PULMONARY AND VASCULAR EFFECTS OF FATTY ACIDS: EVIDENCE FROM ENDOTHELIAL CELL AND RODENT EXPOSURE STUDIES
Virginia Leigh Bass
A dissertation submitted to the faculty of the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the
Department of Environmental Sciences and Engineering.
Chapel Hill 2019
ABSTRACT
Virginia Leigh Bass: Pulmonary and Vascular Effects of Fatty Acids: Evidence from Endothelial Cell and Rodent Exposure Studies
(Under the direction of Michael Madden)
Adverse pulmonary and cardiovascular health effects of air pollutant exposure are well recognized. Biodiesel, and other bio-based emission sources contain fatty acid (FA) components capable of entering the body through particle inhalation. FA can also be
endogenously released by the stress response seen with irritant pollutant exposures. Bioactive FA can interact with cells lining the lungs and blood vessels, potentially inducing pulmonary injury and endothelial dysfunction. Oleic acid (OA) is a common particle-associated FA. Although OA has long been used as an experimental model for inducing ARDS-like
symptoms, the mechanism by which OA causes pulmonary injury is not entirely understood. It has been previously observed that particles in the lung are capable of accumulating iron from cellular sources. As a potential mechanism of cellular injury, we wanted to investigate the role of iron in the effects of OA and OA metabolites on vascular and pulmonary systems. We hypothesized that OA exposure would induce changes in vascular mediators and vascular function, and induce pulmonary injury, possibly dependent on changes in cellular iron
Second, aortic tissue segments were exposed to OA or metabolites (200µM) prior to
assessing vascular function by isolated vessel myography. Third, Male Wistar Kyoto (WKY) rats (12wks age) were intratracheally instilled with OA and/or FAC (20mg/kg and
0.43mg/kg, respectively), either combined or in sequence, to demonstrate the effects of OA and FAC on pulmonary response and iron homeostasis in vivo. We found that OA, and more potently, a hydroxylated form of OA, induce dose-responsive changes in endothelial
ACKNOWLEDGMENTS
I owe so much gratitude to my mentors and advisors, who have guided me through growth as a scientist, and to my friends and family, whose support assured that I was never alone in this journey. Foremost, I am sincerely grateful to my research advisor, Dr. Michael Madden, for welcoming me into his laboratory and providing mentorship during my time in graduate school. I have benefited greatly from his invaluable experience, good-natured guidance, and generous open door.
I would also like to thank the other members of my committee for their time, suggestions, and criticisms throughout this degree. I have great appreciation for Dr. Avram Gold’s contributions as my committee chair and for his experience, adding insights on this project. I feel very fortunate to have had the commitment and thoughtful input of Dr.
Rebecca Fry and Ilona Jaspers. Finally, my humble thanks go to Dr. Urmila Kodavanti, who first allowed me to become involved in this research, who guided me and motivated me in the years before I began this degree, and encouraged me to have strength. She has been a truly graceful and inspiring teacher.
for being a superb officemate. Lastly, I give thanks to Dr. Samantha Snow, Dr. Andres Henriquez, and Dr. Desinia Miller, the influential and talented graduate students who came before me and went on to inspire me with all that comes next.
PREFACE
Research presented in this dissertation was done in collaboration with other scientists, where noted. For all work, Dr. Michael Madden was the principal investigator and provided experimental design advice and writing guidance. Previously published works are included in this dissertation with permission of the contributing authors.
For Chapter 2, ICP-OES iron measurements were performed by Dr. Andy Ghio and Joleen Soukup. I am grateful to Dr. Ghio for his advice and input in designing the iron uptake experiments. Radioactivity experiments were performed by Dr. Madden. I thank Dr. Katelyn Lavrich and Elise Hickman for advice and training in running the Seahorse assays. Chapter 2 was recently submitted for publication:
Bass, V. L., Soukup, J., Ghio, J., and Madden, M. C. (2019). “Effects of Oleic Acid and Derivatives on Human Endothelial Cell Vasoactive Meditator Production.” Submitted.
For Chapter 3, animal husbandry was conducted by the Environmental Public Health Division’s animal care facility staff at the U.S. Environmental Protection Agency. Tissue collection and preparation was done by Dr. Samantha Snow and Dr. Urmila Kodavanti. Dr. Snow and Dr. Leslie Thompson provided consultation and training on collection and analysis of aortic ring reactivity measurements. Chapter 3 was recently published as:
For Chapter 4, Dr. Urmila Kodavanti oversaw animal use and gave guidance on study design. Mette Schladweiler performed the IT instillations, organized necropsies, and assisted with tissue and blood sample collection. Allen Ledbetter performed whole body
plethysmography on the animals. Judy Richards assisted with analysis of bronchoalveolar lavage fluid (BALF) and serum markers. ICP-OES iron measurements were performed by Dr. Andy Ghio and Joleen Soukup. Lung sectioning and staining was done by Integrated Laboratory Systems in Durham, NC. Photomicroscopy and interpretation of histopathology was done with assistance from Dr. Ghio. Dr. Kodavanti also assisted in reviewing and revising the manuscript. This chapter has been prepared as a publication but is not yet submitted.
TABLE OF CONTENTS
LIST OF TABLES ... xiv
LIST OF FIGURES ... xv
LIST OF ABBREVIATIONS ... xvii
CHAPTER 1: INRODUCTION ... 1
1.1 The impact of air pollution on public health ... 1
Classifying ambient air pollutants ... 3
Adverse health effects of air pollutants ... 4
Fatty acids and air pollutants ... 7
1.2 Pulmonary and vascular effects of fatty acids ... 11
Localization of OA in vivo ... 12
The OA-induced model of lung injury ... 13
Endothelial dysfunction as a determinant of adverse cardiovascular outcomes ... 14
Endothelial dysfunction is associated with air pollutant exposure ... 16
Linking OA and endothelial dysfunction ... 18
1.3 Iron as a mediator of particle effects ... 19
Interactions of non-heme iron ... 20
Significance ... 26
Impact ... 27
CHAPTER 2: EFFECTS OF OLEIC ACID AND DERIVATIVES ON HUMAN ENDOTHELIAL CELL VASOACTIVE MEDIATOR PRODUCTION ... 29
2.1 Introduction ... 29
2.2 Materials and methods ... 31
Materials ... 31
Cell culture ... 31
FA preparations ... 32
Cell viability ... 32
Extracellular flux analysis ... 33
Soluble mediators ... 34
Gene expression ... 34
Iron uptake ... 34
Statistical analysis ... 35
2.3 Results ... 35
Cell viability ... 35
HUVEC mitochondrial response to FA ... 37
Oleic acid-induced soluble mediator release ... 39
Gene expression of endothelial signaling factors ... 40
Oleic acid metabolite-induced cellular iron dysregulation ... 42
2.4 Discussion ... 43
Supplement 2A ... 48
Supplement 2B ... 49
CHAPTER 3: 12-HYDROXY OLEIC ACID IMPAIRS ENDOTHELIUM-DEPENDENT VASORELAXATION ... 50
3.1 Introduction ... 50
3.2 Materials and methods ... 51
Aortic Ring Collection ... 51
Measuring LDH Activity Release ... 52
Aortic Ring Exposure and Functional Assessment ... 52
Statistical Analysis ... 53
3.3 Results ... 53
Aortic Ring Viability ... 53
Aortic Ring Myography ... 53
3.4 Discussion ... 56
CHAPTER 4: PULMONARY OLEIC ACID EXPOSURE INDUCES INJURY AND ALTERS IRON HOMEOSTASIS IN VIVO ... 57
4.1 Introduction ... 57
4.2 Materials and methods ... 59
Animals ... 59
Intratracheal exposures ... 60
Whole-body plethysmography ... 62
Necropsy and tissue collection ... 62
Histopathology ... 63
Statistical analysis ... 64
4.3 Results ... 64
OA increases a measure of air flow limitation ... 64
FAC exacerbates OA-induced lung injury and inflammation ... 66
OA alters BALF iron clearance and markers of iron homeostasis ... 70
OA treatment induces sideromacrophage accumulation in the lung ... 73
4.4 Discussion ... 74
4.5 Supplement ... 81
CHAPTER 5: OVERARCHING HYPOTHESIS AND SIGNIFICANCE ... 83
5.1 Summary ... 83
Investigating FA effects in vitro and ex vivo ... 83
Addressing the proposed iron-mediated mechanism of FA-induced effects ... 85
FA-induced changes in isolated vascular tissue response ... 86
Pulmonary exposure to OA in vivo ... 86
Manipulating the timing of pulmonary iron supplementation relative to OA-exposure ... 88
5.2 Conclusions ... 89
