Abstract
The cell cycle is a highly controlled mechanism composed of G1, S, G2, and M phases,
with the transitions between these phases regulated in part by cyclin dependent kinases
(CDKs). CDK inhibitors (CKIs) including p21 and p27 are key proteins that arrest the cell in G1
to prevent division when the cell is under stress or possesses DNA damage. Additionally, p27
expression is upregulated in quiescence (G0), when the cell is not actively cycling. p27 is
targeted for degradation at the G0/G1 transition, and again at the G1/S transition when p21 is
also degraded, though the order of cell cycle protein degradation at G1/S, and its impact on
normal cell proliferation, is unknown. The purpose of this study is to develop a fusion protein
biosensor that mirrors endogenous p27 expression in mammalian cells. We generated two
constructs containing the p27 gene fused C-terminally with Yellow Fluorescent Protein (Venus
(Ven)). We transfected cells with one such construct and compared p27-Ven levels by
performing time course synchronizations and serum starvation experiments visualized via
immunoblot. Contrary to our expectations, p27-Ven was not degraded at the G1/S transition, nor
were levels upregulated in G0. We therefore cannot currently consider our fusion protein a
reliable biosensor. Interestingly, in other studies, functional N-terminally tagged p27 biosensors
have been developed. We thus produced Ven-p27 constructs and will investigate them as
potential alternatives. Once we develop an effective biosensor, we will produce a
high-resolution temporal map of protein degradation primarily via single cell analysis using live cell
imaging. By perturbing CKI and other cell cycle protein degradation with drugs and siRNAs, we
Introduction
Cells proliferate by progressing through a series of phases known collectively as the cell
cycle. The transitions between the phases are tightly controlled by cell cycle checkpoints, which
are periods when the cell is kept from dividing until certain intracellular and extracellular
conditions are met. Many protein classes, such as cyclins and cyclin-dependent kinases (CDKs),
form crucial complexes that provide signals to downstream effectors, which permit the transition
to the next phase of the cycle and induce events such as centrosome duplication and DNA
replication via protein phosphorylation (Murray, 2004). The ability of cyclin-CDK complexes to
promote cellular growth is directly regulated by the levels of CDK inhibitor proteins (CKIs).
CKIs prevent cell cycle progression beyond a particular checkpoint by binding to cyclin-CDK
complexes and blocking their enzymatic activity (Coqueret, 2003). In this way, CKIs serve a
critical function as tumor suppressors and control the rate of proliferation and responsiveness of
cell cycle progression to extracellular factors such as substrate adhesion and growth factors (He,
Wang, Chen, & Guan, 2012). A significant majority of human cancers (90%) have an altered
expression of certain CKIs, CDKs, and cyclins within the transformed cells; this altered
expression causes cell proliferation to progress rapidly and without regulation (Bonelli, Tuccillo,
Borrelli, Schiattarella, & Buonaguro, 2014).
The CKI p27 has demonstrated an important role in both cell differentiation and
proliferation, with increased levels of p27 seen following differentiation signals and decreased
levels seen following growth factor signaling (J. Slingerland & Pagano, 2000). The protein p27
inhibits cyclin E/CDK2 and cyclin A/CDK2 complexes, leading to cellular arrest at G1 (Lee &
Kim, 2009). After p27 degradation occurs, these complexes are responsible for S phase onset
end of G1 (J. M. Slingerland et al., 1994), though p27 is not degraded entirely at this point in the
cell cycle. It is known that the protein exists at reduced levels throughout the cell cycle in the
majority of proliferating cells in cell lines such as BALB/c 3T3 (Agrawal, Dong, Wang, Kayda,
& Pledger, 1995). High expression of p27 is seen in cells that are in G0 (quiescence), and
degradation of p27 occurs in cells that re-enter the cell cycle at G1 (Oki et al., 2014).
Although the timing and order of fluctuations in p27 expression relative to other proteins
at high temporal resolution is unknown, the mechanisms by which p27 accumulates and degrades
are well understood. A major regulator of p27 expression is the ubiquitin proteasome system,
which is responsible for ubiquitinating 80-90% of intracellular proteins (Shen, Schmitt, Buac, &
Ping Dou, 2013). Three types of enzymes mediate this process—E1, E2, and E3—and serve to
activate, conjugate, and ligate ubiquitin, respectively, in a sequence of events that results in a
protein substrate attached to a poly-ubiquitin chain. Once ubiquitinated, proteins are guided to
the 26S proteasome complex for removal of ubiquitin chains before protein degradation occurs.
The E3 enzyme in particular becomes part of the ligase complex SCFSKP2, which facilitates p27 degradation (Shen et al., 2013). Another major component of this complex is S-phase
kinase-associated protein 2 (SKP2) and is a well-known protooncogene and F-box protein. SKP2 plays
a role in mediating p27 degradation at the G1/S transition (Bochis, Irimie, Pichler, & Neagoe,
2015). Further, p27 degradation is regulated by SKP2 at the S/G2 transition, as p27 accumulates
at this transition in mice lacking the Skp2 gene (Nakayama et al., 2004). Unlike during the G1/S
and S/G2 transitions, SKP2 is not present in the cell to degrade p27 at the G0/G1 transition, yet
p27 is nevertheless degraded (Hara et al., 2001). Explaining this discrepancy, a pathway has
been discovered linking p27 degradation to Kip1 ubiquitylation-promoting complex
exported from the nucleus (Kotoshiba, Kamura, Hara, Ishida, & Nakayama, 2005). Nuclear
export and subsequent cytoplasmic localization occurs due to phosphorylation of the Serine 10
residue of p27. However, cytoplasmic localization does not appear to be necessary for p27
proteolysis as was once thought (Rodier et al., 2001).
A large amount of research has been done regarding proteins present in the S and M
phases of the cell cycle, but the cell cycle transitions have gone relatively unexplored, leaving
the exact accumulation and degradation of cell cycle proteins at critical transitions unknown. In
this study, we seek to address this void, focusing on the CKI p27. We generated two plasmid
DNA constructs containing the p27 gene (CDKN1B) C-terminally tagged to Yellow Fluorescent
Protein (Venus (Ven)). Fluorescent tagging is an appropriate method to use for obtaining
information regarding protein accumulation and degradation timing, as we are able to easily
visualize the fluctuations in fluorescence via a number of microscopy techniques and eventually
live cell imaging. After suspecting that the p27-Ven fusion protein was not a reliable biosensor
to mirror endogenous p27 expression, and with the knowledge that others in our field have also
struggled with C-terminally tagged fusion proteins (Oki et al., 2014), we generated two
N-terminally tagged Ven-p27 constructs that will undergo rigorous testing to verify if they are more
effective and dependable biosensors.
