MAIA, JESSICA M. Joint Analysis of Multiple Gene Expression Traits to Map Expression Quantitative Trait Loci. (Under the direction of Professor Zhao-Bang Zeng).
The goal of this dissertation is to address the issue of how to meaningfully find
quan-titative trait loci (QTL) for correlated traits. It has been shown in the literature that a joint
QTL analysis of multiple traits can have more power and be more precise than single trait
QTL analysis when traits are correlated. Phenotypic correlation arises from environmental
correlation, genetic correlation, or both. We wish to characterize the extent of the genetic
correlation among traits.
First, we use a canonical transformation, in the form of principal component analysis,
to combine many correlated traits into one, and apply single trait QTL analysis to it. We
analyzed two different data sets: one from Saccharomyces cerevisiae, and another from
eucalyptus. The traits analyzed in both data sets were gene expression levels generated in
microarray experiments.
Subsequently, we implemented a novel multiple trait mapping method based on
Multi-ple Interval Mapping to functionally related clusters previously studied in Saccharomyces
cerevisiae. Treating RNA abundance as a phenotypic trait, we quantified the extent of
the phenotypic variance due to genetic variance, and found additional QTL, previously
undetected, which were functionally related to the clusters being studied.
The last part of our research contains a study of QTL for individual amino acid
biosyn-thetic pathways of Saccharomyces cerevisiae. In the first part of this chapter, we look at the
QTL topology for all individual amino acid biosynthetic pathways, finding a major
tran-scriptional regulatory region for traits in these pathways. In the second part, we look at the
QTL topology of some individual amino acid biosynthetic pathways in detail, paying close
by
Jessica Mendes Maia
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Doctor of Philosophy
Bioinformatics
Raleigh, North Carolina
2007
Approved By:
Dr. Trudy Mackay Dr. Jung-Ying Tzeng
Dr. Zhao-Bang Zeng Dr. Dahlia Nielsen
Dedication
Biography
Jessica Maia was born in Long Beach, California. She grew up in Brazil, in the small
historic town of Mariana, in the state of Minas Gerais. She has two younger brothers, Eric
Acknowledgements
I would like to thank my advisor, Dr. Zhao-Bang Zeng, and the members of my committee:
Dahlia Nielsen, Trudy MacKay, and Jung-Ying Tzeng for fruitful discussions and academic
Contents
List of Figures ix
List of Tables xi
1 Introduction 1
1.1 Foreword . . . 1
1.2 QTL Analysis Review . . . 2
1.2.1 Genetic Maps . . . 3
1.2.2 QTL Studies . . . 3
1.2.3 Yeast Expression QTL Studies . . . 5
1.2.4 Eucalyptus Expression QTL Studies . . . 9
1.3 Review of Quantitative Trait Locus Analysis Methods . . . 12
1.4 Single Trait QTL Mapping Methods . . . 12
1.4.1 Single Marker Analysis . . . 12
1.4.2 Interval Mapping . . . 14
1.4.3 Composite Interval Mapping . . . 17
1.4.4 Multiple Interval Mapping . . . 19
1.5 Threshold for QTL detection . . . 20
1.6 Thesis chapters . . . 21
2 Using Principal Component Analysis for Expression Quantitative Trait
Lo-cus Mapping 27
2.1 Abstract . . . 27
2.2 Introduction . . . 28
2.3 Materials and Methods . . . 31
2.3.1 Expression Data Sets . . . 31
2.3.2 Linkage map . . . 31
2.3.3 Principal component Analysis . . . 32
2.3.4 Clustering Analysis . . . 32
2.3.5 Gene annotation . . . 33
2.3.6 QTL Analysis . . . 34
2.4 Eucalyptus Results . . . 35
2.4.1 Clustering and QTL Analyses . . . 35
2.4.2 Eucalyptus Cluster Annotation . . . 37
2.4.3 Eucalyptus Principal Component Analysis Results . . . 38
2.5 Yeast Results . . . 41
2.6 Discussion . . . 47
2.7 Acknowledgements . . . 49
2.8 References . . . 50
Appendix . . . 54
3 Multiple Trait Multiple Interval Mapping 77 3.1 Abstract . . . 77
3.2 Introduction . . . 78
3.3 MT-MIM Model . . . 80
3.4 Likelihood . . . 82
3.5 MT-MIM Strategy . . . 84
3.6 Model Selection . . . 84
3.7 Hypothesis testing . . . 87
3.8 Data Analysis . . . 88
3.8.1 Cluster E . . . 89
3.8.2 Cluster G . . . 92
3.8.3 Cluster H . . . 96
3.9 Discussion . . . 100
3.10 Materials and Methods . . . 103
3.10.1 Linkage map . . . 103
3.10.2 Data . . . 103
3.10.3 Missing Markers . . . 103
3.10.4 QTL Analysis . . . 103
3.11 Acknowledgements . . . 104
3.12 References . . . 105
Appendix . . . 108
Genetic Correlation and Heritability . . . 109
Cluster E - Genetic Correlations . . . 110
Cluster G - Genetic Correlations . . . 111
Cluster H - Genetic Correlations . . . 113
4 Multiple Trait Multiple Interval Mapping for Pathway Analysis in Yeast 115 4.1 Abstract . . . 115
4.2 Introduction . . . 116
4.3 QTL Analysis . . . 118
4.3.1 Distribution of QTL per Pathway . . . 118
4.3.3 Leucine Pathway . . . 127
4.3.4 Isoleucine Pathway . . . 127
4.3.5 Arginine Pathway . . . 128
4.3.6 Lysine Pathway . . . 128
4.3.7 Methionine Pathway . . . 128
4.3.8 The Role of GCN4 . . . 129
4.4 Phenotypic and Genetic Correlation . . . 130
4.5 Discussion . . . 135
4.6 Materials and Methods . . . 137
4.6.1 Data . . . 137
4.6.2 Pathways . . . 137
4.6.3 QTL Analysis . . . 138
4.7 References . . . 139
Appendix . . . 142
Pathway Gene Fuction . . . 143
List of Figures
Figure 1.1 Eucalyptus cross design. . . 10
Figure 2.1 QTL locations for all eucalyptus traits . . . 37
Figure 2.2 QTL locations for all eucalyptus genes . . . 55
Figure 2.3 QTL locations for principal components of all eucalyptus genes . . 56
Figure 2.4 QTL location for the first 12 principal components of all genes . . . 57
Figure 2.5 QTL locations for genes in cluster G2 . . . 58
Figure 2.6 QTL locations for genes in cluster G3 . . . 59
Figure 2.7 QTL locations for genes in cluster G4 . . . 60
Figure 2.8 QTL locations for genes in cluster G5 . . . 61
Figure 2.9 QTL locations for principal components of cluster G3 . . . 62
Figure 2.10 QTL locations for genes in cluster G6 . . . 63
Figure 2.11 QTL locations for genes in cluster G7 . . . 64
Figure 2.12 QTL location for principal components of cluster G7 . . . 65
Figure 2.13 QTL locations for genes in cluster G8 . . . 66
Figure 2.14 QTL location for principal components of cluster G8 . . . 67
Figure 2.15 QTL locations for genes in cluster G9 . . . 68
Figure 2.16 QTL location for principal components of cluster G9 . . . 69
Figure 2.17 QTL locations for genes in cluster G10 . . . 70
Figure 2.18 QTL location for principal component 6 of cluster G10 . . . 71
Figure 2.20 QTL location for principal component 7 of cluster G13 . . . 73
Figure 2.21 QTL locations for genes in cluster G15 . . . 74
Figure 2.22 QTL location for principal components of cluster G15 . . . 75
Figure 2.23 QTL locations for genes in cluster G16 . . . 76
Figure 4.1 Distribution of QTL per chromosome . . . 121
Figure 4.2 Length and position of pathway QTL in chromosome III . . . 123
Figure 4.3 Phenotypic correlations . . . 131
Figure 4.4 Genetic correlations for pathway traits . . . 133
List of Tables
Table 1.1 Backcross QTL and marker genotype frequencies and effects . . . . 14
Table 1.2 QTL genotype probabilities given three marker genotype classes . . . 15
Table 1.3 QTL genotype probabilities given two marker genotype classes . . . 16
Table 2.1 Distribution of QTL for each eucalyptus cluster . . . 36
Table 2.2 Principal components per cluster with at least one QTL . . . 39
Table 2.3 Principal component QTL of traits with QTL located in a particular linkage group . . . 41
Table 2.4 Cluster B QTL location . . . 43
Table 2.5 Cluster E QTL location . . . 44
Table 2.6 Cluster F QTL location . . . 45
Table 2.7 Cluster H QTL location . . . 46
Table 3.1 Example: QTL positions . . . 85
Table 3.2 Example (continued): close linkage model . . . 86