5.3 Recommendations ... 91
5.4 Contributions to knowledge in the field of public health and environmental toxicology ... 93
APPENDIX 2: HUVEC SEAHORSE MITO STRESS TEST
NORMALIZATION ... 97
APPENDIX 3: AORTIC RING VIABILITY ASSESSMENT AND
REACTIVITY TESTING ... 102
APPENDIX 4: AORTIC RING IRON UPTAKE ANALYSIS ... 105
LIST OF TABLES
Table 2.1 HUVEC gene expression changes. ... 41
Table 2.2 Custom primer sequences used in RT-PCR. ... 47
Table 3.1. Aortic ring tissue viability. ... 53
Table 4.1. Markers of iron transport and storage in bronchoalveolar lavage
fluid (BALF). ... 71
Table 4.2. Markers of iron transport and storage in serum. ... 81
LIST OF FIGURES
Figure 1.1. Composition of the particle phase from cooked meat emissions
by weight. ... 9
Figure 1.2. Serum FA fold-increases in WKY rats after one or two days of ozone exposure ... 11
Figure 1.3. Distribution of oleic acid in rat tissues 30 minutes after i.v. administration. ... 13
Figure 1.4. Iron concentrations epithelial cell mitochondrial fractions after exposure to wood smoke particles (WSP). ... 21
Figure 1.5. Test OA oxygen group variance. ... 22
Figure 1.6. Proposed mechanism of FA-induced cell damage ... 25
Figure 1.7. Summary of experiments and proposed mechanisms of this project ... 28
Figure 2.1. Comparison of HUVEC viability ... 36
Figure 2.2. FA exposed HUVEC mito stress test extracellular flux ... 38
Figure 2.3. FA exposed HUVEC soluble mediator release ... 39
Figure 2.4. HUVEC iron uptake following incubation with FA ... 42
Figure 2.5 Comparison of OA vehicles by HUVEC LDH activity release dose-response ... 48
Figure 2.6 OA cellular association differs with vehicle utilized. ... 49
Figure 4.1. Study dosing protocols. ... 61
Figure 4.2. PenH assessed by plethysmography. ... 65
Figure 4.3. Markers of pulmonary inflammation, injury, and leakage. ... 67
Figure 4.4. BALF neutrophil counts. ... 69
Figure 4.5. Lung tissue iron concentrations measured by ICP-OES. ... 73
Figure 4.6. Macrophage morphology and iron enrichment. ... 74
Figure 4.7. OA and FAC induce formation of sideromacrophages in BALF cells. ... 82
Appendix 1 Figure 1. HUVEC LDH activity release dose-response to OA and derivatives ... 96
Appendix 2 Figure 1. Site-specific inhibitors act on the electron transport chain ... 98
Appendix 2 Figure 2. The mitochondrial stress test involves sequential addition of site-specific inhibitors ... 98
Appendix 2. Figure 3. Protein normalized bioenergetic parameters of FA exposed HUVEC. ... 100
Appendix 3. Figure 1. EC[50] and Hill slope comparison for non-linear regression analysis of dose-response curves ... 104
Appendix 4. Figure 1. Aortic ring iron uptake assay. ... 105
LIST OF ABBREVIATIONS
12-OH OA 12-hydroxy oleic acid
2-OH OA(Na2+) 2-hydroxy oleic acid sodium salt
AAALAC Association for Assessment and Accreditation of Laboratory Animal Care International
ACh Acetylcholine
ANOVA Analysis of variance
ARDS Acute respiratory distress syndrome
ATP Adenosine triphosphate
BALF Bronchoalveolar lavage fluid
BSA Bovine serum albumin
COPD Chronic obstructive pulmonary disorder
COX-2 Cyclooxygenase-2
CPM Counts per minute
CVD Cardiovascular disease
DE Diesel exhaust
DMEM Dulbecco's Modified Eagle Medium
DOE Department of Energy
EDN1 Endothelin-1
EGM-2 Endothelial growth media-2 eNOS Endothelial nitric oxide synthase ERK Extracellular signal-regulated kinase-1
ET-1 Endothelin-1
EtOH Ethanol
FA Fatty acid
FAC Ferric ammonium citrate
FCCP Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone GGT Gamma-glutamyl transferase
HAP Hazardous air pollutant HBSS Hank’s balanced salt solution ICAM-1 Intercellular adhesion molecule 1
ICP-OES Inductively coupled optical emissions spectroscopy iNOS Inducible nitric oxide synthase
IT Intratracheal
IV Intravenous
KCl Potassium chloride
KHB Krebs-Henseleit buffer LCFA Long chain fatty acid
MBCD Methyl beta-cyclodextrin
Me-OA Methyl oleate
MUFA Monounsaturated fatty acid
NAAQS National ambient air quality standards
NF-kB Nuclear factor kappa-light-chain-enhancer of activated B cells
NMMAPS National Morbidity, Mortality and Air Pollution Study
NO Nitric oxide
NOS3 Endothelial nitric oxide synthase
OA Oleic acid
OCR Oxygen consumption rate
PBS Phosphate buffered saline solution
PE Phenylephrine
Penh Enhanced pause
PGF2! Prostaglandin F2 alpha
PM Particulate matter
PM2.5 Fine particulate matter PUFA Polyunsaturated fatty acid ROS Reactive oxygen species
Rt Relaxation time
SEM Standard error of the mean
SNP Sodium nitroprusside
US EPA United States Environmental Protection Agency
UV Ultraviolet
CHAPTER 1: INRODUCTION
1.1 The impact of air pollution on public health
Recently, estimates of excess deaths due to ambient air pollution exposures have been reassessed, and new global exposure models have placed the number of mortalities due to fine particulate matter and ozone pollution for the year 2015 at 8.8 million excess deaths worldwide (Burnett et al., 2018; Lelieveld et al., 2019). This number reflects the assertion of the World Health Organization that air pollution exposure is among the top 5 global risk factors for premature death (Cohen et al., 2017). Air pollution has also been identified for its significant contribution to the burden of disease worldwide, and is now understood to be a major risk factor for development of cardiovascular disease. Despite some progress with air pollutant reduction over the last 50 years in the United States, air quality remains a critical global determinant of public health.
Historically, air pollution began to be accepted as a cause of adverse health effects based on several notable instances of severe air pollutant exposures in the mid 20th century. During this time, air contaminants came to be viewed as a threat to public health in industrial and urban communities. These early events informed the epidemiological studies of air pollution that first correlated increased hospital admissions and deaths with high air pollutant levels.
In one such instance, in 1948, Donora, Pennsylvania was in the midst of a
to settle on the town for four days (Tarr, 2005). The sudden decrement in air quality caused 20 fatalities, and treatment for respiratory problems was sought by nearly half of the
residents of the small town. Reported health effects from what was later viewed as one of the major public health disasters of that time period persisted among Donora residents in the years that followed.
Another incident that inspired increased concern for public protection from air pollutants occurred in London in 1952. “The Great Smog”, as it was termed, was a heavy fog mixture primarily composed of coal combustion emissions and sulfur dioxide (Polivka, 2018). The smoke particles in the air formed a haze so thick that vehicles could not navigate on the roads and London commerce was brought to a standstill. Over 4,000 premature deaths in the city during that time were attributed to the smog, a number now estimated at closer to 12,000. In response to the incident, the Clean Air Act of 1956 was passed in the United Kingdom, which would restrict the burning of coal in urban areas.
Today in the United States, although the population continues to grow and vehicle use has not diminished, regulatory controls and reductions in industry have had a significant impact on reduction of the six criteria air pollutants defined by the National Ambient Air Quality Standards (NAAQS) (US EPA, 1999). The areas of the United States that still often exceed these thresholds generally correspond with present urban and industrial centers. Though the United States and Western Europe are no longer experiencing the magnitude of industrial growth seen in prior centuries, developing parts of world are still realizing the consequences of rapid industrialization and population growth.
air pollution. The 2008 Olympic Summer Games in Beijing were a catalyst for the country to address its environmental conditions. China’s Olympic preparations were marred by intense air pollution warnings which put pressure on the government to enact more meaningful regulations. Despite shutting down factories, restricting car usage and slowing down
construction, Beijing was unable to attain more than a 10–15% reduction in total atmospheric aerosols (Witte et al., 2009). This modest reduction was attributed to meteorology and regional particulate levels, demonstrating the complexity of air pollutant transport affecting broad geographic areas and the continued difficulty of reducing environmental air pollutants worldwide.