Once we determine which fusion proteins, if any, are effective biosensors, we will create
a high-resolution temporal map of protein degradation order at the G1/S and S/G2 transitions
using these as well as other constructs coding for cell cycle proteins such as p21 and
Cdc6. Future experiments will involve altering the level and order of protein expression and
degradation impacts cell growth. This knowledge will be useful in further investigation of
diseases such as cancer that are driven by deviation from normal cellular proliferation control.
Materials and Methods
Plasmid Preparation
We generated multiple C-terminally and N-terminally tagged fluorescent plasmid
constructs (p27-Ven and Ven-p27, respectively) via two reaction cloning protocols: Gateway®
BP and LR cloning (Thermo Scientific, DE) and single-step isothermal DNA recombination
(Gibson et al., 2009). Using PCR and Q5® high fidelity DNA polymerase (NEB, MA), we
amplified the CDKN1B gene sequence in the presence of forward and reverse primers specific to
each construct design (see Appendix). Electrophoretic gels used to separate DNA for subsequent
purification steps or verification of cloning success were composed of 1% agarose. We
conducted gel extraction and column purification steps using the appropriate QIAQuick® DNA
purification protocols (QIAGEN, MD). For Gateway® cloning, we subcloned the DNA
fragments into the donor vector pDONR221 using the Gateway® BP reaction protocol and used
electrocompetent DH5αE. coli cells in transformation.
After performing the BP reaction and transformation, we plated on kanamycin plates and
incubated overnight. We performed PCR on colonies using standard techniques to screen for
successful transformations. We then amplified and isolated the plasmid via a QIAQuick®
mini-prep protocol (QIAGEN, MD). At this point, we subcloned the entry vectors into destination
vectors (see Appendix) using the standard Gateway® LR reaction protocol and transformed into
DH5αE. coli. pLenti PGK Neo DEST (w531-1) and pLenti PGK Puro DEST (w529-2) were
2009). pcDNA5/FRT/TO-Venus-Flag-Gateway (1124) was a gift from Jonathon Pines
(Addgene plasmid # 40999). The vectors will henceforth be referred to as the following:
Destination Vector Name New Vector Name Associated Expression Vector Name
pcDNA5/FRT/TO-Venus-Flag-Gateway (1124) pDESTJC13 pEXPJC13-p27-Ven
pLenti PGK Puro DEST
(w529-2) pLenti-puro pLenti-puro-Ven-p27
pLenti PGK Neo DEST
(w531-1) pLenti-neo pLenti-neo-Ven-p27
We employed the same screening methods after completing the LR protocol though
selected with ampicillin rather than kanamycin. After construct screening and purification, we
confirmed cloning success through DNA sequencing via UNC sequencing facilities (UNC-CH,
NC). We generated all constructs except pEXPJC17-p27-Ven using the above method. For
pEXPJC17-p27-Ven, a nourseothricin derivative of pEXPJC13-p27-Ven, we subcloned the
nourseothricin resistance gene into pEXPJC16-p27-Ven (another derivative of
pEXPJC13-p27-Ven) after removing its puromycin resistance gene using a single-reaction isothermal cloning
method (Gibson et al., 2009). We employed the same general methods for purification,
transformation, and verification as with the Gateway® cloning scheme.
Cell Cultures and Stable Cell Line Generation
We maintained all cell lines in Dulbecco's modified Eagle's medium (DMEM) (Sigma) +
10% fetal calf serum (FBS), L-Glutamine, and penicillin/streptomycin. To generate our stable
cell lines, we transfected pEXPJC13-p27-Ven into human embryonic kidney HEK 293T
mammalian cells obtained from American Type Cell Collection using a standard transfection
protocol with polyethyleneimine (PEI). After transfection and fluorescence confirmation, we
developed pEXPJC17-p27-Ven and went forward with usage of that construct for its preferred
U2OS-TRex cells, which were a gift from John Aster, for further experimentation (Malecki et al.,
2006). TRex cells utilize the Flp-In™ T-REx™ System which enables the use of FRT sites
within the plasmid to perform recombinase-mediated DNA recombination and generate stable,
inducible expression cell lines (Thermo Scientific, DE). Our pEXPJC17-p27-Ven construct is
under a doxycycline-inducible system (dox). For the pLenti constructs, we employed a standard
lentiviral transfection protocol to transfect the cells using pVSVG and ΔNRF plasmids. We then
performed a transduction into the human telomerase reverse transcriptase immortalized retinal
pigment epithelial cell line hTERT-RPE1 using a standard protocol with 8 µg/mL polybrene.
The expression of the Ven-p27 fusion protein generated in these cells is not
doxycycline-inducible but rather constitutively expressed.
Microscopy
We imaged all cells using a Carl Zeiss Axiovert 40 CFL inverted microscope with a 10X
objective and 10X lens. We visualized fluorescence using an X-Cite® 120Q wide-field
fluorescence microscope excitation light source. We utilized ImageJ software to alter DIC
images to grayscale and fluorescence images to yellow from their original green for improved
viewing and better understanding of our results. Because we did not quantify our images, we
also used ImageJ to enhance contrast and decrease background noise so that dimly fluorescing
cells could be better observed for qualitative analysis.
Protein Lysate Preparation
We prepared whole cell lysates by washing and collecting cells in trypsin and then
centrifuging the cells at 2,000 rpm for 2 minutes. After removing the trypsin via pipetting, we
this process once and then resuspended the pellet in 2X Laemmli sample buffer (SDS) + 10% β
-mercaptoethanol (BME). We then boiled the lysates for 5 minutes before immunoblotting.
Immunoblot Analysis
For all of our immunoblots, we used 10% polyacrylamide gels and loaded 2.5 µL of
Precision Plus Protein™ Dual Color Standards ladder (BioRad, CA) and 15-20 µL of protein
lysate. We electrophoresed the gels at 170V for 70 minutes and transferred at either 110V for 75
minutes or 20V overnight. We transferred all blots to PVDF (Thermo Scientific, DE) except for
the serum starvation and fusion protein comparison blots, for which we used nitrocellulose
membranes (GE Healthcare Life Sciences, UK). Before blocking, we added Ponceau S solution
to confirm uniform protein loading concentration (Sigma). We blocked for 1-2 hours in 5%
nonfat dry milk/Tris buffered saline with Tween® (TBST).