Table 3.3 Example (continued): pleiotropic model . . . 86
Table 3.4 MIM effects - cluster E . . . 90
Table 3.5 MT-MIM cluster E initial model . . . 91
Table 3.6 Cluster E MT-MIM and MIM additive effects . . . 91
Table 3.7 Cluster E heritabilities . . . 92
Table 3.9 MT-MIM cluster G initial model . . . 95
Table 3.10 Cluster G MT-MIM and MIM additive effects . . . 95
Table 3.11 Cluster G heritabilities . . . 96
Table 3.12 MIM H cluster . . . 97
Table 3.13 Cluster H initial MT-MIM model . . . 98
Table 3.14 Cluster H - MT-MIM and MIM additive effects . . . 99
Table 3.15 Cluster H heritabilities . . . 100
Table 3.16 Cluster E genetic correlations . . . 111
Table 3.17 Cluster G genetic correlations - Part I . . . 112
Table 3.18 Cluster G genetic correlations - Part II . . . 112
Table 3.19 Cluster H genetic correlations - Part I . . . 113
Table 3.20 Cluster H genetic correlations - Part II . . . 114
Table 4.1 Distribution of QTL per pathway. . . 119
Table 4.2 Percentage and number of QTL per chromosome. . . 122
Chapter 1
Introduction
1.1
Foreword
Recently, quantitative trait locus analysis has been applied to microarray experiments,
treating RNA transcript abundance as a phenotypic trait (Brem et al., 2002; Yvert et al.
2003; Schadt et al. 2003; Kirst et al. 2005; Li et al. 2006). A quantitative trait locus (QTL)
for a gene expression trait is a regulatory region which has a polymorphism in the
segre-gating population. Expression QTL studies, which examine thousands of gene expression
levels, differ drastically from previous QTL studies which analyzed fewer number of traits.
The increase in the number of phenotypes presents new challenges to the realm of
quantitative trait locus mapping methods such as automating QTL software to perform
thousands of genotype vs. phenotype associations, and establishing QTL detecting
thresh-olds which take into account the multiplicity of hypothesis testing. In addition, groups of
into account when finding QTL for correlated traits.
This dissertation addresses the issue of finding quantitative trait loci for correlated gene
expression traits. It has been shown in the literature that a joint QTL analysis of multiple
traits can have more power and be more precise than single trait QTL analysis when traits
are correlated (Jiang and Zeng 1995; Knott and Haley 2000; Sorensen et al. 2003).
We approach the challenge of finding common expression QTL (eQTL) among traits in
two ways. First we find QTL for each expression trait individually, and scan the genome for
shared QTL among traits. Some of these shared QTL represent transcriptional regulatory
regions common to many traits. We then group gene expression levels which share a QTL
according to function or using cluster analysis. Given groups of related genes, most of
which share a QTL, we use a novel multiple trait QTL mapping method to estimate genetic
correlation among traits due the QTL they share.
Our second approach involves reducing the number of expression traits by dimension
reduction. We reduce the number of gene expression levels using principal components
analysis. We then apply single trait QTL mapping to these principal component traits,
hoping that the QTL we find for these principal components have similar location to QTL
which were found for each of the expression traits individually.
1.2
QTL Analysis Review
Using fine-scale molecular maps to find regions in the genome associated with a trait of
interest has been done successfully for many years (Lander and Botstein 1989; Haley and
Knott 1992; Jansen 1993; Zeng 1994). In this section, we review a few QTL studies and
some quantitative trait locus mapping methods, starting with the simplest method, which is
1.2.1
Genetic Maps
The goal of quantitative trait locus studies is to find regions of the genome associated
with a trait of interest. For these types of studies, measurements of the trait of interest
(phenotypic measurements) are needed as well as a genetic map.
Genetic maps show the order of markers on a chromosome, and the distance between
markers as a fraction of the recombination frequencies between them. One of the most
common mapping functions were introduced by Haldane (1919) and Kosambi (1944).
Hal-dane’s mapping function assumes that crossovers occur randomly and independently from
one another. Haldane’s mapping function is given by:
m=−ln(1−2c)
2 ,
wherecis the observed recombination frequency, andm is the map distance in Morgans. Kosambi’s mapping function allows for small interference and is given by:
m = 1 4ln
1 + 2c 1−2c
,
wheremandcare the same as in the Haldane’s mapping function.
1.2.2
QTL Studies
QTL studies have been done on a number of traits in different species such as bristle
number in Drosophila (Payne 1918; Thoday 1961), leanness in pigs (Smith and Bampton
1977), seed and pigment weight in beans (Sax 1923), heterosis in maize (Stuber et al.
1992), prolificacy in sheep (Pipe and Bindon 1982), among many others. The common
thread about these traditional QTL studies is that the number of traits being studied is
small.
More recently, QTL mapping methods have been used to find transcriptional regulatory
mapping has been applied to microarray experiments in order to better understand the
na-ture of regulatory regions of gene expression levels (Brem et al. 2002; Schadt et al. 2003;
Li et al. 2006). Using QTL mapping with mRNA abundance is treated as a phenotypic
trait, one can identify gene expression regulatory regions for each trait separately. This
allows for the study of patterns of cis vs. trans regulation in the entire data set (Yvert et al.
2003; Kirst et al. 2005).
Some expression QTL studies have a goal to better understand transcriptional
regula-tory regions. For example, Brem and colleagues (2002) and Yvert and colleagues (2003)
studied transcriptional regulatory patterns in yeast, revealing whether cis or trans
regula-tion is more prevalent. This issue is at the heart of whether transcripregula-tional regularegula-tion occurs
at the site of the gene whose RNA abundance is being studied (cis-regulation), or at some
other regulatory region (trans-regulation). Gibson and Weir (2005) discuss some
quantita-tive aspects of eQTL studies and summarize the extent of cis vs. trans regulation in various
experiments.
Other eQTL papers tie the eQTL regions found by analyzing gene expression levels, to
a phenotypic trait such as lignin biosynthesis in eucalyptus (Kirst et al. 2004), or fat pad
mass and obesity in mice (Schadt et al. 2003). For example, Kirst and colleagues (2004)
applied correlation analysis to each of the gene expression levels and diameter growth.
Some expression traits highly correlated with diameter growth also shared QTL with the
diameter growth trait.
Li et al. (2006) studied eQTL by environment interactions in C. elegans. RNA
abun-dance was measured in 80 samples at two different temperatures, at which there would be
differences in body size, lifespan and other characteristics. Li and collaborators (2006)
found that a significantly greater percentage of trans-acting genes showed eQTL by
envi-ronment interaction compared to cis-acting genes.
for eucalyptus and yeast, which are two data sets which will be studied further in this
dissertation.
1.2.3
Yeast Expression QTL Studies
The budding yeast data set which we used in our analysis was published in several
installments (Brem et al. 2002; Yvert et al. 2003; Brem and Kruglyak 2005). Each
installment expanded the number of segregants in the population. Subsequently, there have
been several studies which analyzed this same data set. Next we will describe the first two
papers which made the expression traits publicly available (Brem et al. 2002; Yvert et al.