Classifying ambient air pollutants
The EPA has defined six criteria air pollutants for targeted reduction after characterizing the components of smog mixtures. The criteria pollutants are particulate matter (PM), ozone (O3), sulfur dioxide (SO2), nitrogen oxides (NOx), which includes nitrogen dioxide (NO2), carbon monoxide (CO) and lead (Pb). NAAQS delineate acceptable levels of these six criteria pollutants for an area by time periods, such as acute and/or
extended durations of, e.g., 1 hr and 1 yr for NO2. Primary standards are established based on human health and secondary standards are based on ecological and environmental factors. Additional air pollutants are generally termed Hazardous Air Pollutants (HAPs) or Air Toxics. Both HAPs and NAAQS are regulated based on toxicological evidence to protect human health because those primary standard limits are stricter than secondary standards. Further discussion of air pollutants in this dissertation will relate to human health.
components. Within mixtures, air pollutants can more generally be divided into particulate matter (PM) and gaseous categories. The gaseous pollutants include NOx, and SOx which when exposed to UV radiation react to form secondary pollutants like ozone in the troposphere.
PM is the most abundant pollutant by mass and is also strongly correlated with much of the burden of mortality associated with air pollutant exposure. The unique chemical characteristics of particulate pollution include its high surface area and diverse composition. PM may contain metallic, organic, inorganic salts, and elemental carbon species. East coast, Northeast, and Midwest-derived PM tends to contain more sulfates, whereas west coast and southwest particulates are characteristically more nitrogen rich (US EPA, 2009). The widely varying compositions of PM make it hard to attribute the biological effects from an exposure to specific components.
PM is classified for NAAQS regulation by its size, which determines the ability to penetrate airways and possibly enter the bloodstream. PM2.5 includes particles up to 2.5μm aerodynamic diameter and is considered the fine to ultrafine fraction; these particles are better able to penetrate into the deep lung with a deposition rate of approximately 40 percent (Kim, 2009). Coarser particulates, from 2.5-10 microns are classified as PM10, and most are generally removed from the air stream before reaching the distal lung (US EPA, 2009).
Adverse health effects of air pollutants
Air pollutant exposures can induce pulmonary, as well as cardiovascular and
metabolic alterations, leading to adverse health effects (Brook et al., 2013; Chen et al., 2008; Pope et al., 2002; Pope et al., 2004). Pulmonary effects impair respiration and cause
alterations of the lung (Snow et al., 2014; Takenaka et al., 1999). These effects can also make the respiratory system more susceptible to infections (Jaspers et al., 2005; Tsai et al., 2014). Regardless of composition, exposure to PM2.5 is associated with short-term and long-term health effects beyond the respiratory system. Cardiovascular health effects and
associated mortalities have been shown in numerous epidemiological studies (Pope et al., 2006). These effects are supported by findings from controlled exposures of both humans and animals, which experimentally demonstrate the links between air particulate pollution and adverse cardiovascular events.
Particulate pollution may peak for short periods over hours or days. Short-term
cardiovascular effects of PM2.5 have been measured in studies of 10µg/m3 increases over a 24 hour period, which have been associated with a 0.4-1% increase in cardiovascular mortality (Burnett et al., 1998). A 0.2% increase per 10µg/m3 increase in PM10 was estimated in the National Morbidity, Mortality and Air Pollution Study (NMMAPS) (United States
Environmental Protection Agency, 2004). Myocardial infarction was estimated to increase by 0.65% with every additional 10µg/m3 PM10. Hospitalizations showed a similar, nearly linear trend, which was amplified in asthmatics and patients with COPD or recent pneumonia (Zanobetti et al., 2003). Long-term PM2.5 exposure, measured in the same 10µg/m3 increases over months to a year, has similarly been associated with cardiovascular mortality, at a higher rate: 1.06-1.76%, or approximately 800,000 people per year in the US (Brook et al., 2010).
(Dockery et al., 1993) and the American Cancer Society studies (Pope et al., 1995), which were cohort-based investigations linking disease outcomes to air pollutant exposures. Findings from the Harvard Six Cities study marked the beginning of the field of
environmental cardiology. Large-scale time-series studies, such as the National Morbidity, Mortality and Air Pollution Study (NMMAPS), show the association between particulate pollution and cardiovascular events in data from 20-100 cities and hundreds of counties. Studies of temporal and spatial distributions of PM estimate that the potential health-related benefits of hypothetical air quality improvements would yield an increase in life expectancy (Fann et al., 2012).
Currently, the mechanism behind these effects is thought to stem from a combination of possible pathways. First, ultrafine particulates may pass into the bloodstream directly from the lung where soluble organic compounds may be carried throughout the body, causing vascular effects and increased coagulation. Alternatively, particles may enter the lung and react there, causing systemic inflammation and oxidative stress. Oxidation products in the lung can trigger a cellular response elsewhere, leading to the release of histamine, increased cytokines and downstream systemic effects. Finally, the autonomic nervous system is shown to translate irritant effects in the lung to systemic effects by neurohormonal pathways, which link cardiovascular reactions to inhalation of particles. Stimulation of C-fibers and vagal nerve pathways in the lung can alter heart rate, cause arrhythmias, vasoconstriction, increased blood pressure, endothelial dysfunction or further inflammation (Brook, et al., 2010).
Given the demonstrated link between PM and hospital admissions, as well as
of cardiovascular morbidity and mortality is attributable to PM in ambient air. While studies are still needed to define the mechanisms behind these effects, much is known that can help public health professionals understand the increased risks of poor air quality especially within susceptible populations. Studies have shown that reductions of air pollutants can result in fewer deaths (Corrigan et al., 2018) and it is likely that increased awareness
combined with stronger regulation of air contaminants will have a positive impact on adverse health effects and disease outcomes .
Fatty acids and air pollutants
Long chain fatty acids (FA) are aliphatic monobasic acids greater than 14 carbons in length. OA is an 18-carbon monounsaturated omega-9 fatty acid (18:1 cis-9). OA is a component of dietary and endogenous circulating fatty acids. In plasma, free FA are
normally found in low concentrations with postprandial increases. FA can also be found on the surface of particulate pollution or be released in circulation following exposure to irritant type pollutants that induce a neurohormonal stress response (Miller et al., 2016; Miller et al., 2015; Snow et al., 2017). OA is highly representative of FA in ambient airborne pollution.
over the course of a month (Ren et al., 2016). Of this, over half of the lipids identified were long chain FA.
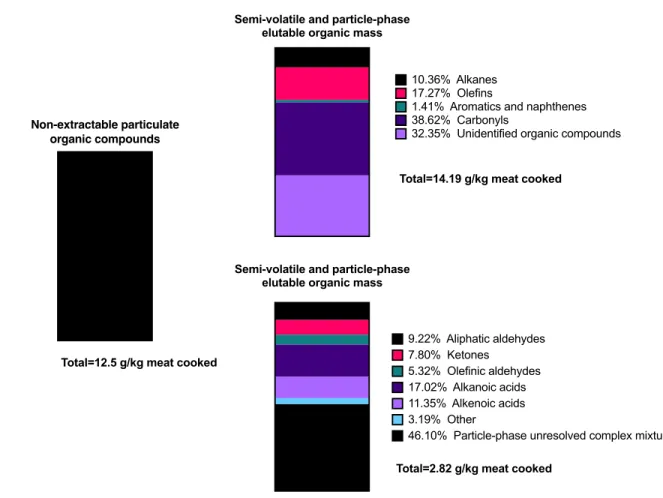
Figure 1.1 Composition of the particle phase from cooked meat emissions by weight. Alkanoic acids make up 17.02% of the semi-volatile and particle phase organics by weight. Figure adapted from Schauer et al. (1999a).
Cooking emissions, like frying oil or grilling exhausts from commercial and
residential cooking processes can have significant FA content (Li et al., 2017; Poudel et al., 2017; Rogge et al., 1991; Veres et al., 2013; Zhao et al., 2015). Other PM sources
containing FA include tobacco smoke (Gao et al., 2006), diesel exhaust (Mills et al., 2011; Schauer et al., 1999b), marine aerosols (Gantt et al., 2013; Hawkins et al., 2010; O'Dowd et al., 2004), and biofuels emissions (Bunger et al., 2016).