After blocking, we added primary antibodies in 2.5% milk/TBST for 1 hour then washed
with TBST 3 times for 5 minutes each. When probing for p27, we used rabbit polyclonal p27
primary antibody (C-19, Santa Cruz Biotechnology, sc-528) at 1:3,000. For our cyclin A
immunoblot, we probed with cyclin A polyclonal primary antibody (C-19, Santa Cruz
Biotechnology, sc-596) at 1:2,000. We added donkey anti-rabbit HRP secondary antibody
(Jackson ImmunoResearch Laboratories, PA) to all blots at 1:10,000 in 1.25% milk/TBST for 1
hour. We washed all blots in TBST 3 times for 5 minutes each before preparing blots for
visualization using Amersham ECL Western Blotting Detection Reagent (GE Healthcare Life
Sciences, UK).
Time Course Synchronizations
We conducted two time course synchronization experiments: a thymidine-nocodazole
for both of these experiments. In the thymidine-nocodazole block and release experiment, we
blocked cells in thymidine to synchronize at the beginning of S phase. Upon release, we allowed
the cells to progress through one cell cycle before blocking the cells with nocodazole, which
subsequently synchronized the cells at the G2/M phase transition. In the double thymidine block
and release experiment, we synchronized cells at the G1/S transition for lysate collection,
enabling us to visualize p27-Ven and endogenous p27 expression in S phase rather than G1.
For our thymidine-nocodazole block and release experiment, we plated cells at 25%
confluence in 60 mm dishes for 3 doxycycline conditions (0, 5, and 20 µg/mL). After 24 hours
had elapsed, we treated the cells with 2.5 mM thymidine for 24 hours. We then washed the cells
twice with 1X PBS and added DMEM + 10% FBS + 100 ng/mL nocodazole + doxycycline for
16 hours. At this point, we collected each well’s media into one of three 50 mL conicals based
on doxycycline concentration and centrifuged at 2,000 rpm for 5 minutes. We aspirated the
liquid and resuspended each conical in DMEM + 10% FBS. Before replating the liquid, we
washed the cells twice with 1.5 mL 1X PBS. We collected each time point by trypsinization
following our protein lysate protocol for a total of nine time points.
For our double thymidine block and release experiment, we plated cells at roughly 20%
confluence in 6-well dishes with multiple doxycycline conditions. One day after plating, we
blocked cells in 2.5 mM thymidine for 18 hours. We released the cells by washing twice with
1X PBS and replaced the media with DMEM + 10% FBS. After 8 hours, we blocked again in
2.5 mM thymidine and added doxycycline for 18 hours before releasing the cells using the same
protocol. We collected two time points following our protein lysate protocol.
Serum Starvation
JC17-p27-Ven, RPE-TRex as a control for p27 expression in normal cells (a gift from the
Jackson lab), and U2OS-TRex-JC13-GFP as a control for cells containing an expression plasmid.
We plated cells into 6-well dishes in 2 mL DMEM + 10% FBS and had multiple conditions per
cell line (Figure A1). The day after we plated the cells, we removed the medium and replaced
with either DMEM + 10% FBS or 0% FBS following our conditions. After 24 hours of
starvation, we added 10 µg/mL doxycycline to the appropriate p27-Ven wells to induce fusion
expression. Before collecting each well as a protein lysate, we visualized the cells using
differential interference contrast (DIC) microscopy and a YFP filter.
Results
! The primary goal of this study is to develop and verify fusion protein biosensors that
mirror endogenous protein expression. Our overall expression strategy is to develop plasmid
DNA constructs that contain the genetic sequence for our desired fusion protein, p27-Ven. The
fusion protein sequence bears a promoter region that is doxycycline-inducible. In utilizing as
doxycycline-inducible system, we can determine an appropriate doxycycline concentration to use
going forward in which we can induce the same level of fusion protein p27 expression as
endogenous p27 expression. We elected to use a U2OS cell line because we are particularly
interested in protein dynamics within transformed cells and have previously used U2OS cells
when developing other cell lines we hope to compare to our p27-Ven lines.
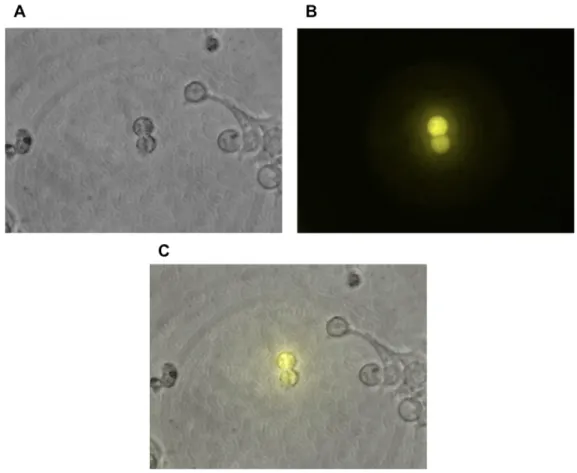
pEXPJC13-p27-Ven expresses in HEK 293T cells
To develop an effective fusion protein biosensor, we first generated a C-terminally
tagged doxycycline-induced plasmid construct: pEXPJC13-p27-Ven. We then transfected the
plasmid into HEK 293T mammalian cells to confirm our construct’s ability to fluoresce in vivo
HEK 293T cells fluoresced, indicating successful cloning and construct development (Figure 1,
A and B). p27-Ven did not appear localized in a specific region of the cell due to overexpression
of the protein hindering qualitative analysis of the composite image (Figure 1C). The result of
the transfection enabled us to go forward in transducing U2OS cells with this construct.
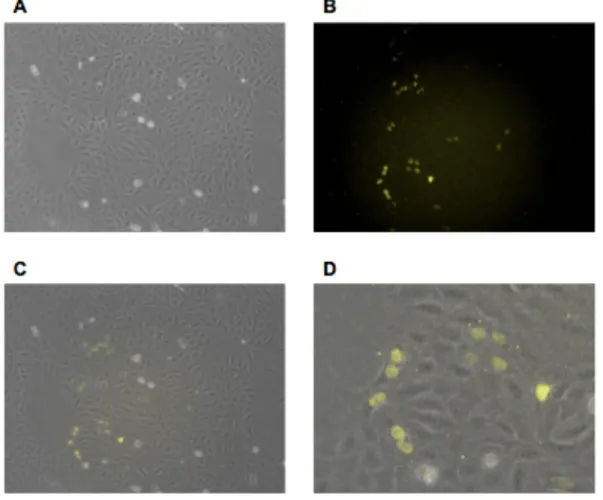
pEXPJC17-p27-Ven expresses in U2OS cells
After confirming that our p27-Ven fusion protein was capable of producing visible
fluorescence, we designed and constructed a plasmid with the more effective drug resistance of
nourseothricin rather than hygromycin. By utilizing a stronger drug resistance marker, we
decreased the length of time required for selection to generate stable cell lines, which provided
us with the ability to generate multiple cell lines that bear this construct more efficiently than
would have been possible using a hygromycin resistance system. We designed and produced the
plasmid construct pEXPJC17-p27-Ven to accomplish this objective. Once the construct was
generated, we performed a transfection using HEK 293T cells before transducing U2OS
cells. We confirmed that the transduction was successful and that pEXPJC17-p27-Ven exhibited
fluorescence via microscopy with a YFP filter after inducing expression with doxycycline
(Figure 2B). Fluorescence levels appeared variable across individual cells that exhibited
fluorescence, though a sizeable proportion of the cells were fluorescing (Figure 2, A and B). The
fluorescence appeared localized to the nuclei of the fluorescing cells due to the rounded shape of
the fluorescence and composite images that were generated (Figure 2, C and D). From these
data, we concluded that U2OS cell machinery was effectively translating the fusion protein and
that the protein was capable of fluorescing as expected. The protein is also localizing in the
same manner as is expected of functioning endogenous p27, though the functionality of the
fusion protein cannot be confirmed to mirror endogenous p27 based on these conclusions.