2003) and some others which we find relevant.
Brem and colleagues (2002) studied transcriptional regulation in Saccharomyces
cere-visiae. Gene expression levels and marker genotypes were observed on two parental strains
and their progeny. The parental strains are from a laboratory strain (BY) and a wild strain
(RM). The progeny consists of 40 haploid samples.
Gene expression levels for 6,215 genes or expression traits were measured in the parental
strains with 6 replications. Twenty five percent of genes were differentially expressed
between the parental strains at a p-value < 0.005. These results were found using the Wilcoxon-Mann-Whitney test, permuting the data set to obtain a significance threshold.
Gene expression levels were also observed for 40 haploid progeny samples obtained
from the cross between the parental strains. Heritability for these expression traits was
computed for the progeny as a function of the ratio of the parental expression variance
to the segregant expression variance with the formula: (segregant variance - parent
vari-ance)/segregant variance. The expression traits were shown to have median heritability of
84%.
(2002) performed single marker analysis on 6,215 traits with 3,312 markers. A total of 570
expression traits showed associated with one or more markers with a p-value<5×10−5.
Twenty percent of differentially expressed genes showed linkage to at least one marker
with a p-value<5×10−5.
There are 262 gene expression traits which are linked to at least one marker in the
genome (p-value < 5× 10−5) which are not differentially expressed between parental
strains (p-value<0.005). Brem and colleagues (2002) stipulate that these linkages could be false positives; or that in a given parent, many alleles with opposing effects regulate
gene expression, which would lower the expression difference between parents; or that
difference in expression levels between parents exists but there is lack of power to detect
QTL.
There are 1220 expression traits differentially expressed between parents (p-value <
0.005) which are not linked to any marker. Brem and colleagues (2002) argue that is
be-cause transcription is regulated by multiple loci, each with a small effect. For only about
20% of the genes differentially expressed is the single marker effect big enough to be
detected. Thirty-six percent of 570 traits with an eQTL are cis-regulated. Brem and
col-leagues define trait to be cis-regulated if the marker linked to it is within 10kb of that trait.
In the second yeast expression QTL paper published by the same group as Brem et al.
(2002), Yvert and colleagues (2003) expanded the initial data set, by increasing the number
of segregants in the cross to 86 samples. Yvert and colleagues (2003) found expression
QTL, using single marker analysis, for the expression of all genes in the yeast genome.
They found that 75% of all QTL were trans-acting QTL, and that most of these QTL were
not enriched for transcriptional factors.
In addition, Yvert and colleagues (2003) used hierarchical clustering to define gene
expression clusters. Yvert and colleagues (2003) focused on clusters in which gene
genes are expected to have pair wise correlation greater than 0.725 by chance. In
chap-ter 3, we re-analyze a subset of cluschap-ters shown in this paper, finding additional QTL and
genes under the QTL peaks which have similar functions to genes in a cluster. Yvert and
colleagues (2003) positionally cloned two trans-acting regulators; each regulator contains
a polymorphism and affects the transcription of functionally related genes in that cluster.
Brem and Kruglyak (2005) conclude that most QTL they detect for the yeast data set
they use have weak effects. The data set is the same as in Brem et al. (2002) and Yvert et al.
(2003), except that the number of segregants was increased to 112 samples and the number
of markers is 2,957. Single marker analysis was performed to detect QTL, and permutations
were used to declare QTL at 5% false discovery rate (Storey and Tibshirani 2003). Brem
and Kruglyak (2005) claim to find epistasis for about 16% of highly heritable transcripts
(h2 >0.687). The heritability of each transcript,h2, was computed ash2 = (σ2
s −σp2)/σ2p,
where σ2
p, σs2 are the pooled variance among parental measurements, and the phenotypic
variance among segregants.
Storey and collaborators (2005) used data from the same yeast experiment as Brem
and Kruglyak (2005) to come up with a new scheme to estimate epistasis between QTL.
Storey et al. (2005) performed QTL analysis of 6,216 yeast expression traits, using 3,312
markers and 112 haploid segregants. Storey et al. (2005) claim their sequential search is
more powerful to find main QTL effects and epistatic effects as compared to an exhaustive
2-dimentional scan.
The exhaustive 2-dimentional scan works the following way: for every expression trait,
every pair of markers is fitted to a model which includes two QTL main effects and an
epistatic effect. The model is:
expression= baseline level+locus1 effect + locus2 effect + epistatic effect + noise. (1.1)
In contrast with the 2-dimentional scan, Storey et al. (2005) used a sequential genome
are:
M0: expression=baseline level + noise
M1: expression=baseline level + locus1 + noise
M2: expression=baseline level + locus1 + locus2 + epistasis + noise.
In step one, one selects the QTL which shows the most improvement in the goodness of
fit of model M1 compared with model M0. Then Storey et al. (2005) select a second
QTL which is the one that shows the greatest improvement in the goodness of fit of M2
compared with model M1. A total of 170 QTL pairs were found to be significant under
model M2 at a false discovery rate of 10%.
Zou (2006) using the same data set, was able to find more gene expression traits which
had 2 QTL only but less epistatic interactions than Storey et al. (2005) by performing a
sequential genome scan slightly different than what is presented in Storey et al. (2005).
Zou’s method used Multiple Interval Mapping (Kao et al. 1999) on one expression trait at
a time. Zou’s strategy is as follows:
(1) search for the first QTL and add it into the model if its effect is significant at type I
error rate of 10%, obtained through permutations;
(2) if a QTL is found in step 1, add one QTL at a time into the model conditional on the
existing QTL in the model (given that the QTL effect is greater than the threshold);
(3) search for epistatic interactions between QTL found in steps 1 and 2;
(4) delete QTL from the model that are not significant at a type I error rate of 5%.
The main difference between the models of Zou (2006) and Storey et al. (2005) is
that Zou searches for epistatic interactions only after all the QTL main effects have been
added into the model. In addition, QTL are only added into the model if their effects are
significant. Surprisingly, the number of expression traits controlled by only 2 QTL at a false
discovery rate of 10% found by Zou is 729 compared with 170 of Storey et al. (2005).
trait locus analysis to genes expression levels of a eucalyptus inbred cross. The next section
describes some results for this data set.
1.2.4
Eucalyptus Expression QTL Studies
Kirst and colleagues (2005) studied transcriptional regulation of genes in an Eucalyptus
pseudobackcross: E. grandis × F1 hybrid (E. grandis × E. globulus). E. globulus has
high wood density and relatively slow growth; E. grandis has lower wood density but
faster growth. Crosses between these two strains have shown ample genetic and phenotypic
Copyright © 2007 by the Genetics Society of America
Kirst, M. et al. Genetics 2005;169:2295-2303
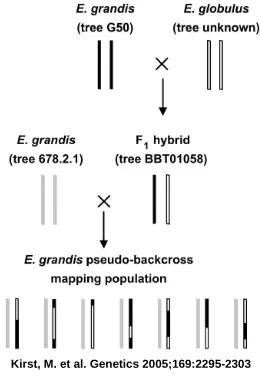
FIGURE 1.-- Mating design of the E. grandis pseudobackcross mapping population
Figure 1.1: Eucalyptus cross design.
Kirst and colleagues (2005) set out to discover the transcriptional regulation differences
between individuals in the same species and individuals in two related species. They used
the two marker maps to find expression QTL for 91 progeny samples. One map is that of the
F1 hybrid (tree BBT01058) and the other of the E. grandis backcross parent (tree 678.2.1)
(Figure 1.1). In the E.grandis marker map, there were a total of 96 AFLP fragments, in
12 major linkage groups; in the F1 hybrid map, there were a total of 122 fragments which
Of the 2,608 genes considered, 1373 (53%) were differentially expressed. Using
Com-posite interval mapping (Zeng 1994), Kirst and colleagues (2005) identified eQTL for 811
genes using the F1 hybrid map, and 451 eQTL using the E.grandis map. These eQTL were
significant using a type I error rate of 10% obtained through permutations.