Biodiesel is produced by transesterification of FA from plant or animal sources; however, the conversion reaction is always incomplete, leaving behind un-esterified FA in
Non-extractable particulate organic compounds
Total=12.5 g/kg meat cooked
Semi-volatile and particle-phase elutable organic mass
Total=14.19 g/kg meat cooked
10.36% Alkanes 17.27% Olefins
1.41% Aromatics and naphthenes 38.62% Carbonyls
32.35% Unidentified organic compounds
Semi-volatile and particle-phase elutable organic mass
Total=2.82 g/kg meat cooked
9.22% Aliphatic aldehydes 7.80% Ketones
5.32% Olefinic aldehydes 17.02% Alkanoic acids 11.35% Alkenoic acids 3.19% Other
pollutant constituents. There is no legal regulatory standard for the un-esterified FA content in biofuels, so concentrations in PM vary. Biodiesel can be enriched with particular FA to engineer fuel properties (Knothe, 2005; Moser). Additionally, composition of biodiesel by source material varies. For instance, German biodiesel is most commonly made from rapeseed oil, which is 90% unsaturated FA, and of that 62.2% OA. Combustion byproducts from this fuel differ from biofuels made of coconut oil, which is only 7.9% unsaturated FA and 6.4% OA (Bunger, et al., 2016). FA, including OA, can become airborne and be present in ambient air gas and PM phases. Inhalation of organic-derived particles may permit OA and other FA to enter systemic circulation in the blood stream, allowing exposure of the vascular system.
FA can also be increased in circulation in response to air pollutant inhalation not from a particle source. Endogenous release of FA has been shown in response to cigarette smoking (Kershbaum et al., 1961), biodiesel exposure (Surowiec et al., 2016), NO2 (Ward-Caviness et al., 2016), acrolein (Snow, et al., 2017), and ozone exposure (Miller, et al., 2016) (Figure 2). This increase is likely related to a neurohormonally regulated stress response to these
Figure 1.2. Serum FA fold-increases in WKY rats after one or two days of ozone exposure, relative to filtered air exposed rats. The blue box indicates the data for OA. Figure adapted from Miller, et al. (2015).
1.2 Pulmonary and vascular effects of fatty acids
Translocation of FA containing particles into the circulation could allow for particles to directly interact with vascular endothelial cells and initiate vascular oxidative stress, inflammation, and affect endothelial-mediated responses. The direct contact of the endothelium with the blood means that it is vulnerable to damage from, and able to sense changes in, circulating factors. The endothelium can then either respond or transmit signals to neighboring cell types, such as vascular smooth muscle cells. Extra-pulmonary
translocation of particles has been shown to be related to particle size, charge, and surface modifications, and could occur as a result of increases in pulmonary epithelial permeability
X Air-1D Air-2D
myristate (14:0) 1.31 1.49
myristoleate (14:1n5) 1.33 1.84 pentadecanoate (15:0) 1.37 1.39
palmitate (16:0) 1.59 1.8
palmitoleate (16:1n7) 1.51 2.47
margarate (17:0) 1.51 1.68
10-heptadecenoate (17:1n7) 1.31 1.47
stearate (18:0) 1.33 1.36
oleate (18:1n9) 1.61 2.38
cis-vaccenate (18:1n7) 1.65 1.46 nonadecanoate (19:0) 1.27 1.38 10-nonadecenoate (19:1n9) 1.4 1.63 eicosenoate (20:1n9 or 11) 1.29 1.39 dihomo-linoleate (20:2n6) 1.6 1.45 mead acid (20:3n9) 1.77 1.51 arachidonate (20:4n6) 1.22 1.23 docosadienoate (22:2n6) 1.42 1.07
adrenate (22:4n6) 1.88 1.26
O3-2D Long Chain Fatty Acids O3-1D
1 day | 2 day FA concentration fold
pass directly into alveolar capillaries or the lymphatic system (Harmsen et al., 1985). Additionally, FA can potentially dissociate from PM, and pass through the airway epithelial barrier. Leukotriene-B4, a FA derived from arachidonic acid metabolism, was shown to pass into the blood after installation in human bronchioles (Martin et al., 1989). Studies reporting vascular and pulmonary effects of OA exposure suggest possible negative health effects of exposure through multiple non-dietary routes.
Localization of OA in vivo
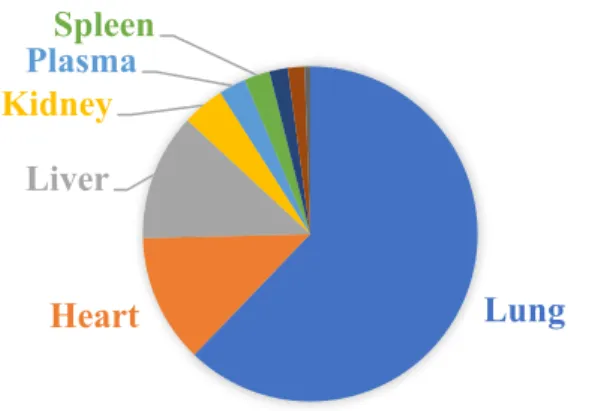
A study using intravenously administered 14C-OA in rats traced 14C mainly to the lung (85%) at 30 min post- administration. The concentration of OA was estimated to be 3.24 μmol/g in lung tissue, which was almost ten times greater than the next highest
concentrations, found in the heart and liver tissue (Beilman, 1995) (Figure 3). Concentrations of OA in the lung remained significantly elevated relative to other tissues through 12hr post-administration. 14C was also found in high concentrations in BAL fluid, but was not
Figure 1.3. Distribution of oleic acid in rat tissues 30 minutes after i.v. administration. Figure adapted from findings of Beilman (1995).
The OA-induced model of lung injury
OA has long been used as a means of inducing lung inflammation, edema, and immune response in animal models for the purpose of studying acute respiratory distress syndrome (ARDS), its etiology, and possible therapeutic strategies. When administered intravenously or intratracheally in animal models, OA induces inflammation, neutrophil infiltration, and pulmonary edema in the lung, making it a useful model of ARDS (Matute-Bello et al., 2008; Schuster, 1994). Given IV, OA appears to localize to pulmonary
capillaries, where it may induce the responses associated with ARDS symptoms. The
mechanisms linking tissue damage and OA exposure remain unclear. Based on the ability of OA to induce pulmonary injury and ARDS through intravenous administration, there has also been suggestion that it may directly interfere with cell membrane integrity, though this hypothesis is currently less accepted, based on studies of lipid membrane dynamics and free FA aggregation patterns in solution (Beilman, 1995; Cistola et al., 1988). Studies of the effects of OA in the context of the ARDS model have shown that i.v. administration of OA
Lung
Heart Liver Kidney
PlasmaSpleen
cyclooxygenase inhibitor indomethacin (Hageman et al., 1989). Cyclooxygenase inhibitors are not shown to reduce OA-induced lung injury, however, as alveolar edema, hemorrhage, and interstitial infiltrate were still noted following indomethacin or cobra venom
pretreatment in rats (Dickey et al., 1981). OA has also been shown to induce protein kinase C-dependent increases in ET-1 expression in vitro (Park et al., 2003). Loss of cell barrier integrity is another previously reported effect of OA in the ARDS model, which has been attributed to OA-induced increases in intracellular calcium (Wang et al., 1994).
Increased free circulating FA levels are associated with cardiovascular risk (Pilz et al., 2008). OA levels are elevated in the serum of patients with low to intermediate levels of cardiac lesions (Duarte et al., 2016). FA oxidation products, including hydroxy FA have been found in advanced atherosclerotic plaques (Waddington et al., 2003). Endogenous production of OA in response to stress could induce similar distributions of OA to tissues and with similar effects. When released from adipose tissue stores by lipolysis, OA is mainly found bound to serum albumin or in its free un-esterified form in circulation (Spector et al., 1969). Assays have shown that radiolabeled OA can be traced to the heart, liver, lung, spleen, kidney, muscle, intestine, adrenals, bloods lymph, adipose, mucosal, and dental tissues. Normally, the FA:albumin molar ratio in the blood is 0.7:1, but this ratio can be markedly raised up to 8:1 in disease states such as diabetes mellitus or nephrotic syndrome.
Endothelial dysfunction as a determinant of adverse cardiovascular outcomes
FA can be elevated following air pollutant exposures by two possible routes: inhalation and translocation of PM constituents, or endogenous elevation in response to an inhaled stressor. These routes localize increased levels of FA in the lung and blood,
lung tissue (Beilman, 1995). In the deep airways, the alveolar region where the a large portion of inhaled fine PM is likely to deposit (Kim, 2009), thin pulmonary epithelial cells and endothelial cells form single cell layer interface between air and blood. Particles passing through this cell layer into the blood can occur passively if cell barriers are weakened,
increasing lung permeability, or by transport. FA that enter the body through the lung carried on the surface of inhaled particles or dissolved in the extracellular fluid, can pass through the pulmonary epithelial cell layer into pulmonary vascular beds, where they may interact with vascular endothelial cells locally, or enter circulation and contact the endothelial layer
systemically. By either route, inhalation or endogenous release, increased circulating FA will reach the interface between the blood and the vascular system where endothelial cells reside.