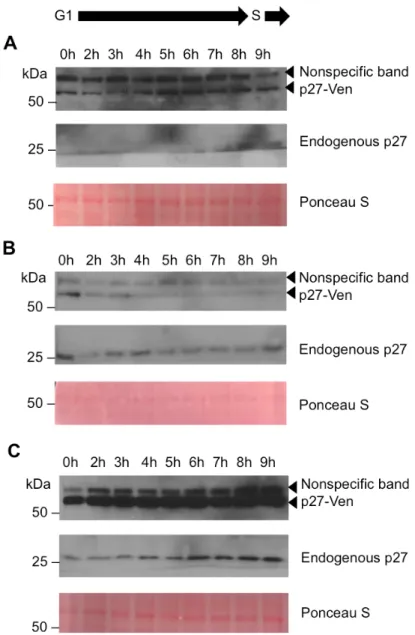
p27-Ven and endogenous p27 express abnormally in cell synchronizations
Once we generated a stable cell line and established that p27-Ven fluoresces in U2OS
cells, we aimed to verify whether or not the fusion protein exhibited similar dynamics as
endogenous p27. We first performed a thymidine-nocodazole block and release synchronization
to visualize the expression of p27-Ven and endogenous p27 as the cells synchronously moved
through G1 into S phase (Figure 3). Blocking initially in thymidine synchronizes the cells at the
beginning of S phase. After releasing the cells from thymidine and allowing the cells to progress
through one cell cycle from that point, we blocked in nocodazole, which serves to improve
synchronization efficiency by synchronizing the cells at the G2/M phase transition. Upon
releasing the cells from nocodazole, we expected to observe both endogenous p27 and p27-Ven
expression decrease as the cells progressed from G1 into S phase.
In these as well as subsequent blots, a prominent nonspecific band appeared directly
above the fusion protein p27-Ven. We were able to establish which band was the nonspecific
band by comparing a U2OS-GFP cell line to our p27-Ven line and probing for p27. This blot
also confirmed that we had doxycycline-inducible Ven expression (Figure A2). Leaky
p27-Ven expression was observed in the cells that received 0 µg/mL doxycycline, as the fusion
protein should only be induced in the presence of doxycycline (Figure 3A). This leaky
transcription of the genes encoding p27-Ven in the absence of doxycycline stimulation. Further,
endogenous p27 was not visible on the blot until late in the time course due to the band being
close to the bottom of the gel and transferring off of the membrane during immunoblotting.
Regardless of this error, the endogenous p27 level appeared nearly equivalent to that of the
fusion protein at the same time points (7h, 8h, and 9h). We had expected that endogenous p27
levels would decrease within this timeframe, though any explanation for this discrepancy
between our expected and actual results would be purely speculative until this experiment is
repeated and endogenous p27 expression is fully observed.
Although previous studies have demonstrated that p27 is degraded at the G1/S transition
(J. M. Slingerland et al., 1994), p27-Ven did not appear to be degraded at the G1/S transition in
the leaky 0 µg/mL doxycycline cells (Figure 3A). Similarly, in the cells that received 20 µg/mL
doxycycline, p27-Ven levels remained constant throughout the time course (Figure 3C). It
appeared that p27-Ven expression decreased very slightly in the 5 µg/mL doxycycline cells over
the time course (Figure 3B). However, contrary to our expectations for endogenous p27
expression, expression remained constant in both the 20 µg/mL and 5 µg/mL doxycycline cells,
if not increasing in the 20 µg/mL doxycycline time course (Figure 3, B and C). Possible
explanations for this trend include that the cells did not effectively synchronize or that the fusion
protein was not degraded with the same kinetics in which endogenous p27 was degraded.
We attempted to use cyclin E to confirm when the G1/S transition occurred during the
time course but grossly overestimated the antibody dilution factor, which resulted in an
overprobed blot (image not shown). Nevertheless, the G1/S transition was expected to occur
during this timeframe based on previous studies with U2OS cells (Grant et al., 2013; Laoukili et
course synchronization three times (twice when using a 6 hour time course and once with a 9
hour time course (Figure 3)). These results caused us to grow concerned that by inducing
p27-Ven expression in the cells, we were potentially impacting the ability of the cells to move into S
phase effectively due to the role p27 plays in cell cycle inhibition at G1 phase.
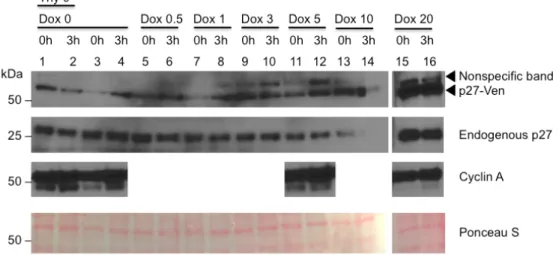
Figure 4. Immunoblot of double thymidine time course synchronization. U2OS-JC17-p27-Ven cells were blocked in thymidine for 18 hours, were released for 8 hours, and then blocked again in thymidine for 18 hours before being collected at various time points. Thy 0 cells received no thymidine treatment and were thus an unsynchronized control. Variable doxycycline
concentrations were used (0, 0.5, 1, 3, 5, 10, and 20 µg/mL). Cyclin A was used to confirm that the cells were in S phase for select doxycycline concentrations (0, 5, and 20 µg/mL). Ponceau S served as a loading control.
