a few years ago, cases of
positive selection have been difficult to demonstrate. A large-scale database
search performed by Endo et al. (1996) identified only 17 out of 3,595 genes
that might have undergone adaptive evolution. Endo et al. (1996) considered
a gene to be under positive selection if the average dw w as greater than ds in
more than half of the pairwise sequence comparisons. This approach
computes the co ratio as an average across all amino acid sites and over time;
although popular, it has little power. For example, Crandall et al. (1999) found
that the approach of pairwise comparison failed to detect positive selection in
the protease gene of HIV-1 despite clear evidence of parallel evolution.
Crandall et al. (1999) suggested that the ty ratio averaged over sites was a poor
indicator of positive selection. Indeed, the assumption that all sites in a
sequence are under equal selective pressure is unrealistic. Typically adaptive
evolution occurs at only a few sites as most amino acids in a protein are under
structural and functional constraints with dw and, hence co, close to 0 (e.g.,
Golding and Dean, 1998). Moreover, those few sites m ight not be clustered in
a sequence, since sites that are far apart in a primary sequence can be
clustered in the three-dimensional structure. Thus calculating co as an average
over all amino acid sites substantially reduces the power to detect positive
selection, and even sliding w indow analysis m ight not detect positive
Darwinian selection in many genes (e.g., Endo et al., 1996).
Much effort has been taken to account for variable selective pressures
across sites to improve the power of the methods for detecting positive
selection. Most of such methods do not take a priori know ledge about the kind
of selective pressure acting on particular sites (e.g.. Bush et al., 1999; Fitch et
Yamaguchi-Kabata and Gojobori, 2000; Yang et al., 2000a). In a classical study of the MHC
class I (Hughes and Nei, 1988) all amino acid residues were partitioned into
those that belong to the antigen-recognition region and those that are outside.
The evidence of positive selection was obtained by comparing and ds rates
in each partition. Yang and Swanson (2002) implem ented an ML method for
pre-partitioned data sets and applied it to the human MHC class I and
abalone sperm lysin genes previously analyzed by H ughes and N ei (1988)
and Lee et al. (1995) respectively. To account for heterogeneity among the site
partitions, they used a separate œ ratio for each partition.
Note that all m ethods mentioned above assume a constant fy ratio for
all lineages, and thus have low power of detecting positive episodic or
directional selection but have been successful in detecting recurrent
diversifying selection.
1.3.1
Methods based on ancestral reconstruction
Most non-likelihood methods that account for variable selective pressures
reconstruct sequences of the extinct ancestors; then, at each site, count
changes along the tree to identify sites with an excess of nonsynonym ous
substitutions. Fitch et al. (1997) analysed 254 sequences of the hemagglutinin
(HA) gene of human influenza A and found possible sites under positive
selection. They used parsimony ancestral reconstruction and tested whether
the proportions of nonsynonym ous and synonym ous substitutions at each
sites deviated from binomial expectations calculated using the total numbers
of non-synonym ous and synonym ous substitutions. Effectively, this meant
that the comparison was made with averages over all sites, w hich is not a
stringent enough criterion as the average of the co ratio over all sites in a gene
is usually < 1. Thus, this method might have high false positive rate. Later,
Bush et al. (1999) examined a larger hemagglutinin data set using a slightly
modified m ethod of Fitch et al. (1997). The authors attempted to correct for
uncertainties introduced by variation in the tree topology, and used a more
stringent criterion whereby expected substitution rates were calculated across
sites excluding the codons with a significant excess of nonsynonym ous
substitutions over synonym ous. A more systematic approach w as developed
by Suzuki and Gojobori (1999). They estimated numbers of synonym ous and
nonsynonym ous sites and the substitutions using ancestral sequences
reconstructed by parsimony and tested whether there w as a significant
deviation from the neutral expectation ” ( o = l " . Data sets of human HLA, V3
region of HIVI env gene, and the H A gene of human influenza A were
examined for positive selection. At least 200 sequences were used in each
analysis. N ote, only 3 codons were suggested to be under positive selection in
influenza H A gene; that is in contrast to 25 codons suggested by the method
of Fitch et al. (1997). A method conceptually very similar to that of
Suzuki-Gojobori m ethod was applied to analyze 186 sequences from HIV l gpl20
(Yamaguchi-Cabata and Gojobori, 2000).