As the cell type that lines blood vessels, endothelial cells are the first site of transduction for blood borne signals or hazards. Endothelial cells are one of the initial cell types that can be injured by toxicants in the blood. Damaged endothelial cell can affect signaling intermediaries that control vascular functions for the whole vascular tissue. The endothelial cell layer typically serves as a signal mediator between the extracellular
environment and the adjacent smooth muscle layer, to regulate vascular tone and dilation or contraction of the vessel (Félétou, 2011) .
Endothelial dysfunction can be detected by measuring changes in the levels of signaling intermediaries produced by endothelial cells or by changes in how vascular tissues respond to stimuli. Endothelial cells control vascular tone by mediating NO producing signaling cascades, which includes responding to and regulating production of thromboxane, endothelin, prostaglandins, and cyclooxygenase enzymes. Endothelial-mediated
and relaxation of smooth muscle. Impaired blood vessel relaxation is recognized as a classic problem with endothelial dysfunction, as is an increase in adhesion molecules, and reduced endothelial repair (Sun et al., 2009).
Isolating the response of specific functional pathways in the vasculature aids in determination of which functional mechanisms are impacted by an exposure. Some functional effects occurring in endothelial dysfunction include reduced
endothelium-mediated production of NO, reduction in smooth muscle vasorelaxation, or reduced receptor response to endothelial mediated vasoconstriction (Shepherd et al., 1991). Loss of smooth muscle responsiveness to systemic cues can lead to proliferation of smooth muscle cells and fibrotic tissue in the intima and media of vessels, which defines the early stages of
atherosclerosis development. Through these mechanisms, decrements in endothelium function can contribute to hypertension and vascular diseases. Endothelial dysfunction is a disease state that can promote vasospasm, thrombosis, intimal growth, inflammation, and plaque rupture leading to tissue ischemia, atherothrombotic, and myocardial infarction. Because these are some of the leading causes of air pollutant related morbidity and mortality, the relationship between air pollutants and endothelial dysfunction bears investigation.
Endothelial dysfunction is associated with air pollutant exposure
(2007). A study by Hansen et al. (2007) showed that diesel exhaust PM, given IP to apoE(-/-) mice with slight atherosclerosis, resulted in endothelial dysfunction in aortic rings collected one hour after the exposure. A study using total suspended particles collected by filters in an industrial area of Germany producing iron, steel and coke, showed that addition of 300 μg/ml ofparticles increased relaxation of preconstricted rat aortic rings 25% through a mechanism that was only partially endothelium-dependent and may also involve effects on the smooth muscle (Knaapen et al., 2001). In a cross-sectional study, carotid artery intima-media thickness, a positive association was shown between accelerated thickening of the blood vessels and proximity to highways and PM2.5 (Kunzli et al., 2010). Together, these findings indicate the relationship between particulate exposure and the effects leading to development of vascular disease.
more impairment of endothelial-dependent vascular relaxation, indicating that PM composition may be determinant of endothelial response (Aragon et al., 2016).
Possible pathways that produce cardiovascular outcomes related to PM include those that depend on the antioxidant capacity of the cellular environment and the concentration and turnover of oxidants. Vascular oxidative stress is thought to influence vascular permeability and leukocyte adhesion, accompanied by changes in signal transduction and redox regulated transcription factors like NF-kB (Lum et al., 2001). In a study showing the importance of
antioxidant capacity in maintaining vascular function, aged and superoxide dismutase 2-deficient mouse aortic rings exposed to ultrafine PM ex vivo were observed to have a reduced vasorelaxation response thought to derive from reduced antioxidant production due to age and reduced superoxide-dismutase production, respectively (Carter et al., 2018). A diet high in olive oil fed to spontaneously hypertensive rats was shown to reduce maximum contractile response and enhance endothelium dependent-relaxation in aortic rings, an effect thought to stem from the antioxidant components of olive oil (polyphenols) and not seen with a high oleic acid sunflower oil diet lacking these compounds (Herrera et al., 2001).
Linking OA and endothelial dysfunction
The mechanism by which OA causes lung injury and ARDS-like symptoms is not entirely understood, but it may involve damage to endothelial cells. Examination by microscopy of lung tissue from rats with experimental lung injury showed endothelial cell blebbing within 10min of administering roughly 240µg OA IV (Beilman, 1995).
dysfunction. Endothelial dysfunction is characterized by any change in the normal responses of endothelial cells or endothelial responsive tissues to external stimuli. In one study, rabbits infused with a high OA lipid emulsion (4 ml/kg) developed lung injury, accompanied by high levels of vasoactive lipids PGE2 and PGF2a, in both venous and arterial plasma. OA
exposure may contribute to acute oxidant stress in the vascular endothelium, creating endothelial dysfunction with a role in the pathophysiology of various vascular diseases. The source of endothelial dysfunction, may relate to production of reactive oxygen species, which function as signaling molecules, but can also induce cell injury and death at higher
concentrations.
As an early factor in the progression towards CVD, endothelial dysfunction produced by an exposure is an important indicator of the potential to cause adverse cardiovascular outcomes. Evidence that links PM exposures with endothelial dysfunction and studies showing endothelial effects of OA exposure together indicate the potential for FA as a component of air pollutant exposure to contribute to associated CVD.
1.3 Iron as a mediator of particle effects
Interactions of non-heme iron
It has been demonstrated in vitro and in vivo that particles in the lung accumulate iron, which may be derived from cellular or blood sources (Ghio et al., 2000). One reasoning for this observation states that iron has electropositive properties and are attracted to oxygen-containing functional groups, such as alcohol, epoxide, aldehyde, or carboxyl groups (Ghio et al., 2013). These groups are frequently found on the surface of particles derived from
biomatter combustion, including biodiesel exhaust, cigarette smoke, or wood particles. FA, like OA, possess a carboxyl end-group, and represent a possible particle constituent that may be responsible for this iron sequestering effect.
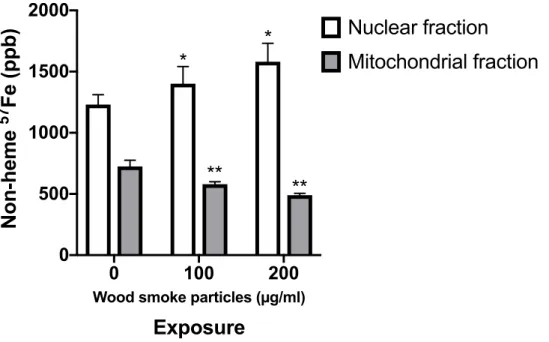
Figure 1.4. Iron concentrations epithelial cell mitochondrial fractions after exposure to wood smoke particles (WSP). BEAS-2B cells were incubated with 57Fe FAC and exposed to 100 or 200 μg/mL WSP for 15 min then 57Fe was measured by ICPMS. Wood smoke particles decreased iron found in the mitochondrial fraction isolated from cells. * = significantly different from control exposed nuclear fraction. ** = significantly different from control exposed mitochondrial fraction. Figure adapted from Ghio, et al. (2015).
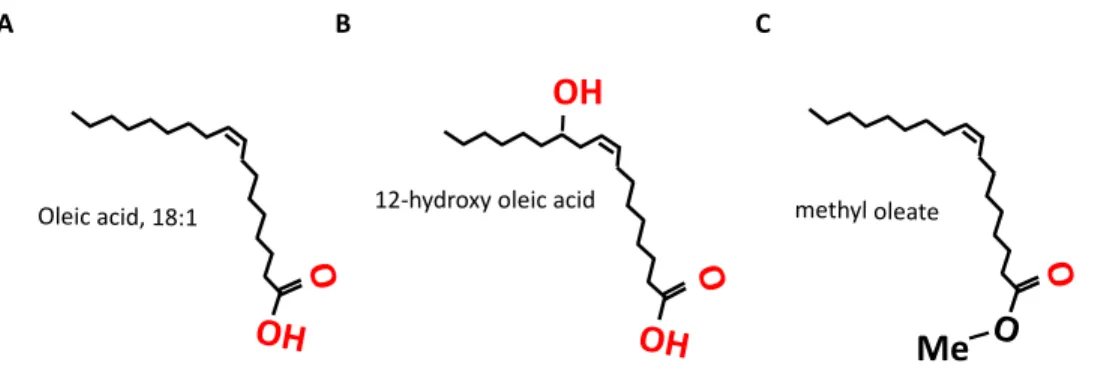
Because the mechanism by which FA are reported to chelate iron in chemico involves affinities between the iron species and oxygen containing groups, it is helpful to compare the iron binding affinities of varying structures of FA. For this purpose, the structurally simple and environmentally abundant FA OA is a good model. Hydroxylation of the FA carbon chain provides a good model of a FA with additional oxygen groups, while esterification of the carboxyl end reduces the available oxygen groups (Figure 1.5). Comparing the effects of these modifications will suggest the relative importance of oxygen group availability in sequestering iron from cells and show variation in toxic effects in endothelial cells.