In an attempt to observe p27-Ven expression in S phase, we performed a double
thymidine block and release experiment. Because thymidine blocks cells at the beginning of S
phase, this experimental design enabled us to look for changes in p27-Ven and endogenous p27
expression at the start and into S phase. We expected to see both p27-Ven and p27 levels remain
at a low level as the cells progressed through S phase, depending on the exact timing of p27
degradation relative to the cell cycle.
Endogenous p27 expression remained constant across time points as well as relatively
constant across doxycycline concentrations, as expected (Figure 4). p27-Ven levels fluctuated
inconsistently; the 0, 1, and 5 µg/mL doxycycline cells (lanes 3 and 4; 7 and 8; and 11 and 12,
respectively) demonstrated a possible increase in p27-Ven, while the unsynchronized 0 µg/mL
doxycycline cells (lanes 1 and 2) demonstrated a decrease in p27-Ven. The remaining cells
determine if the cells were in S phase, when cyclin A levels are high. Cyclin A levels remained
constant and high across time points and doxycycline concentrations.
Endogenous p27 levels decrease in serum-starved U2OS cells
Because of the inconsistencies regarding the anticipated and actual results of the
expression of p27-Ven in U2OS cells, we suspected that our fusion protein might not be an
effective biosensor for endogenous p27 expression at the G1/S phase transition. Despite this
suspicion, we recognized that we could attempt to employ the biosensor to examine p27
expression as cells transition between G1 and G0 phase. To determine if p27-Ven expression
was comparable to endogenous p27 expression in G0, we performed a serum starvation
experiment (Figure 5). In this experiment, we compared U2OS cells with p27-Ven to U2OS-
cells with GFP, which provided a control for any impact the addition of a fluorescent protein
might have on cells, as well as for any differences across U2OS cell lines. We also compared
our p27-Ven cell lines to an RPE cell line, which provided a positive control for endogenous p27
expression in non-transformed cells. Additionally, we compared wells that were allowed to
become confluent with cells to those that were not allowed to do so, as confluence and contact
inhibition can trigger cells to enter G0 even in the presence of FBS (Hayes et al., 2005; Vinals &
Pouyssegur, 1999). In addition, we examined the effect doxycycline induction had on p27-Ven
expression and compared serum-starved cells to cells receiving FBS. We expected to observe
high p27-Ven and endogenous p27 levels in serum-starved cells as well as confluent cells in
comparison to non-serum-starved cells and non-confluent cells.
After immunoblotting and exposing the blot for a prolonged amount of time, we only
that were not confluent upon collection (Figure 5A). Because of this, we focused our attention
on determining whether p27 endogenous expression was consistent with our expectations.
Although there was a slight increase in p27 expression in the serum-starved cells that
were neither confluent nor doxycycline-induced, there was a dramatic decrease in p27 expression
in the serum-starved confluent cells within this group when compared with the FBS+ confluent
cells (Figure 5A). This was inconsistent with our expectations. Further, this trend was also seen
in doxycycline-induced cells, which was expected since doxycycline should not impact p27
endogenous expression.
While the p27-Ven cell line demonstrated results in the serum starvation that conflict
with dependable results in other studies related to p27 expression in G0, the endogenous p27 in
the controls used in this experiment behaved as expected. In the U2OS-GFP cells, serum
starvation yielded roughly equivalent expression of endogenous p27 as was seen when we forced
cells to grow to confluence (Figure 5A). The FBS+ cells that were not confluent demonstrated
low levels of endogenous p27. These results were also demonstrated in the RPE cells as
predicted. This experiment was repeated three times with small adjustments in methodology
until the results in this paper were obtained. Each experiment indicated results similar to those
demonstrated.
Prior to collection of the protein lysates used in the serum starvation immunoblot, we
imaged the U2OS-p27-Ven cells under DIC and with a YFP filter to visually characterize the
fluorescence. We opted to complete this step in the event that few cells were expressing greater
amounts of p27-Ven than other cells and thus biasing our immunoblot data. Cells treated with
10 µg/mL doxycycline and supplied FBS had roughly equivalent numbers and intensities of
doxycycline (Figure 5, B and C, E and F). Variable intensity of fluorescence between cells was
slightly more pronounced in the FBS+ cells than in the serum-starved cells, though variability
was present in both cell populations. p27-Ven localization was nuclear in both cell populations
(Figure 5, D and G). Variable fluorescence intensity yet consistently nuclear localization was
expected results based on previous visualization of this cell line. However, we did not expect to
see a population of cells in the FBS+ cells that were fluorescing much brighter than the brightest
fluorescing cells in the serum-starved population.
Ven-p27 as a reliable marker for endogenous p27 expression
Because of our difficulties verifying the p27-Ven fusion protein as a biosensor, we
reached the decision that this fusion protein is likely not a reliable biosensor around which we
should base future research endeavors. We also discovered that a study by Oki et al. has
produced both C- and N-terminally tagged mutant p27 fusion proteins, with only the N-
terminally tagged fusion protein being declared a successful biosensor (Oki et al., 2014). Thus,
we developed an N-terminally tagged p27 fusion protein, Ven-p27, to have a better chance of
successfully creating a biosensor for endogenous p27 expression (Figure 6A). While the fusion
proteins generated by Oki et al. involve a defective p27K- mutant that does not possess CDK
activity, we designed our fusions using wild-type p27 so that they might function as biosensors
for endogenous p27 expression. In designing a new construct with this scheme, we shifted away
from U2OS cells and focused on generating stable RPE cell lines. We chose to use a
non-transformed cell line in the event that the U2OS cells were not responding as expected because
they were transformed and thus behaved aberrantly. We also moved from using a
doxycycline-inducible system to a lentiviral system to enable faster experimentation. We developed and then
Although no fluorescence was visualized using microscopy with a YFP filter in the
hTERT-RPE1 transduced cells (image not shown), immunoblot results demonstrated that the
transductions were in fact successful for both the pLenti-neo-Ven-p27 and pLenti-puro-Ven-p27
constructs. Ven-p27 levels were low in these constructs and roughly equivalent to the level of
fusion protein created in the uninduced JC17-p27-Ven cells (Figure 6B). Control
U2OS-JC17-p27-Ven cells with and without doxycycline both demonstrated fluorescence, with leaky
expression seen in uninduced cells and greater expression in induced cells. Localization of the
fusion protein was nuclear and was consistent with previous microscopy (Figure 6, C-H).