Naturally, all the above m ethods require large samples so there are
reconstruction can reduce the accuracy of the results produced by these
methods. For divergent samples of sequences, the parsimonious ancestral
reconstruction is highly unreliable, rendering analyses of such data
unfeasible. Moreover, parsimony considers only the reconstructions with a
m inimum amount of changes and so can seriously underestimate the total
number of changes (as shown by Nielsen, 2002). N ot only can the
parsimonious solution be unlikely but there could be non-parsimonious
reconstructions that are much more likely (Nieslen, 2002). Furthermore, the
above m ethods cannot easily account for molecular biases, for example in
data sets with high transition/ transversion ratio bias the numbers of
transitions w ould be underestimated. Finally, none of the above-mentioned
m ethods offer a mathematically rigorous solution to measuring the
confidence in sites inferred to be under positive selection.
Recently N ielsen and Huelsenbeck (2002) proposed a statistically
rigorous alternative that avoids problems associated w ith using parsimony
and relying on a single reconstruction. Referring to a reconstruction as a
mapping of mutations on a phylogeny, the authors formulate numbers of
nonsynonym ous and synonym ous changes as functions of the mapping, so
they can be inferred for any mapping directly. Since the correct m apping is
unknown, the authors suggest computing the conditional (on data)
expectation of nonsynonym ous and synonym ous changes by which all
mapping possibilities are considered and weighted accordingly. This requires
calculating a posterior probability of a mapping, which is evaluated in
Bayesian framework. While the ML method requires a phylogeny to be
known, the method of Nielsen and Huelsenbeck (2002) takes it as one of the
nuisance parameters which is integrated out via Markov chain Monte Carlo
(MCMC) technique. The current implementation assumes general
time-reversible nucleotide m odel and uniform parameter priors. H owever, using a
codon m odel of evolution w ould be more appropriate for incorporating
differences in rates of nonsynonym ous and synonym ous substitutions.
Hypotheses are tested by comparing the posterior and posterior predictive
(expected) distributions of the statistics of interest using posterior predictive
P-values (Meng, 1994). This new method was applied to the 28 sequences of
the H A gene from human influenza A. The same data set w as previously
analyzed by Yang et al. (2000a) and is a subset of sequences originally
analyzed by Fitch et al. (1999). Remarkably, Nielsen and Huelsenbeck (2002)
detected sites under positive selection that were essentially the same as those
identified by the ML method (Yang et al., 2000a). The method of posterior
predictive P-values can be easily extended and adapted to address a variety of
problems but its statistical properties ought to be studied further. This is out
of the scope of the study presented here, and I w ill be focusing on evaluating
the performance of ML m ethods that account for variable selective pressure
across the sequence (Nielsen and Yang, 1998; Yang et al., 2000a). These
methods are described in detail in the next section (Section 1.3.2). Note, ML
approach does not rely on the ancestral reconstruction and so does not suffer
calculated for each site provide an intuitive measure of the confidence in a
prediction.
1.3.2 Models implemented within maximum likelihood framework
Nielsen and Yang (1998) and Yang et al. (2000a) developed codon-based
models that account for variation of the co ratio among sites. In these models
the CO ratio takes values from a number of discrete site classes or from a
continuous distribution. ML models of among site co-variation are
implemented in the ML framework and can be used: (i) to test for presence of
codon sites affected by positive selection, and (ii) to identify such sites when
they exist using Bayesian inference. Although ML m odels can be
computationally intense for large number of lineages, they do not rely on
ancestral reconstruction like methods described in the previous section, and
so can tolerate divergent data.
The ML approach involves the likelihood ratio test (LRT) of two nested
models, one of which does not account for sites with cy > 1 w hile another
does. A gene is considered to be under positive selection if (i) the LRT is
significant, and (ii) at least one of the ML estimates of cy is > 1. When the ML
parameter estimates and the LRT indicate presence of sites under positive
selection, the empirical Bayesian approach can be used to predict them
(Nielsen and Yang, 1998; Yang et al., 2000a). One com putes the posterior
probability that a site belongs to each co class of the m odel given the data at