Combining the eQTL data from both maps, 1067 traits had a total of 1655 eQTL. A total
of 821 gene expression traits had only one significant eQTL. Kirst and colleagues (2005)
estimated epistatic interaction via Multiple Interval Mapping (Kao et al. 1999). Epistatic
interactions were significant for 310 genes in the F1 hybrid map, and 285 genes in the E.
grandis map.
A total of 195 genes had eQTL in both marker maps, and 13 of these genes were located
in homologous regions. This suggests that most eQTL were trans-acting. Kirst et al. (2005)
argue that if cis-regulation were more prevalent, then we would see more homologous
eQTL. This result is the similar to yeast eQTL study of Yvert et al. (2003) which found
that trans-regulation is more prevalent than cis-regulation in yeast. Transcriptional hotspots
were found using both eucalyptus maps.
In another paper, Kirst et al. (2004) tied gene expression regulatory regions to regions
which regulate growth variation, previously detected by QTL mapping. The experimental
cross is the same one described previously in Kirst et al. (2005). QTL analysis for diameter
growth and for each of the 2,608 genes was done for 91 samples using Composite Interval
Mapping (Zeng 1993, 1994). Two significant QTL (experiment α = 0.01) for diameter growth were identified.
Subsequently, Kirst and colleagues (2004) applied correlation analysis to each of the
2,608 gene expression levels and diameter growth. A total of 37 gene expression levels
were correlated with growth (individual test significant threshold of 0.0001), most of which
were negatively correlated with diameter growth.
phenyl-propanoid pathways. High lignin content in a tree can be detrimental to growth (Kirst et
al. 2004). Then Kirst and colleagues (2004) confirmed that diameter growth and lignin
content were negatively correlated by sampling 8 individuals from the backcross progeny.
Kirst and colleagues (2004) also found common QTL for diameter growth and expression
levels of genes in lignin biosynthesis.
1.3
Review of Quantitative Trait Locus Analysis Methods
1.4
Single Trait QTL Mapping Methods
1.4.1
Single Marker Analysis
To find associations between a trait and a marker in a population, with marker classes
M/M,M/m, andm/mat a given loci, one can perform a parametric test such as thet-test or a non-parametric test such as the Wilcoxon-Mann-Whitney test. These tests can find
significant difference between trait means of different marker groups.
LetµM M,˜ µM m,˜ andµmm˜ be the observed trait means for individuals with marker geno-typesM/M, M/m, andm/mat a given locus respectively. And letnM M, nM m,andnmm
be the sample size of the marker classes, ands2
M M, s2M m, ands2mm be the sample variance
for each class.
Next we will give thetstatistic for a backcross and an F2population (Zeng 2000). For
additive marker effect is:
t = qµM M˜ −µmm˜ s2( 1
nM M +
1 nmm)
, where (1.2)
(1.3)
s = (nM M −1)s 2
M M+ (nmm−1)s2mm
nM M +nmm−2 . (1.4)
The test for dominance effect in the F2 population is:
t2 = ˜
µM m−µM M/˜ 2−µmm˜ /2 q
s2( 1 nM m +
1
4nM M +
1
4nmm)
, where (1.5)
(1.6)
s2 = (nM M−1)s 2
M M + (nM m−1)s2M m+ (nmm −1)s2mm nM M+nM m+nmm−3
. (1.7)
In a backcross population, there are only two marker classes, denoted here byM Mand
M m. The test statistic for a backcross population is:
t= qµ˜M M −µ˜M m s2( 1
nM m +
1 nM M)
, (1.8)
where
s2 = (nM M −1)s 2
M M+ (nM m−1)s2M m
nM M +nM m−2 . (1.9)
Single marker analysis for QTL mapping has been used for many years. Two problems
with single marker analysis are: it does not estimate QTL position and the difference in
trait means of marker classes is confounded with the recombination frequency between the
QTL and its flanking markers.
Table 1.1: Backcross QTL and marker genotype frequencies and effects (Zeng 2000)
QQ Qq
M M Frequency 1−r r
Effect µ+a µ+d
M m Frequency r 1−r
Effect µ+a µ+b
Below we will see how the difference in the trait means between marker classes in a
back-cross population is confounded by the recombination frequencyr:
µM M−µM m = [(1−r)(µ+a) +r(µ+d)]−r[(r(µ+a) + (1−r)(µ+d)](1.10)
= (1−2r)(a−d). (1.11)
Next we will find an improvement to single marker analysis, by a QTL mapping method
named interval mapping.
1.4.2
Interval Mapping
The precision of quantitative trait locus mapping methodology has increased
signifi-cantly since Lander and Botstein (1989) proposed Interval Mapping (IM), which can
esti-mate a QTL effect based on a marker map. Nonetheless, interval mapping was the
foun-dation for future QTL mapping methods, and the paper which introduced it (Lander and
Botstein 1989) was very influential.
First, let’s establish the possible probabilities of a QTL genotype based on its flanking
marker genotypes. LetrMiQ be the recombination fraction between markerMi and QTL
Qand letrMiMi+1 be the recombination fraction between markersMi andMi+1. The next
population with three marker classes such as anF2population.
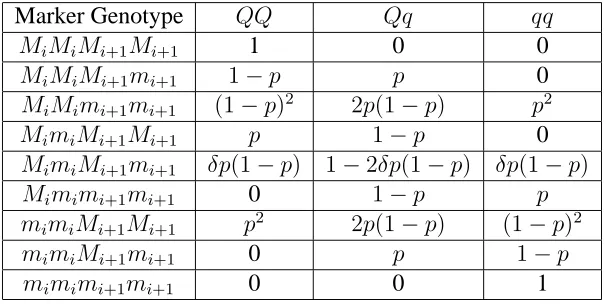
Table 1.2: QTL genotype probabilities given marker genotypes (Jiang and Zeng 1995)
Marker Genotype QQ Qq qq
MiMiMi+1Mi+1 1 0 0
MiMiMi+1mi+1 1−p p 0
MiMimi+1mi+1 (1−p)2 2p(1−p) p2
MimiMi+1Mi+1 p 1−p 0
MimiMi+1mi+1 δp(1−p) 1−2δp(1−p) δp(1−p)
Mimimi+1mi+1 0 1−p p
mimiMi+1Mi+1 p2 2p(1−p) (1−p)2
mimiMi+1mi+1 0 p 1−p
mimimi+1mi+1 0 0 1
wherep=rMiQ/rMiMi+1, andδ=r 2
MiMi+1/[(1−rMiMi+1)
2 +r2
MiMi+1]. Double
recombi-nation is ignored.
For a backcross population, the putative QTL genotype can take on two values,QQand
Table 1.3: QTL genotype probabilities given marker genotypes (Zeng 2000)
Marker Genotype QQ Qq
MiMiMi+1Mi+1
(1−rMiQ)(1−rQMi+1)
1−rMiMi+1 ≈1
rMiQrQMi+1
1−rMiMi+1 ≈0
MiMimi+1Mi+1
(1−rMiQ)rQMi+1
rMiMi+1 ≈1−p
rMiQ(1−rQMi+1) rMiMi+1 ≈p miMiMi+1Mi+1
rMiQ(1−rQMi+1) rMiMi+1 ≈p
(1−rMiQ)rQMi+1
rMiMi+1 ≈1−p miMimi+1Mi+1
rMiQrQMi+1
1−rMiMi+1 ≈0
(1−rMiQ)(1−rQMi+1)
1−rMiMi+1 ≈1
wherep=rMiQ/rMiMi+1.