Discussion
In this study, we sought to develop fluorescent p27 plasmid constructs to serve as
biosensors for endogenous p27 expression in mammalian cells. To this end, we generated
C-terminally and N-C-terminally tagged fluorescent p27 constructs and investigated their
functionality as biosensors. We focused on the protein p27 due to its involvement in not only
cell cycle proliferation but also in cancer. It has been shown that p27 has a significant impact on
cell proliferation, with p27 degradation and deregulation being linked to poor prognoses in
multiple forms of cancers (Chiarle et al., 2000). Cancers such as colon, breast, nasopharyngeal,
and others demonstrate lower nuclear p27 levels in more advanced stages of cancer, indicating
an inverse correlation between p27 expression and a desirable prognosis (Alkarain, Jordan, &
Slingerland, 2004; Chu, Hengst, & Slingerland, 2008; He et al., 2012; Liu et al., 2013; J.
Slingerland & Pagano, 2000).
We successfully developed hygromycin resistant pEXPJC13-p27-Ven and transfected it
into HEK 293T cells to validate fusion protein fluorescence. Instead of further utilizing
pEXPJC13-p27-Ven, we modified the construct to obtain a construct with the more favorable
drug resistance of nourseothricin (pEXPJC17-p27-Ven). We confirmed the success of this
cloning and subsequent transduction after visualizing fluorescent U2OS cells.
In studying the kinetics of our fusion protein, we saw that both the fusion protein and
course experiment. Although we repeated this experiment multiple times in an attempt to obtain
a greater protein yield and confirm visualization of the G1/S transition, we never saw p27-Ven or
endogenous p27 levels fluctuate. Our double thymidine block also indicated our fusion protein
might not be functioning properly. Endogenous and fusion protein p27 levels remained constant
and elevated in this experiment. Additionally, cyclin A levels remained constant, which
demonstrated the cells were in S phase and low p27 levels should have been observed.
There are a number of reasons that may explain these unanticipated results. In both
experiments, the cells may not have synchronized very well. Additionally, in the
thymidine-nocodazole experiment, the cells may have had an abnormally prolonged G1 phase due to the
addition of p27-Ven, which is a cell cycle inhibitor. We also considered the possibility that a
small proportion of cells were overexpressing the fusion protein in any given experiment. This
cell population may be distorting the data because immunoblotting averages protein expression
across a population. Also conceivable is that the U2OS cells do not regulate p27 properly as a
consequence of their transformed status.
In the future, we plan to utilize single cell analysis via live cell imaging to avoid
continually experiencing difficulties when interpreting immunoblot data. We will also repeat
these experiments and include a control cell line such as untransfected U2OS cells to confirm
that the cells are indeed cycling. In this study, our cyclin E blot was very overprobed, leading to
possible misinterpretation of the blot as well as no clear indication that the cells actually
transitioned from G1 to S phase. Due to this error, we will work to successfully probe future
thymidine-nocodazole blots to look for cyclin E or another protein such as p21 that experiences a
change in expression at the G1/S transition. Although we were able to include a nonspecific
results as formal loading controls. We were unable to include these controls in this study
because we did not image our blots when staining with Ponceau S during experimentation.
Like our time course synchronization experiments, our investigation of p27-Ven and
endogenous p27 expression via serum starvation yielded unexpected results. Interestingly, we
saw minimal p27-Ven expression via immunoblotting yet clearly saw fluorescence in some cells
when imaging the cells using a YFP filter. This disparity may be explained by our previous
suggestion that if few cells were expressing the fusion protein, average fusion protein expression
would appear decreased. However, fluorescence in this experiment generally appeared
consistent with fluorescence seen in past experiments. Further experimentation is necessary
before we can identify and understand the cause of this low fusion protein expression.
By examining endogenous p27 expression, we concluded the endogenous p27 in
U2OS-JC17-p27-Ven cells was not responding as expected to the serum starvation, yet the endogenous
p27 in U2OS-JC13-GFP and RPE cells was responding appropriately and similarly across the
cell lines. The contrast in endogenous p27 levels across the two U2OS lines suggests that some
attribute of p27-Ven or simply its presence is influencing endogenous p27 kinetics and/or U2OS
cell cycling. Based on our findings, it is possible that the fusion protein disrupted expected
cellular events, though there are other conditions that might explain these results. One caveat to
these data is our lack of a truly successful replicate due to low lysate concentration and excessive
nonspecific antibody binding in previous replicate attempts. With this factor in mind, we
concluded that p27-Ven should not be utilized as a biosensor for endogenous p27 expression
unless further replicates prove these data incorrect.
In light of inconclusive results and time constraints, we developed novel N-terminally
influenced by Oki et al., who created mutant N- and C-terminally tagged p27 fusion proteins,
with only the N-terminally tagged fusion functioning properly (Oki et al., 2014). We have
successfully cloned pLenti-puro-Ven-p27 and pLenti-neo-Ven-p27 constructs and are working to
generate stable hTERT-RPE1 cell lines containing these constructs. While our transduction of
hTERT-RPE1 cells with these constructs was deemed successful via immunoblot, the fusion
protein was produced at similar levels to that seen in uninduced expression of the C-terminally
tagged construct in U2OS cells. We concluded that because fluorescence was not visualized via
microscopy in the hTERT-RPE1 cells but was seen in the uninduced U2OS cells, it is possible
that a greater number of RPE cells were generating the fusion protein to a lesser extent than the
few leaky U2OS cells. Thus, we are working to generate stable cell lines that produce greater
levels of Ven-p27 and can be visualized not only via immunoblot but also via microscopy.
We hope that by changing from C- to N-terminally tagged fusions as well as from a
transformed to a non-transformed cell line, we will develop an effective biosensor system for
endogenous p27 expression. Future studies will utilize verified p27 fusion protein constructs in
mammalian cells to produce a high-resolution temporal map of protein degradation primarily via
single cell analysis using live cell imaging. With this map, the effect of protein degradation
order on cellular proliferation and genomic stability can be better understood.
Acknowledgements
I thank Dr. Jean Cook for providing helpful insight regarding modifying and approving
experimental designs and the final editing of this paper. I thank Dr. Gavin Grant not only for
designing all primers used in this study and constructing pEXPJC17-p27-Ven, but also for
serving as a mentor, training me in laboratory techniques, and assisting me in troubleshooting. I
helpful discussion. I thank John Aster for the gift of U2OS-TRex cells, Eric Campeau for the
gift of the pLenti-neo and pLenti-puro vectors, and Jonathon Pines for the gift of the
pDESTJC13 vector. Lastly, I thank my BIOL 692H peers who reviewed this manuscript and
References
Agrawal, D., Dong, F., Wang, Y. Z., Kayda, D., & Pledger, W. J. (1995). Regulation of cyclin E
and p27kip during mitosis in BALB/c 3T3 cells. Cell Growth & Differentiation!: The
Molecular Biology Journal of the American Association for Cancer Research, 6, 1199–
1205.