0 100 200
0 500 1000 1500 2000
Wood smoke particles (µg/ml)
Exposure
Non-heme
57
Fe (ppb)
Nuclear fraction
Mitochondrial fraction
*
*
**
Figure 1.5. Test OA oxygen group variance. A) 18:1 cis-9 OA, B) 12-OH OA, and C) methyl ester of OA. Hydroxylation adds, and methylation reduces, available oxygen groups on the FA. Oxygen containing groups that could show an electronegative affinity for iron are shown in red.
In cells, mitochondrial respiration plays a central role in bioenergetics, biosynthesis, and signal transduction (Chandel, 2015). Mitochondrial respiration is thought to be devoted to different primary biological functions of cell types in vivo, with the role of the
mitochondria corresponding to cell phenotype. Mitochondria content of endothelial cells is relatively low (e.g., 2-6% of cell volume in rat endothelial cells) compared to other cell types, like hepatocytes (28%) or cardiac myocytes (32%) (Tang et al., 2014). This relatively low volume of mitochondria correlates with the lower energy requirements of vascular tissue. Redox-based signaling is one of several key roles of mitochondria and, barring a need for high energy production, this may be a particularly important function of endothelial mitochondria (Dromparis et al., 2013).
The direct contact of endothelial cells with the blood means that they are uniquely vulnerable to damage delivered systemically. Additionally, close proximity to the air space in the lung makes these pulmonary endothelial cells particularly vulnerable to damage from inhalation exposures. FA-induced changes in cellular iron metabolism may cause disruption of key iron-containing enzymes.
12-hydroxy oleic acid
O OH OH
methyl oleate
O
O Me
O Oleic acid, 18:1
OH
Mitochondria are rich in iron, which is required for proper function of several iron-containing enzymes, like cytochrome c oxidase and iron-sulfur proteins (e.g. aconitase, complexes I, II, and III). Disruption of these enzymes can impair mitochondrial membrane function and increase intracellular reactive oxygen species (ROS), causing oxidative stress. Changes in intracellular iron availability due to FA exposure might impact mitochondrial
function, which may determine the degree of cellular injury (Yao, et al., 2005). The evidence that supports a mitochondrial source of cellular damage includes measurement of increased reactive oxygen species (ROS) produced by in vitro exposure of endothelial cells to OA and similar long-chain FA, such as palmitic acid. In a recent study of the mechanism of FA cytotoxicity in endothelial cells, OA was shown to increase ROS production, with reversible effects with the addition of inhibiting agents specific to mitochondrial complex II. By demonstrating that a targeted reversal of ROS production could be accomplished, the study concluded that either additional antioxidant effects and/or the prevention of site-specific mitochondrial uncoupling could protect endothelial cells exposed to OA.
Both FA and iron overload are associated with proinflammatory pathways.
Studies of mice given OA IT have shown upregulation of the ERK pathway in lung tissue lysates, concurrent with lung injury and inflammation (Goncalves-de-Albuquerque et al., 2012). Excess oxidative stress has also been shown to upregulate pro-inflammatory mediators via the cycolooxygenase-2 (COX-2) pathway through activation of NF-ΚB and
25
Figure 1.6. Proposed mechanism of FA-induced endothelial cell damage. FFA from an extracellular source cross into the cytosol where they may interact with cellular pools of iron. Mitochondrial iron containing complexes may be disrupted, promoting
uncoupling of respiration or mitochondrial dysfunction, ROS production, and oxidative stress in the cell. This may damage the cell membrane causing the releasing active LDH into the extracellular environment, or impact production of vasoactive mediators.
FFA Ce ll m em br an e Mitochondria Oxidative Stress FFA FFA FFA Mitochondrial dysfunction Membrane damage, cell death FFA FFA Fe ROS
FFA Fe
1.4 Rationale and hypotheses
We hypothesized that FA play a role in the health effects of inhalation exposures to
some air pollutants, especially bio-based combustion derivatives. It is predicted that FA
contribute to pulmonary and vascular effects with a mechanism that may partly involve
alterations to iron metabolism, due to potential effects on mitochondrial function and
endothelial signaling.
Significance
This research will support the mechanism of how some inhaled pollutants induce
pulmonary and systemic effects and what the possible outcome of pollution inhalation will be
in those with underlying cardiovascular disease-associated iron overload. Despite being
commonly associated with particulate air pollutants, the health effects of inhaled FA, and the
mechanism inducing these effects, are not well understood. This project will be one of the
few to examine FA-induced toxicity through a specific pathway related to alterations in iron
metabolism in endothelial cells. Inhalation of OA may permit FA to enter systemic
circulation in the blood stream, allowing exposure of the vascular system. Additionally,
increases in circulating FA in humans are known to increase by endogenous production
following exposure to ozone and other stressors.
A limited number of studies have used in vitro approaches to investigate OA-induced
vascular effects and their mechanisms. Because of their presence in ambient air, along with
evidence linking OA to adverse health effectsFAshould be studied in isolation to better
OA is a highly representative FA associated with air pollutant exposures (Kershbaum,
et al., 1961; Rogge, et al., 1991; Schauer, et al., 1999b; Veres, et al., 2013). In addition,
previously reported effects induced by a variety of FA, including iron-translocation,
inflammation, increased membrane permeability, and vascular response, are reproduced by
OA, indicating that it is also representative of FA as a class (Huang et al., 1992; Shinohara et
al., 1995). In studies using cultured alveolar epithelial cells, transcellular permeability
induced by FA was shown to relate to hydroxyl group availability more so than degree of
unsaturation, with OA (18:1), linoleic (18:2), and linolenic (18:3) having similar potency
(Wang, et al., 1994). A study examining the components of tobacco smoke that permit iron
translocation in solution found that both saturated (stearic and palmitic) and unsaturated FA
were responsible for the effect (Qian et al., 1991), which has also been reported with pure
OA in chemico (Nalini, et al., 1993). Effects of FA are likely to vary by chain length,
saturation, and oxygen group availability; however, we believe OA to be a suitable
approximation of generalized FA properties.
The experimental approaches used in in this work will link FA exposure to changes in
iron metabolism and vascular function. The studies will also demonstrate in vivo the
relationship between OA’s pulmonary effects and changes in iron regulation. A better
understanding of the impact of FA on iron metabolism in lung and vascular tissues will aid
public health efforts to reduce morbidity from FA-associated air pollutants and pollutants
known to induce endogenous FFA release (e.g. PM and O3).
Impact
This study will provide insight into the mechanism and modifiers of the effects of OA
changes, and both pulmonary and systemic in vivo effects with mitochondrial dysfunction,
iron dysregulation, and vascular mediator production (Figure1.7). By utilizing novel in vitro
and in vivo exposure methods, this study will address the mechanism behind the effects of
OA and FA as they relate to the health effects associated with airborne pollutants.
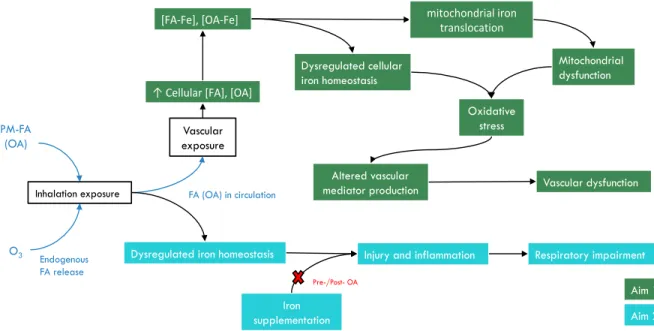
Figure 1.7. Summary of experiments and proposed mechanisms of this project PM-FA
(OA)
O3
Inhalation exposure
Altered vascular mediator production
Injury and inflammation Dysregulated iron homeostasis
Iron supplementation
Vascular dysfunction
Respiratory impairment
Aim 1 Aim 2 Vascular
exposure
Endogenous FA release
FA (OA) in circulation
Dysregulated cellular iron homeostasis
↑ Cellular [FA], [OA]
[FA-Fe], [OA-Fe] mitochondrial iron translocation
Mitochondrial dysfunction
Oxidative stress
CHAPTER 2: EFFECTS OF OLEIC ACID AND DERIVATIVES ON HUMAN ENDOTHELIAL CELL VASOACTIVE MEDIATOR PRODUCTION1
2.1 Introduction
Exposure to air pollution particulate matter (PM) is associated with increased adverse
cardiovascular risk factors, including hypertension, atherosclerosis, and metabolic syndrome
(Dockery, 2001; Pope, et al., 2006). The development of PM-induced biological effects
attributed to the fatty acid (FA) component of PM has not been extensively studied.