For a backcross population, letyj be the phenotypic trait measurement for individuali;
bbe the effect of a single allele substitution at the QTL;x∗be an indicator random variable
of the QTL genotype; b0 be the mean of the model, andej be a random variable which
follows a normal distribution N(0,σ2). Then interval mapping’s linear model is as follows:
yj =b0+bx∗j +ej. (1.12)
The likelihood equation for interval mapping’s the linear model (equation 1.12) is:
L(b0, b, σ2) = n Y j=1
[Gj(0)Lj(0) +Gj(1)Lj]wherej = 1,· · · , n. (1.13)
The likelihood function is given byLj(x) = z((yj −(b0 +bx∗j)), σ2), where z(x, σ2) = (2πσ2)−1/2exp(−x2/2σ2). The function G
j(x) represents the probability of the QTL
genotypexgiven the flanking marker genotypes (Table 1.3) . In the case of a backcross, the QTL genotypes QQ and Qq correspond to x values of 1 and 0 respectively. Lander and Botstein (1989) use the maximum likelihood analysis to obtain estimates of the model
parameters.
One disadvantage of interval mapping is that the additive effects estimated by interval
Zeng 1994; Kao et al. 1999). In addition, interval mapping is not an interval test, that is,
if there is a QTL, regions linked to it might appear to be significant even when there is no
QTL present in those locations (Zeng 1994).
1.4.3
Composite Interval Mapping
An improvement of the precision and estimates of QTL effects of Interval Mapping
came about when Jansen (1993) and Zeng (1994) independently used regression methods
which could take into account the presence of other QTL effects into a linear model.
Com-posite interval mapping (CIM), as labeled by Zeng (1994), used covariate markers to
esti-mate QTL position and additive effects. The linear model for composite interval mapping
is given by
yj =µ+b∗x∗
j + X k6=i,i+1
bkxjk+ej, (1.14)
where:
yj is the trait value of thejthindividual, µis the mean of model,
b∗is the effect of the QTL expressed as a difference in effects between the homozygous
and heterozygote QTL genotype classesQQandQq,
x∗
j is an indicator random variable taking value 0 or 1 with probability depending on the
genotypes of the flanking marker genotypes and the position of putative QTL (Table 1.3),
bkis the partial regression coefficient of the phenotypeyon thekthmarker conditional
on all other markers,
xjkis an indicator random variable for thejthindividual andkthmarker genotype which
takes values 0 or 1 depending on whether the maker type is homozygote or heterozygote,
and
The known parameters arexjk, b0, yj. Using maximum likelihood analysis, the parameters b∗, bk, σ2 are estimated.
Composite interval mapping, unlike interval mapping, is an interval test. If linked
mark-ers to the QTL are included in the summation in equation 1.14, CIM is able to control for
the effects of other linked QTL in the model (Zeng 1993, 1994). In addition, if epistasis
is ignored, the partial regression coefficient bk, depends only on the QTL located in the marker interval being tested for the presence of a QTL (Zeng 1993, 1994).
The likelihood for a backcross population using composite interval mapping is:
L(b∗, B, σ2) = n Y j=1
[p1jφ(
yj −XjB−b∗
σ ) +p0jφ(
yj −XjB
σ )], (1.15)
whereXjB =µ+P
kbkxjk,
p1j is the probability that the markerx∗j is homozygous, and p0j is the probability that the markerx∗j is heterozygous.
Maximum likelihood analysis is used to find estimates for model parameters are computed
using the expectation maximization (EM) algorithm.
Defining a threshold to add or delete a QTL for this QTL mapping method is non-trivial
because the test statistic under the null hypothesis of no QTL is not known. A threshold
for QTL detection depends on the number of markers included into the linear regression
model, the size of the QTL interval being tested in terms of the genetic distance, and on
the sample size. In his 1994 paper, Zeng suggested that for a large sample size, and when
not too many markers are fitted into the model, that the value ofχ2
α/M,2 can be used as an
approximation for the100α%threshold value when there areM intervals in the genome in some marker scenarios.
Another way of finding an appropriate threshold to declare a QTL is to use
permuta-tions. Churchill and Doerge (1994) suggested simulating the null hypothesis of no QTL by
the maker genotypes for each individual fixed.
1.4.4
Multiple Interval Mapping
Kao et al. (1999) proposed a linear model which can fit multiple QTL into a model,
estimating both additive and epistatic effects of QTL which affect a given trait. They named
their model Multiple Interval Mapping (MIM). MIM can be more precise and powerful than
CIM.
The statistical backcross model for MIM (Kao et al. 1999) for traity, individuali, and
mQTL (Q1,· · · , Qm), can be written in the form:
yi =µ+ m X
r=1
arx∗ir+ m X r6=k
δrk(wrkx∗irx∗ik) +εi, (1.16)
whereµis the mean of the model,
ar is the additive effect of QTLr,
x∗
ir represents the putative QTL genotype for individuali, QTLr, δrkis the indicator variable for epistasis between QTLrand QTLk,
wrkis the epistatic effect between QTLrand QTLk, and
εiis the error term for individuali, which we assume is distributed asN(0, σ2).
The likelihood equation for MIM with a model nsamples, mQTL (Q1,· · · , Qm),
lo-cated in positions (p1,· · · , pm) forθ = (p1,· · · , pm, a1,· · · , am,· · · , wjk,· · · , σ2)is:
L(θ|X, Y) = n Y i=1 2m X j=1
pijφ((yi−µij)/σ), (1.17)
wherepij is a variable containing information about the probability of QTL genotypes. There are different ways of searching for QTL with MIM. One way is an iterative
effect, then tests for epistasis between QTL in the model. After that one would then re-test
QTL in the model for significance, and then optimize QTL positions. These steps would
be performed until no QTL can be added into the model according to a particular QTL
de-tection threshold or stopping criteria such as the information criteria (IC) (Stuart and Ord
1991; Miller 1990).
MIM has some advantages over other QTL mapping methods because unlike interval
mapping and composite interval mapping, with MIM epistatic interactions can be modeled
explicitly; MIM can also give better QTL position estimates because it searches
simultane-ously for QTL in multiple marker intervals.
1.5
Threshold for QTL detection
In classical QTL analysis, the number of traits analyzed is small compared to the
thou-sands and sometimes tens of thouthou-sands of traits analyzed in expression QTL studies. Even
though the statistical methodology of traditional QTL studies is used in the realm of eQTL
studies, figuring out significance levels for various QTL detecting methods is non-trivial.
One way of finding an appropriate threshold to declare a QTL is to use permutations.
Churchill and Doerge (1994) suggested simulating the null hypothesis of no QTL by
per-muting the trait values among individuals in the segregating population, while keeping the
maker genotypes for each individual fixed. One would then record the test values with the
permuted samples and compare them to test values obtained by applying a QTL mapping
method to the data of interest.
Zou et al. (2004) proposed a re-sampling method using the result that the score statistic
is asymptotically equivalent to the likelihood ratio statistic. This method has a much lower
computation burden than the permutation scheme proposed by Churchill and Doerge (1994)
In order to address this issue of testing many null hypotheses, Storey and Tibshirani
(2003) came up with an estimate of the positive false discovery rate (FDR). With this FDR
estimate, one can compute the expected proportion of genes which falsely were declared to
have one or more QTL, given the total number of expression traits with at least one QTL.
The FDR estimate of Storey and Tibshirani (2003) is applicable to eQTL studies where
thousands of QTL are claimed to be significant. Zou (2006) adapted Storey’s false
dis-covery rate methodology to multiple interval mapping (MIM). Model selection is an active
research area in QTL methodology development. Finding an appropriate threshold level
for which to add or delete QTL into a model is still very challenging.
1.6
Thesis chapters
In this first chapter, we described a few expression QTL studies, focusing on yeast and
eucalyptus expression QTL experiments. Then we reviewed a few QTL mapping methods
such as single marker analysis, interval mapping, composite interval mapping and multiple
interval mapping.