Alkarain, A., Jordan, R., & Slingerland, J. (2004). p27 deregulation in breast cancer: prognostic
significance and implications for therapy. Journal of Mammary Gland Biology and
Neoplasia, 9, 67–80. doi:10.1023/B:JOMG.0000023589.00994.5e
Bochis, O. V, Irimie, A., Pichler, M., & Neagoe, I. B.-. (2015). The Role of Skp2 and its
Substrate CDKN1B ( p27 ) in Colorectal Cancer, 24(2), 225–234.
Bonelli, P., Tuccillo, F. M., Borrelli, A., Schiattarella, A., & Buonaguro, F. M. (2014).
CDK/CCN and CDKI Alterations for Cancer Prognosis and Therapeutic Predictivity.
BioMed Research International, 2014, 361020. doi:10.1155/2014/361020
Campeau, E., Ruhl, V. E., Rodier, F., Smith, C. L., Rahmberg, B. L., Fuss, J. O., … Kaufman, P.
D. (2009). A versatile viral system for expression and depletion of proteins in mammalian
cells. PLoS ONE, 4(8). doi:10.1371/journal.pone.0006529
Chiarle, R., Budel, L. M., Skolnik, J., Frizzera, G., Chilosi, M., Corato, A., … Inghirami, G.
(2000). Increased proteasome degradation of cyclin-dependent kinase inhibitor p27 is
associated with a decreased overall survival in mantle cell lymphoma. Blood, 95, 619–626.
Chu, I. M., Hengst, L., & Slingerland, J. M. (2008). The Cdk inhibitor p27 in human cancer:
prognostic potential and relevance to anticancer therapy. Nature Reviews. Cancer, 8(4),
Coqueret, O. (2003). New roles for p21 and p27 cell-cycle inhibitors: a function for each cell
compartment? Trends in Cell Biology, 13(2), 65–70. Retrieved from
http://www.ncbi.nlm.nih.gov/pubmed/12559756
Gibson, D. G., Young, L., Chuang, R., Venter, J. C., Hutchison, C. a, & Smith, H. O. (2009).
Enzymatic assembly of DNA molecules up to several hundred kilobases. Nature Methods,
6(5), 343–5. doi:10.1038/nmeth.1318
Grant, G. D., Brooks, L., Zhang, X., Mahoney, J. M., Martyanov, V., Wood, T. a., … Whitfield,
M. L. (2013). Identification of cell cycle–regulated genes periodically expressed in U2OS
cells and their regulation by FOXM1 and E2F transcription factors. Molecular Biology of
the Cell, 24, 3634–3650. doi:10.1091/mbc.E13-05-0264
Hara, T., Kamura, T., Nakayama, K., Oshikawa, K., Hatakeyama, S., & Nakayama, K. I. (2001).
Degradation of p27Kip1 at the G0-G1 Transition Mediated by a Skp2-independent
Ubiquitination Pathway. Journal of Biological Chemistry, 276(52), 48937–48943.
doi:10.1074/jbc.M107274200
Hayes, O., Ramos, B., Rodríguez, L. L., Aguilar, A., Badía, T., & Castro, F. O. (2005). Cell
confluency is as efficient as serum starvation for inducing arrest in the G0/G1 phase of the
cell cycle in granulosa and fibroblast cells of cattle. Animal Reproduction Science, 87(3-4),
181–192. doi:10.1016/j.anireprosci.2004.11.011
He, W., Wang, X., Chen, L., & Guan, X. (2012). A crosstalk imbalance between p27(Kip1) and
its interacting molecules enhances breast carcinogenesis. Cancer Biotherapy &
Radiopharmaceuticals, 27(7), 399–402. doi:10.1089/cbr.2010.0802
of the interaction between p27 and Kip1 ubiquitylation-promoting complex, the ubiquitin
ligase that regulates proteolysis of p27 in G1 phase. The Journal of Biological Chemistry,
280(18), 17694–700. doi:10.1074/jbc.M500866200
Laoukili, J., Kooistra, M. R. H., Brás, A., Kauw, J., Kerkhoven, R. M., Morrison, A., …
Medema, R. H. (2005). FoxM1 is required for execution of the mitotic programme and
chromosome stability. Nature Cell Biology, 7(2), 126–136. doi:10.1038/ncb1217
Lee, J., & Kim, S. S. (2009). The function of p27 KIP1 during tumor development. Experimental
& Molecular Medicine, 41(11), 765–71. doi:10.3858/emm.2009.41.11.102
Liu, Z., Long, Y., Zhang, Y., Huang, W., Long, X., Yang, H., … Fang, W. (2013). Nuclear p27
expression confers a favorable outcome for nasopharyngeal carcinoma patients. Disease
Markers, 35(6), 925–32. Retrieved from
http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3881392&tool=pmcentrez&ren
dertype=abstract
Malecki, M. J., Sanchez-Irizarry, C., Mitchell, J. L., Histen, G., Xu, M. L., Aster, J. C., &
Blacklow, S. C. (2006). Leukemia-associated mutations within the NOTCH1
heterodimerization domain fall into at least two distinct mechanistic classes. Molecular and
Cellular Biology, 26(12), 4642–4651. doi:10.1128/MCB.01655-05
Murray, A. W. (2004). Recycling the cell cycle: cyclins revisited. Cell, 116(2), 221–34.
Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/14744433
Nakayama, K., Nagahama, H., Minamishima, Y. a, Miyake, S., Ishida, N., Hatakeyama, S., …
Nakayama, K. I. (2004). Skp2-mediated degradation of p27 regulates progression into
http://www.ncbi.nlm.nih.gov/pubmed/15130491
Oki, T., Nishimura, K., Kitaura, J., Togami, K., Maehara, A., Izawa, K., … Kitamura, T. (2014).
A novel cell-cycle-indicator, mVenus-p27K−, identifies quiescent cells and visualizes G0–
G1 transition. Scientific Reports, 4. doi:10.1038/srep04012
Pellegata, N. S. (2012). MENX and MEN4. Clinics (São Paulo, Brazil), 67 Suppl 1(2), 13–8.
doi:10.6061/clinics/2012(Sup01)04
Rodier, G., Montagnoli, A., Di Marcotullio, L., Coulombe, P., Draetta, G. F., Pagano, M., &
Meloche, S. (2001). p27 cytoplasmic localization is regulated by phosphorylation on Ser10
and is not a prerequisite for its proteolysis. EMBO Journal, 20(23), 6672–6682.
doi:10.1093/emboj/20.23.6672
Shen, M., Schmitt, S., Buac, D., & Ping Dou, Q. (2013). Targeting the ubiquitin - proteasome
system for cancer therapy. Expert Opinion Therapeutic Targets, 17(9), 1091–1108.
doi:10.1517/14728222.2013.815728.Targeting
Slingerland, J. M., Hengst, L., Pan, C. H., Alexander, D., Stampfer, M. R., & Reed, S. I. (1994).
