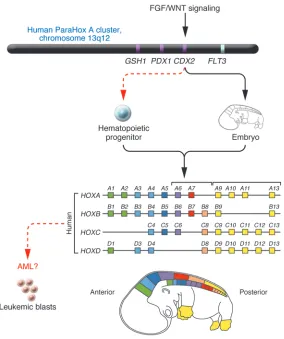
HOX deregulation in acute myeloid leukemia
Full text
Figure
Related documents
The Montana Court felt that the Washington decree" came within the Sistare exception-that it empowered the Washing- ton Court to revise past-due installments,
The legal implications of the current situation are far reaching. The specific issues that require analysis are: 1) lack of a fair election for the Kashmiri
Figure 4. First, the undifferentiated hiPSCs were detached using two methods: mechanical and enzymatic dissociation. The suspension cell culture period was called
In this study, to improve the electrical efficiency of solar panels that operate in non-optimal conditions, an active water cooling system has been built on top of
M.D., [email protected] Children’s Advocacy Center of Southwest Florida Child Protection Team. 3830 Evans Avenue
(2012), this yeast, despite being very utilized in fermentation processes of wine production, is isolated in a smaller number when a direct isolation technique is used,
Here, the Global 200 Business School Report’s CSR rating shows a dominance of schools in North America and Europe excelling, through the eyes of MBA employers, in producing
The purpose of these activities is to present the Republic of Macedonia as an attractive destination for foreign investments through the promotion of business advantages, and