In the second chapter, we test the usefulness of principal component analysis in the
realm of QTL mapping. We transform the original traits using principal component
anal-ysis, and examine whether the QTL found for these principal component traits match the
location of QTL for individual traits. We analyzed two different data sets: one from
Sac-charomyces cerevisiae, and another from eucalyptus. The traits analyzed in both data sets
were gene expression levels generated in microarray experiments.
In the third chapter, we implement a novel multiple trait mapping method based on
mul-tiple interval mapping. We propose a way to find an initial model and to test for pleiotropy
vs. close linkage. We then apply this method to functionally related clusters previously
The fourth chapter is an analysis of transcriptional variation of gene expression levels
in yeast individual amino acid biosynthetic pathways. We look at the number of QTL
and the extent of pleiotropy in each pathway. We also estimate genetic correlations and
heritabilities for genes in individual amino acid biosynthetic pathways.
In this dissertation, we treated RNA abundance as a phenotypic trait for two
experi-mental crosses: one from eucalyptus and the other from yeast. Both crosses are modeled as
a backcross in the QTL experiment design. We analyzed a subset of Kirst and colleagues
(2005) eucalyptus data. We used their cDNA arrays consisting of 2,610 gene expression
levels measured on 88 samples of a eucalyptus pseudo-backcross: E. grandis x F1 hybrid
(see Figure 1.1). The average marker spacing is about 1 marker for every 10 centiMorgans.
The final version of the yeast data set we used was a subset of data set published in
Brem and Kruglyak (2005). This data set comes from a cross between a wild (BY) and
laboratory (RM) strains of Saccharomyces cerevisiae. The expressions of 6,195 genes
were measured in 112 haploid samples using a platform of custom open reading frames
(Yvert et al. 2003). In chapter 2, only 86 yeast samples were used which was the number
of available samples at that time. In chapters 3 and 4, 112 yeast samples are used in the
1.7
References
Brem, R.B., G. Yvert, R. Clinton and L. Kruglyak, 2002 Genetic Dissection of
Transcrip-tional Regulation in Budding Yeast. Science 296: 752-755.
Brem, R.B., and L. Kruglyak, 2005 The landscape of genetic complexity across 5,700 gene
expression traits in yeast. PNAS 102:1572-1577.
Churchill, G.A., and R.W. Doerge, 1994 Empirical threshold values for quantitative trait
mapping. Genetics 138: 963-71.
Gibson, G., and B. Weir, 2005 The quantitative genetics of transcription. Trends Genet.
21: 616-623.
Haldane, J.B.S., 1919 The combination of linkage values, and the calculation of distance
between the loci of linked factors. J. Genetics 8: 299-309.
Haley, C.S., and S.A. Knott, 1992 A simple regression method for mapping quantitative
trait loci in line crosses using flanking markers. Heredity 69: 315-24.
Jansen, R.C., 1993 Interval mapping of multiple quantitative trait loci. Genetics 135:
205-211.
Jiang, C., and Z.-B. Zeng, 1995 Multiple Trait Analysis of Genetic Mapping for
Quantita-tive Trait Loci. Genetics 140: 1111-1127.
Jiang, C., and Z.-B. Zeng, 1997 Mapping quantitative trait loci with dominant and missing
markers in various crosses from two inbred lines. Genetica 101: 47-58.
Kao, C.-H., Z.-B. Zeng and R. D. Teasdale, 1999 Multiple interval mapping for quantitative
Kirst, M., A.A. Myburg, J.P. De Leon, M.E. Kirst, J. Scott J, and R. Sederoff, 2004
Co-ordinated genetic regulation of growth and lignin revealed by quantitative trait locus
anal-ysis of cDNA microarray data in an interspecific backcross of eucalyptus. Plant Physiol.
135:2368-2378.
Kirst, M., C.J. Basten, A.A. Myburg, Z-B. Zeng and R.R. Sederoff, 2005 Genetic
Ar-chitecture of Transcript-Level Variation in Differentiating Xylem of a Eucalyptus hybrid.
Genetics 169: 2295-303.
Knott, S.A., and C.S. Haley, 2000 Multitrait least squares for quantitative trait loci
detec-tion. Genetics 156: 899-911.
Kosambi, D.D., 1944 The estimation of map distances from recombination values. Ann.
Eugen. 12: 172-175.
Lander, E.S., and D. Botstein, 1989 Mapping mendelian factors underlying quantitative
traits using RFLP linkage maps. Genetics 121: 185-99. Erratum in: Genetics 1994, 136:
705.
Li, Y., O.A. Alvarez, E.W. Gutteling, M. Tijsterman, J. Fu, et al., 2006 Mapping
deter-minants of gene expression plasticity by genetical genomics in C. elegans. PLoS Genet.
29:e222.
Miller, A.J., 1990 Subset Selection in Regression. Chapman and Hall, London.
Payne, F., 1918 The effect of artificial selection on bristle number in Drosophila
am-pelophila and its interpretation. Proc. Natl. Acad. Sci. U.S.A. 4: 55-58.
Piper, L.R., and R.M. Bindon, 1982 Genetic segregation for fecundidy in Booroola Merino
Congress on Sheep and Beef Cattle Breeding. Dunmore Press, Palmerston North,
Aus-tralia.
Sax, K., 1923 The association of size differences with seed-coat pattern and pigmentation
in Phaseolus vulgaris. Genetics 8:552-560.
Schadt, E.E., S.A. Monks, T.A. Drake, A.J. Lusis, N. Che, et al., 2003 Genetics of gene
expression surveyed in maize, mouse and man. Nature. 422: 269-270.
Smith, C., and P.R. Bampton, 1977 Inheritance of reaction to halothane anaesthesia in pigs.
Genet. Res. 29: 287-292.
Sorensen, P., M.S. Lund, B. Guldbrandtsen, J. Jensen and D. Sorensen, 2003 A comparison
of bivariate and univariate QTL mapping in livestock populations. Genet Sel Evol. 35:
605-622.
Storey, J.D., and R. Tibshirani, 2003 Statistical significance for genomewide studies. Proc.
Natl. Acad. Sci. U.S.A.100: 9440-9445.
Storey, J.D., Akey, J.M. and L. Kruglyak, 2005 Multiple locus linkage analysis of genomewide
expression in yeast. PLoS Biol. 3:e267.
Stuart, A., and J.K. Ord, 1991 Kendall’s Advanced Theory of Statistics. Oxford Univ.
Press, New York, 5th Ed., Vol. 2.
Stuber, C.W., S. E. Lincoln, D. W. Wolff, T. Helentjaris and E. S. Lander, 1992
Identifica-tion of Genetic Factors Contributing to Heterosis in a Hybrid From Two Elite Maize Inbred
Lines Using Molecular Markers. Genetics 132: 823-839.
Yvert, G., R.B. Brem, J. Whittle, J.M. Akey, E. Foss, et al., 2003 Trans-acting regulatory
variation in Saccharomyces cerevisiae and the role of transcription factors. Nat. Genet. 35:
57-64.
Zeng, Z.-B, 1993 Theoretical basis for separation of multiple linked gene effects in
map-ping quantitative trait loci. PNAS 90:10972-10976.
Zeng, Z.-B, 1994 Precision Mapping of Quantitative Trait Loci. Genetics 136: 1457-1468.
Zeng, Z.-B, 2000 Statistical Methods for Mapping Quantitative Trait Loci. Book manuscript.
Zou F, J.P. Fine, J. Hu, and D.Y. Lin., 2004 An efficient resampling method for assessing
genome-wide statistical significance in mapping quantitative trait Loci. Genetics
168:2307-2316.