A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor
beta-arrested epithelial cells. Molecular and Cellular Biology, 14(6), 3683–94.
doi:10.1128/MCB.14.6.3683.Updated
Slingerland, J., & Pagano, M. (2000). Regulation of the Cdk Inhibitor p27 and Its, 17(October
1999), 10–17.
Vinals, F., & Pouyssegur, J. (1999). Confluence of vascular endothelial cells induces cell cycle
exit by inhibiting p42/p44 mitogen-activated protein kinase activity. Mol Cell. Biol., 19(4),
Appendix
Resistance
Marker Forward Primer Seq 5’ to 3’ Reverse Primer Seq 5’ to 3’
pEXPJC13 -p27-Ven
DNA Sequence
hygromycin attB-F
p27-ATGGGGGAC AAGTTTGTAC AAAAAAGCA GGCTTCATGT CAAACGTGC CGAGTGTC p27-attB-R ATGGGGGAC CACTTTGTAC AAGAAAGCT GGGTACGTT TGACGTCTTC TGAGG pEXPJC17 -p27-Ven DNA sequence
nourseothricin attB-F
p27-ATGGGGGAC AAGTTTGTAC AAAAAAGCA GGCTTCATGT CAAACGTGC CGAGTGTC p27-attB-R ATGGGGGAC CACTTTGTAC AAGAAAGCT GGGTACGTT TGACGTCTTC TGAGG pLenti- neo-Ven-p27 DNA sequence
neomycin Hin-FP
F-Ent-CTTTAAAGGA ACCAATTCAG TCGACAAGC TTTCGCCACC ATGGTGAGC AAGGGCGAG R-P2A-Mlu-p27 GTTAGTAGCT CCGCTTCCA CGCGTCGTT TGACGTCTTC TGAGG pLenti- puro-Ven-p27 DNA sequence
puromycin Hin-FP
F-Ent-CTTTAAAGGA ACCAATTCAG TCGACAAGC TTTCGCCACC ATGGTGAGC AAGGGCGAG R-P2A-Mlu-p27 GTTAGTAGCT CCGCTTCCA CGCGTCGTT TGACGTCTTC TGAGG
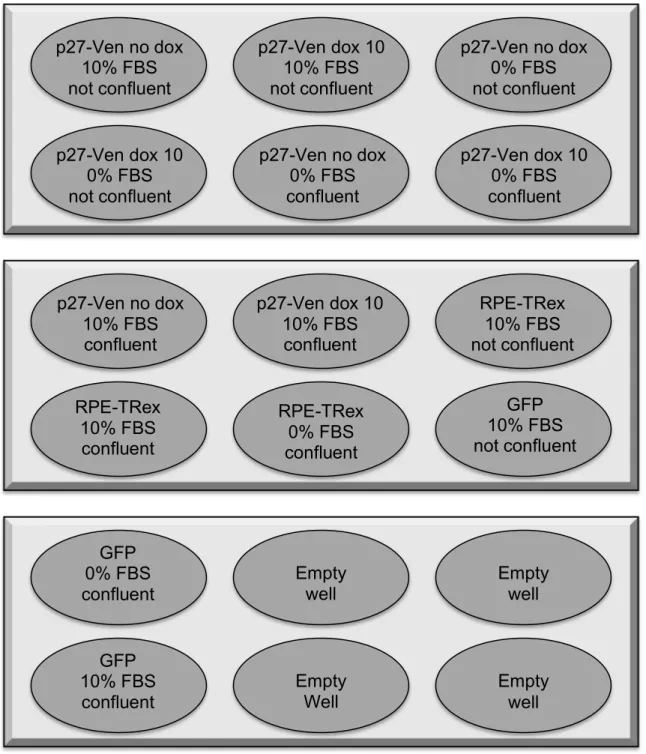
Figure A1. Diagram of 6-well dish layout for serum starvation experiment. “Not confluent” wells were roughly 60-80% confluent by the time of collection, whereas “confluent” wells were 100% confluent.
p27-Ven no dox 10% FBS not confluent
p27-Ven dox 10 10% FBS not confluent
p27-Ven no dox 0% FBS not confluent
p27-Ven dox 10 0% FBS not confluent
p27-Ven no dox 0% FBS confluent
p27-Ven dox 10 0% FBS confluent
p27-Ven no dox 10% FBS
confluent
Figure A2. Immunoblot demonstrating doxycycline-inducible control of p27-Ven. By comparing U2OS-GFP and p27-Ven cell lines, we established that a nonspecific band was present on the blot after probing with p27 primary antibody. This band was
determined to be the top band because it did not increase with increasing doxycycline in either the U2OS-GFP or p27-Ven cell line and is present across both lines. Further, p27-Ven levels increase with increasing levels of doxycycline added, indicating the fusion protein is doxycycline-inducible.
Figure A3. Destination vector pcDNA5/FRT/TO-Venus-Flag-Gateway (1124)
(pDESTJC13) used for cloning pEXPJC13-p27-Ven and pEXPJC17-p27-Ven. Image generated in Vector NTI® (Thermo Scientific, DE).
pDESTJC13 7 61 7 bp
HygR(noATG) AmpR
Ve nus
ccdB CmR TATA/Te tOp2
Flag3X CM V
FRT
attR1
Figure A4. Destination vector pLenti PGK Puro DEST (w529-2) used for cloning pLenti-puro-Ven-p27. Image generated in Vector NTI®.
Figure A5. Destination vector pLenti PGK Neo DEST (w531-1) used for cloning pLenti-neo-Ven-p27. Image generated in Vector NTI®.
pLenti PGK Puro DEST (w529-2)
9599 bp
AmpR
PuroR
CamR
truncHIV-1 3 LTR HIV-1 5 LTR HIV-1 psi pack
RRE
attR1 ccdB
attR1 WPRE truncHIV-1 3 LTR
HIV-1 5 LTR
lacZ a AmpR promoter
lac promoter
pBR322 origin SV40 origin
f1 origin
pLenti PGK Neo DEST (w531-1)
97 7 8 bp
CamR NeoR
AmpR
HIV-1 5 LTR truncHIV-1 3 LTR HIV-1 psi pack
RRE
attR1 ccdB
attR1 WPRE truncHIV-1 3 LTR
HIV-1 5 LTR
lacZ a AmpR promoter
lac promoter
pBR322 origin SV40 origin