Non-esterified (i.e., free) FA, like oleic acid (OA), are bioactive andhave been shown to impair the
endothelium-dependent vasodilation mediator nitric oxide in cultured bovine pulmonary artery
endothelial cells (Davda et al., 1995). Elevation of circulating FA levels have been associated
with endothelial dysfunction and endothelial nitric oxide system dysregulation in
insulin-resistant and healthy subjects (Steinberg et al., 2000; Steinberg et al., 1997). Typical sources of
circulating OA include diet and endogenous metabolism. Air pollution sources that contain a
significant level of FA are generally derived from biomass combustion, such as cigarette smoke,
diesel, biofuels (Knothe, 2005), or meat cooking emissions (Nolte et al., 1999). Additionally,
preparation of biofuels commonly involves the conversion of long chain FA from vegetable and
animal fat triglycerides to mono-alkyl esters, though the conversion is generally incomplete. As
a result, biofuels contain a mixture of lipid compounds, both alkyl-esterified (typically methyl)
and unesterified (Schauer, et al., 1999b). In efforts to improve the cost and fuel efficiency of
biodiesel, it has been suggested that source oils may be engineered with certain FA enrichments,
including oleic acid (Knothe, 2005).
PM from the aforementioned pollution sources contain long chain FA on their surfaces
that can enter the lung, and thereby translocate to the circulatory system, where they may induce
vascular effects. It has been shown that the FA leukotriene B4 a can translocate from the airway
lumen into the blood, demonstrating a potential route of exposure of the vasculature via
inhalation (Martin, et al., 1989). Additionally, increases in circulating FA are known to occur by
endogenous production following exposure to air pollutants like ozone, or cigarette smoke
(Kershbaum, et al.; Miller, et al., 2016). Studies reporting vascular and pulmonary effects of OA
suggest possible negative health effects from exposure through non-dietary routes (Christon et
al., 2005; Davda, et al., 1995; Steinberg, et al., 2000; Steinberg, et al., 1997).
PM have been shown to alter the distribution of iron in the lung following inhalation
(Ghio, et al., 2015), and this effect has been observed with oleic acid in chemico, as well (Yao, et
al., 2005). Little mechanistic work has addressed the potential contribution of iron chelation
associated with particle exposures to biological effects (Kallianpur, 2005; Nalini, et al., 1993;
Penzo et al., 2002; Qian, et al., 1991; Schönfeld et al., 2001; Scorrano et al., 2001; Yao, et al.,
2005).
We wanted to test whether the exposure of human endothelial cells to free OA could alter
mitochondrial function responses and production of secondary mediators involved in the control
of vascular tone. OA exposure may induce endothelial dysfunction which may be a potential
2.2 Materials and methods
Materials
OA, conjugated OA-albumin from bovine serum (OA-BSA), methylated OA (me-OA),
methyl-β-cyclodextrin (MBCD), oligomycin, carbonyl
cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), rotenone, antimycin A, glucose, sodium pyruvate,
and ferric ammonium citrate (FAC) were purchased from Sigma-Aldrich (St. Louis, MO).
12-hydroxy OA (12-OH OA) was purchased from Cayman Chemical (Ann Arbor, MI).
Two-hydroxy OA sodium salt (2-OH OA[Na+]) was purchased from Avanti Lipids (Alabaster, AL).
1-14C-oleic acid (58.2 mCi/mmol) was purchased from Perkin-Elmer (Boston, MA). Seahorse XF
DMEM medium, XF24-well microplates, and XF Flux Paks were purchased from Agilent
Technologies (Santa Clara, CA). Gibco L-glutamine was purchased from Thermo Fisher
Scientific (Waltham, MA).
Cell culture
Free OA, 12-OH OA, and me-OA were diluted in anhydrous EtOH to 700mM, and then
further diluted in cell media for use in cell exposures. Conjugated OA-BSA and 2-OH OA(Na+)
were diluted directly in cell media. Equivalent vehicle controls were made with EtOH (<0.14%
EtOH final concentration) or BSA (<33mg BSA/ml) in EGM-2. MBCD was combined with OA
in HBSS (6.37 mM OA, 38.2 mM MBCD) at a maximal binding ratio of ~1:6 OA:MBCD
(Brunaldi et al., 2010) and maximal MBCD concentration for water solubility, then sonicated
and filter sterilized for dilution in dose response testing.
1-14C-OA uptake by HUVEC was determined by incubating cells with ~105 CPM
cells digested overnight in 1M NaOH. Cellular uptake was assessed by measuring cell-associated
14C as a percentage of the total 14C recovered in cells plus media.
FA preparations
Free OA, 12-OH OA, and me-OA were diluted in anhydrous EtOH to 700mM, and then
further diluted in cell media for use in cell exposures. Conjugated OA-BSA and 2-OH OA(Na+)
were diluted directly in cell media. Equivalent vehicle controls were made with EtOH (<0.14%
EtOH final concentration) or BSA (<33mg BSA/ml) in EGM-2. MBCD was combined with OA
in HBSS (6.37 mM OA, 38.2 mM MBCD) at a maximal binding ratio of ~1:6 OA:MBCD
(Brunaldi, et al., 2010) and maximal MBCD concentration for water solubility, then sonicated
and filter sterilized for dilution in dose response testing.
1-14C-OA uptake by HUVEC was determined by incubating cells with ~105 CPM
radioactive OA in 100μM unlabeled OA for 4hr at 37°C. OA was added with either BSA or
EtOH vehicle. After 4hr or 24hr, cell media was removed, culture rinsed twice with PBS, and
cells digested overnight in 1M NaOH. Cellular uptake was assessed by measuring cell-associated
14C as a percentage of the total 14C recovered in cells plus media.
Cell viability
To asses FA cytotoxicity, HUVEC were cultured in 96-well plates to 90-100%
confluence, then exposed to EtOH, BSA, or MBCD solubilized OA, 12-OH OA, 2-OH OA(Na+),
me-OA or vehicle controls, in quadruplicate, for 4, 24, or 48hr. Cell supernatants were collected
immediately following exposure for use with the Promega Cytotox-96 cytotoxicity assay
activity release2. In some experiments, cell viability was visualized with standard light
microscopy techniques by exclusion of trypan blue dye.
Extracellular flux analysis
A whole cell mitochondrial stress test was performed (modified from Lavrich et al.
(2018))using the Seahorse XF instrument (Agilent Technologies), which measures extracellular
oxygen consumption rate (OCR). HUVEC were seeded at 40,000 cells per well in XF24
microplates two days prior to the assay and exposed for 24hr prior to assessment, with wells
divided into blanks, vehicle control, 50, 100, and 250μM OA, and 100μM me-OA or 12-OH OA
in EGM-2. Media was replaced with XF Cell Mito Assay Media (XF DMEM pH 7.4 with
10mM glucose, 1mM sodium pyruvate and 2mM L-glutamine) immediately prior to the assay.
The Cell Mito Stress Test Assay was performed using injections of 1μM oligomycin,
1.25μM FCCP, and 0.5 μM rotenone, antimycin-A combination to measure OCR response. ATP
production was measured using oligomycin, which inhibits ATP synthase. FCCP, a potent
protonophore that makes the inner mitochondrial membrane permeable, was used to measure
maximal respiration, and antimycin-a and rotenone, which inhibit complexes I and III to
effectively halt mitochondrial respiration within the context of the assay, allow measurement of
non-mitochondrial respiration. These injections also allow indirect calculation of additional
bioenergetic parameters, including spare respiratory capacity, proton leak, and
non-mitochondrial respiration.
Soluble mediators
Cell supernatants were collected from 96-well plated HUVEC immediately following
24hr exposure to FA and were frozen until use in assays. Prostaglandin F2α (PGF2α) and
endothelin-1 (ET-1) were measured by ELISA (Enzo Life Sciences, Farmingdale, NY and
IBL-America, Minneapolis, MN, respectively) according to manufacture protocols. Dose-response
measurements were compared as percent of control baseline concentration.