Zou W., 2006 Transcriptional regulatory patterns in yeast revealed through expression
Chapter 2
Using Principal Component Analysis for
Expression Quantitative Trait Locus
Mapping
2.1
Abstract
In this chapter, principal component analysis is tested as a dimension reduction tool for
gene expression levels, which we consider as phenotypic traits for this study. We investigate
whether principal components can predict the patterns of association between genes and
the regions which regulate their expression. When principal components are used to take
component analysis is not a reliable tool to summarize gene expression levels because
QTL of principal components do not consistently map to the same regions where the QTL
of genes used to compute these principal components are located. However, the QTL
for the first principal component, whenever it exists, maps to the same location as the
QTL locations for genes used to compute the first principal component. We have tested
our methods on two micrroarray data sets. One from a eucalyptus pseudo-backcross and
another from a cross of wild and lab strain of Sacharomyces cerevisiae.
2.2
Introduction
Quantitative trait locus (QTL) mapping has been applied to microarray experiments in
order to better understand the nature of regulatory regions of gene expression levels (Brem
et al. 2002; Schadt et al. 2003; Li et al. 2006). Using QTL mapping with mRNA
abun-dance is treated as a phenotypic trait, one can identify gene expression regulatory regions
for each trait separately. This allows for the study of patters of cis vs. trans regulation in
the entire data set (Yvert et al. 2003; Kirst et al. 2005).
There are however, many reasons why one would want to apply QTL mapping to a
subset of the expression data or a transformation of it. One could apply QTL mapping only
to traits that are clustered together (Yvert et al. 2003), traits which have a related function
(Andersson-Eklund et al. 2000), or gene expression levels which share regulatory regions
(Brem et al. 2002). Principal component analysis (PCA) has also been used in the past
to summarize phenotypic measurements (Liu et al. 1996; Andersson-Eklund et al. 2000).
Here are some arguments why one would want to summarize a data set, or part of it, with
principal component analysis before QTL mapping:
(1) To capture a large amount of the variation with fewer variables. One could start
2000). Weller et al. (1996) suggested using PCA as a form of dimension reduction tool,
by finding QTL for principal components which capture a large percent of the variance,
and ignoring the principal components (PCs) that represent a “small percent of the total
variance”. Weller et al. (1996) use PCA to summarize three phenotypic traits related to
milk production in dairy cattle.
(2) To avoid issues of multiple testing. Finding a QTL for every trait in the dataset
can have less power to detect traits summarized by principal component QTL, because the
QTL mapping threshold has to be adjusted for the number of genotype-trait associations.
Therefore, if there are fewer principal components than traits in the data set, the number of
genotype-trait associations is smaller.
(3) To combine phenotypic measurements which together are indicative of complex
disease. Arguably, it makes sense to combine traits which are different measurements of
the same phenotype, and find QTL for the combined trait instead of each phenotypic
mea-surement separately. One example is osteochondrosis, a generalized skeletal disease in
pigs (Andersson-Eklund et al. 2000). In this study, eight bone density measurements were
combined by principal components; in a separate principal component analysis, four
os-teochondrosis scores from examination of lesions were combined as well. The correlation
between the bone density and osteochondrosis is small (Andersson-Eklund et al. 2000).
Both sets of traits (bone density and osteochondrosis measurements) were indicative of the
presence of osteochondrosis.
(4) To lessen the computational burden. Although the computation time for finding
associations between or thousands of genes and thousands of markers or marker intervals
takes a few days or less (Zou 2006), setting up scripts which automate the search for QTL
usually takes more time than performing the analysis itself, and requires knowledge of how
to write scripts which automate these tasks.
(Mahler et al. 2002; Andersson-Eklund et al 2000; Lan et al. 2003; Liu et al 1996).
What all of these studies have in common is the fact that principal component analysis is
applied to a small number of traits. The underlying assumption of these studies is that QTL
for traits to be summarized with PCA will be in close proximity to QTL of principal
com-ponents. The main goal of this report is to see if QTL for principal components can predict
QTL locations for individual traits (gene expression levels) that have a common function,
or that are clustered together, or both. Unlike other studies, this study looks at QTL patterns
for individual traits and for principal components in small and large trait clusters.
We treat RNA abundance as a phenotypic trait for two experimental crosses: one from
eucalyptus and the other from yeast. Both crosses are modeled as a backcross in the QTL
experiment design. In the first part of this report, we analyze the eucalyptus data set (Kirst
2004; Kirst et al. 2005). A two-way coupled clustering method is applied to the eucalyptus
gene expression levels. Then QTL analysis is applied to all traits in this data set.
Subse-quently, traits in each cluster which have one or more QTL are annotated. Then principal
component analysis and QTL mapping are performed on traits that are clustered together,
and on traits that have QTL located in common regions.
In the second part of this chapter, we compute principal components for yeast gene
expression levels clustered by Yvert et al. (2003). These clusters are composed mostly of
functionally related genes, which also have a high gene expression pair wise correlation.
We then find QTL for each trait in a cluster, and for the principal components of those
2.3
Materials and Methods
2.3.1
Expression Data Sets
We treated RNA abundance as a phenotypic trait for two experimental crosses: one
from eucalyptus and the other from yeast. Both crosses are modeled as a backcross in the
QTL experiment design. We analyzed a subset of Kirst and colleagues (2005) eucalyptus
data. We used their cDNA arrays consisting of 2,610 gene expression levels measured on
88 samples of a eucalyptus pseudo-backcross: E. grandis x F1 hybrid (E. grandis x E.
globulus). The average marker spacing is about 1 marker for every 10 centiMorgans.
The yeast data set we used was a subset of data published by Brem and Kruglyak
(2005). This data set comes from a cross between a wild (BY) and laboratory (RM) strains
of Saccharomyces cerevisiae. The expressions of 6,195 genes were measured in 86 haploid
samples using a platform of custom open reading frames (Yvert et al. 2003). The marker
density of this cross is on average one marker for every 3kb. There are a total of 2956
markers in this data set. The yeast microarray data was downloaded from Gene expression
Omnibus (Edgar et al. 2002), reference series: GSE1990.
2.3.2
Linkage map
The yeast linkage map was constructed using Mapmaker (Lincoln et al. 1992; Paterson
et al. 1988). The Saccharomyces cerevisiae genotypic data was obtained from direct
communication with Rachel Brem. The eucalyptus F1 hybrid paternal map (Kirst 2004;
Kirst et al. 2005) linkage map was obtained via personal communication with Matias
2.3.3
Principal component Analysis
Principal component analysis is a linear transformation of the data. For example,
imag-ine a data set with 88 samples and 2,610 gene expression levels. Each sample has a trait
measurement for each of the 2,610 traits. If one computes 12 principal components from
this data, each sample now will have only 12 trait measurements. The genotype information
for each sample remains unchanged.
Principal component 1 (PC1) captures a greater variance than any other; let v(PC(i)) be the variance in the data captured by principal component i. If there are k principal com-ponents in the data set, then v(PC(1))>v(PC(2)) >· · ·>v(PC(k)). Principal component analysis was done using proc princomp of SAS version 8.02 and JMP software.
2.3.4
Clustering Analysis
When applied to expression data, one-way clustering analyses assigns traits to groups
based on their similarity or dissimilarity. There are many types of clustering analyses
such as hierarchical methods (bottom-up and top-down methods), which yield nested sets
of clusters, and partitioning methods (K-means clustering, self organizing maps), which
assigns genes to a fixed number of clusters (Chipman et al. 2003).
Two-way clustering analysis seeks to cluster both samples and gene expression levels
at the same time. Two sets of gene expression levels might yield a different clustering of
the samples (Chipman et al. 2003). For the eucalyptus data set coupled two-way clustering
(CTWC) method was used to perform two-way clustering of the data (Getz et al. 2000).
This coupled two-way clustering method has been used in several studies (Alon et al. 1999;
Golub et al. 1999; Godard et al. 2003; Getz et al. 2003). The CTWC method uses subsets
of genes to cluster samples and vice-versa (Getz et al. 2000) because if one uses all traits to
other interesting clusters of samples (Chipman et al. 2003).