Gene expression
Relative gene expression in HUVEC was measured using RT-PCR. Following 4hr
OA-BSA dose-response exposures of HUVEC cultured on 12-well plates, RNA isolation was done
using the Qiagen RNeasy kit (Valencia, CA) and quantified using a Nanodrop™ 1000
Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). cDNA was generated as
previously described (Karoly et al., 2007). Taqman pre-developed assay reagents from Applied
Biosystems (Thermo Fisher Scientific, Waltham, MA): ET-1 (EDN1), endothelial nitric oxide
synthase (NOS3) or custom-made: beta-actin (Actb), cyclooxygenase-2 (COX2), intercellular
adhesion molecule 1 (ICAM1) [Supplement 1]) were used for gene transcript detection by
fluorogenic amplification of cDNA using the StepOnePlus detection system (Thermo Fisher
Scientific, Waltham, MA). Dose response was compared using amplification cycle threshold (Ct)
and each sample was normalized to Actb.
Iron uptake
Confluent HUVEC cultured in 1well plates were incubated with 100μM FA (OA,
2-OH OA, or 12-2-OH OA) or vehicle in EGM-2 for 4hr. Media was aspirated and replaced with
HBSS with or without 200μM FAC for 1hr. following exposure, HBSS was removed and cells
trichloroacetic acid solution at 70 °C. Non-heme iron concentration in the supernatant was
determined using inductively coupled plasma optical emission spectroscopy (ICPOES; Model
Optima 4300D, PerkinElmer, Norwalk, CT) (Ghio, et al., 2015).
Statistical analysis
LDH release relative to vehicle control was compared by multiple t-tests method with
Holm-Sidak correction for multiple comparisons. Multiple comparisons by 2-way ANVOA with
correction were used for soluble mediator release and iron-uptake assays. Gene expression fold
change was compared relative to vehicle control groups with ∆∆Ct and significance was
determined using a student’s t-test . All analyses were done with GraphPad Prism version 7 or 8.
A p-value of <0.05 was considered significant.
2.3 Results
Cell viability
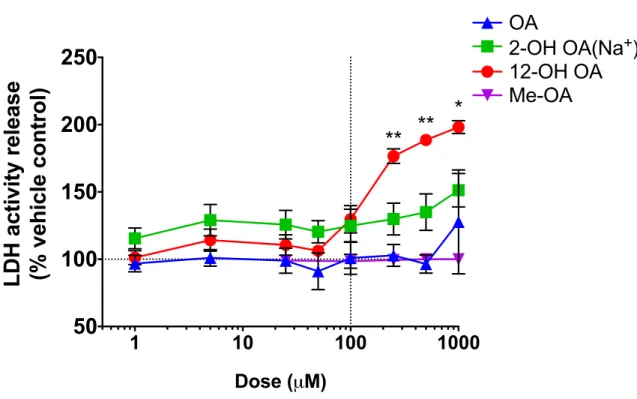
Exposures of HUVEC cultures to FA showed that 12-OH OAsignificantly reduced cell
viability at lower doses than OA after 24hr of exposure (Figure 1). 100μM OA-EtOH and the
OH- metabolites did not significantly increase the release of LDH in HUVEC. HUVEC exposed
Figure 2.1 Comparison of cell viability in response to exposure of HUVEC to OA-EtOH, a hydroxy-metabolite, or me-OA as percent of the vehicle control response. Cell supernatant was collected after 24hr of exposure. Each value is the mean ± S.E.M. of 2-6 replicates. Comparison between OA and 12-OH OA groups done by multiple t-tests, *=p<0.01, **=p<0.0001.
A comparison of OA vehicles (Supplement 2A) showed slightly greater cytotoxicity with
a BSA vehicle, compared to EtOH, at 24hr. Time-course comparison of OA dose-response with
an EtOH vehicle showed that cytotoxicity was also time-dependent between 4 and 48hr of
exposure, with the cytotoxicity of SA relative to EtOH becoming significant at doses greater than
100μM after 48hr of exposure. MBCD, a torus-shaped cyclic oligosaccharide with a
hydrophobic cavity, was shown to induce significantly greater cytotoxicity as a vehicle for OA,
starting at 500μM and 24hr of exposure (Supplement 2A).
Dose-response experiments showed that neither OA-EtOH nor OA-BSA induced
significant cytotoxicity between 4hr and 24hr at 100μM, and either was therefore suitable for use
in assessing further sub-cytotoxic endothelial responses.
1 10 100 1000
50
100
150
200
250
Dose (µM)
LDH activity release
(% vehicle control)
OA
12-OH OA
2-OH OA(Na
+)
Me-OA
**
**
When measuring relative cellular uptake between EtOH and BSA vehicles, 14C-labeled
OA exposures showed that 65% of labeled OA-EtOH became associated with HUVEC, versus
only 8% of OA-BSA at 24hr (Supplement 2B) suggestingdifferentdosimetry of OA dependent
on the vehicle.
HUVEC mitochondrial response to FA3
HUVEC exposed to FA for 24hr prior to assessment by mitochondrial stress test showed
indications of mitochondrial dysfunction at sub-cytotoxic doses of OA and 12-OH OA, but not
me-OA. Basal mitochondrial respiration (Figure 2A), expressed as the starting OCR, was
observed to be decreased in HUVEC that had been dosed with either 100μM 12-OH OA or
250μM OA, relative to control. ATP production was also reduced in these treatment groups
relative to control values (Figure 2B). The addition of FCCP to effectively maximize
mitochondrial respiration showed that cells treated with either 100μM 12-OH OA or 250μM OA
had significantly reduced OCR relative to controls and lower OA doses (Figure 2C). Following
the addition of rotenone and antimycin-A to halt mitochondrial respiration, there was no
difference seen in the relative rates of non-mitochondrial respiration measured (Figure 2D).
Spare respiratory capacity, calculated using basal and maximal respiration measures, and proton
leak also did not vary significantly between treatment groups (Figure 2E and F). Me-OA did not
produce a significantly different response to any of these measures than either the control or 50
and 100μM OA treated groups.
38
Oleic acid-induced soluble mediator release
Dose-response exposure of OA-BSA versus 2- and 12-OH metabolites showed
significantly increased production of PGF2α, primarily in response to 12-OH OA at 100μM
and 250μM compared with OA and control (Figure 3A). Release of ET-1 was reduced by
~50% by 12-OH OA treatment at 100μM (Figure 3B), compared to OA and control, though
all treatments caused a general decline in ET-1 production with increasing dose at 24hr
exposure.
Figure 2.3. FA exposed HUVEC soluble mediator release. Comparison of soluble mediator release by HUVEC exposed to OA and its metabolites. Conditioned media was collected after 24hr exposure to OA-BSA, 2-OH OA(Na+), or 12-OH OA-EtOH or their equivalent
vehicle controls. Mediators were measured by ELISA, as described in methods. A) PGF2α
Gene expression of endothelial signaling factors
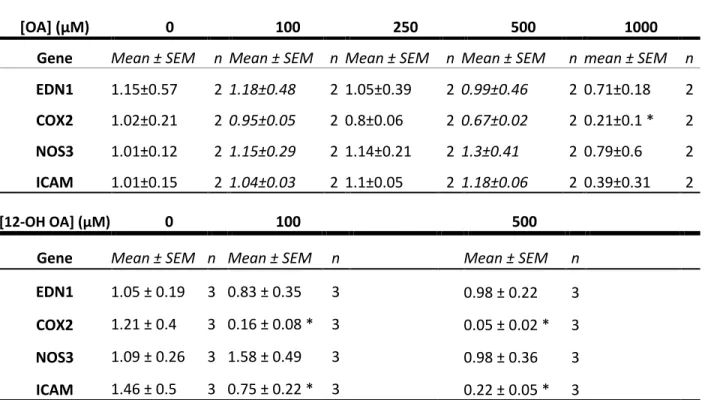
OA exposure caused significant down-regulation of COX2 mRNA with 4hr 1000μM
exposure (Table 1), along with a general trend of down-regulation with increasing OA dose
when compared to the control. 12-OH OA induced an almost 1-fold decrease in COX2
mRNA expression at 500µM, as well as a significant decrease at 100µM. No significant
changes were seen in expression levels of endothelin-1 (EDN1) or endothelial nitric oxide
synthase (NOS3) in response to either exposure. While OA did not change the relative
abundance of ICAM1 mRNA, 12-OH OA caused a significant decrease in expression at both