When applied to the eucalyptus data set, the CTWC method produced a total of 17 gene
expression clusters and 9 eucalyptus sample clusters. Four out of seventeen gene clusters
were excluded from the QTL analysis and principal component analysis because they had
972 or more traits, where the total number of traits is 2610. These four clusters were
excluded because they have at least 37% of all traits in the data set and are likely to contain
genes representing different biological processes. The clustering results were obtained by
uploading our data to an online server (Getz and Domany 2003).
For the Saccharomyces cerevisiae data set, we used four existing gene clusters for
prin-cipal component and QTL analysis. Yvert et al. (2003) clustered genes which are
function-ally related and/or have high phenotypic correlation (pair wise correlation>0.725). Genes in clusters B,E,F (discussed in section 2.5) have similar function and are highly correlated.
Functions of sixteen genes in cluster H are not known, although their gene expression levels
are highly correlated.
2.3.5
Gene annotation
Annotation for gene clusters was done using Blastx (Altschul et al. 1990), which
trans-lates a nucleotide query into a protein sequence, and searches for similarities among the
protein sequence of interest and all protein sequences in the database. The advanced
op-tions for the Blastx searches were set at default. The top three Blastx matches for each gene
in a cluster were considered if they had an E-value less than 1e-10. At an E-value of 1e-10,
“10 hits with scores equal to or better than the defined alignment score, S, are expected to
occur by chance (in a search of the database using a random query with similar length)”
Saccharomyces cerevisiae genes already clustered by Yvert et al. (2003) were used in
the PCA analysis. Out of the four clusters studied, genes in three of those clusters (B,E,and
F) are functionally related. Genes in cluster H have unknown function.
2.3.6
QTL Analysis
QTL mapping was done using the composite interval mapping (Zeng 1993, 1994)
func-tion of QTL cartographer (Wang et al. 2001-2004). For the yeast data set, we set individual
thresholds for QTL detection for each trait that was in one of the four clusters studied. The
5% type I error rate threshold for the yeast data was obtained using Windows QTL
Cartog-rapher with 500 permutations (Churchill and Doerge 1994) for each trait. The thresholds
we found were very similar to those of Zou (2006).
For the eucalyptus data set, we used QTL detection thresholds determined by Kirst
(2004). The empirical experiment wide threshold for type I error rate for QTL detection
using CIM at the 5%level, was determined to be a likelihood ratio (LR) greater than 12.0
(Kirst 2004). This empirical threshold was established using a permutation method
pro-posed by Churchill and Doerge (1994). Twenty genes were picked at random and five
hun-dred permutations were done for each gene. The 5%empirical threshold was determined
for each gene. The most stringent 5% threshold among the 20 genes was used. To
ob-tain a general picture of which regions regulate gene-expression, all eucalyptus traits were
mapped individually to regions in the genome using composite interval mapping (Zeng
2.4
Eucalyptus Results
2.4.1
Clustering and QTL Analyses
Table 2.1 shows the total number of traits in a cluster, and the number of traits which
Table 2.1: The first column of this table contains the cluster name; the second column has the number of traits in a cluster; the third column contains the number of traits which have at least one QTL, and the last column has the ratio of the cluster size and the number of traits which have at least one QTL.
Cluster Cluster Traits with Traits with at least 1 QTL/ Name Size at least 1 QTL Cluster Size
G3 276 26 .09
G7 248 32 .13
G8 166 46 .28
G2 124 41 .33
G10 72 2 .03
G4 64 1 .02
G13 62 18 .30
G9 60 16 .27
G16 46 3 .07
G6 44 6 .14
G5 36 4 .11
G15 26 8 .31
All Traits 2610 620 .24
There is an overlap of significant QTL between the clusters with 248 and 44 traits, and the
clusters with 248 and 36 traits. All the QTL of cluster with 44 traits are in cluster with 248
traits, and all the QTL of cluster with 36 traits are in the cluster with 248 traits. However,
the clusters with 44 and 36 traits do not have any QTL in common. A cluster with more
traits tends to have more QTL; however, the ratio of the number of QTL and the number of
traits per cluster does not seem to depend on the cluster size.
Kirst (2004) found that 811 traits with at least one QTL in the F1 hybrid paternal map
at type I error rate of 10%. We found that at a type I error rate of 5% (Kirst 2004), 620
see the next figure. Each trait was mapped individually using CIM (Zeng 1994). The figure
below shows a few QTL hotspots for all traits in the eucalyptus data set. These hotspots
are located around 0.5 M, 8.5 M, and 10 M. Similar results were obtained by Kirst et al.
(2005).
0 20 40 60 80 100 120 140 160
0 1 2 3 4 5 6 7 8 9 10 11 12
Number of QTLs (LR >12.0)
Position (M) All genes
Figure 2.1: QTL locations for all eucalyptus traits.
Graphs with the QTL locations for all clusters in Table 2.1 are in the Appendix. Next we
annotate each of these clusters to see if genes in them have a common function.
2.4.2
Eucalyptus Cluster Annotation
We annotated all traits in Table 2.1 which had at least one significant QTL, to see if
genes which are clustered together and share a QTL are functionally related. Annotation
Blastx matches for each trait in a cluster were considered if they had an E-value less than
1e-10. The clusters with 6 and 32 QTL (Table 2.1), have each two traits classified as
UDP-glucoses; the cluster with 46 QTL has two heat shock related proteins; the cluster
with 16 QTL has three traits in the dehydrogenase family. The cluster with 41 QTL has
three proteins classified as oxidoreductases, three as dehydrogenases, two as similar to
Arabidopsis thaliana transcription factors, and two classified as histones. Each of the six
remaining clusters did not have genes with a common putative function. Even though some
traits share the same QTL and are clustered together, they do not necessarily have the same
function.
2.4.3
Eucalyptus Principal Component Analysis Results
The data used in this study consists of 2,610 gene expression levels measured in 88
eu-calyptus samples from a pseudo-backcross [E. grandis x F1hybrid (E. globulus x E.grandis)].
To observe if the regions associated with the regulation of individual gene expression, as
detected by QTL mapping, could be predicted by PCA, the principal components of each
cluster were calculated and mapped using composite interval mapping (Zeng 1994).
Principal component analysis was used in the hope that the QTL mapping results of
individual principal components could predict the patterns of association of individual traits
and the regions which regulate their expression. There were two sub-sets of the data for
which principal components were computed and mapped using composite interval mapping
(Zeng 1994): traits that were clustered together and traits that had significant QTL in a
specific region.
component, in parenthesis, is the percentage of the total variance it represents.
Table 2.2: Principal components per cluster with at least one QTL
Cluster Cluster N PC with at Traits with at
Name Size least 1 QTL least 1 QTL
G3 276 3 0 26
G7 248 11 PC 2(15), PC 5(1.8), 32
PC 8(0.96)
G8 166 13 PC2(7.0), PC4(2,7) 46
PC 11(0.76)
G2 124 1 0 41
G10 72 7 PC 6(1.06) 2
G4 64 4 0 1
G13 62 7 PC 7(1.13) 18
G9 60 9 PC 2(5.3), PC 3(3.7), 16
PC 6(1.96), PC 7(1.46)
G16 46 11 0 3
G6 44 6 0 6
G5 36 5 PC 3(2.7), PC 4(2.3) 4
G15 26 6 PC 2(9.28) 8
All genes 2610 28 PC 3 (8.6), PC 6(3), 620
PC 11(1.25), PC 13(.94), PCs 14, 23, 25,26,28
Using principal components to predict the locations of significant associations between
traits in a cluster and the eucalyptus genome, in lieu of finding QTL for each trait in a
cluster separately is problematic. Even though every cluster has principal components
(PCs), sometimes none of the PCs of a cluster have QTL (see clusters G3, G2, G4, G16,
and G6). That is the case for 5 out of 13 clusters (Table 2.2).
For the remaining clusters, most of the principal component QTL do not map to the