Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Development of a Rapid PCR Assay Specific for
Staphylococcus
saprophyticus
and Application to Direct Detection
from Urine Samples
FRANCIS MARTINEAU,1,2FRANC¸OIS J. PICARD,1CHRISTIAN ME´NARD,1PAUL H. ROY,1,3 MARC OUELLETTE,1,2ANDMICHEL G. BERGERON1,2*
Centre de Recherche en Infectiologie, Centre Hospitalier Universitaire de Que´bec (Pavillon Centre Hospitalier de l’Universite´ Laval),1and Division de Microbiologie, Faculte´ de Me´decine, Universite´ Laval,2
Ste-Foy, Que´bec, Canada G1V 4G2, and De´partement de Biochimie, Universite´ Laval, Ste-Foy, Que´bec, Canada G1K 7P43
Received 29 March 2000/Returned for modification 26 May 2000/Accepted 17 June 2000
Staphylococcus saprophyticusis one of the most frequently encountered microorganisms associated with acute
urinary tract infections (UTIs) in young, sexually active female outpatients. Conventional identification methods based on biochemical characteristics can efficiently identifyS. saprophyticus, but the rapidities of these methods need to be improved. Rapid and direct identification of this bacterium from urine samples would be useful to improve time required for the diagnosis ofS. saprophyticus infections in the clinical microbiology laboratory. We have developed a PCR-based assay for the specific detection ofS. saprophyticus. An arbitrarily primed PCR amplification product of 380 bp specific forS. saprophyticuswas sequenced and used to design a set ofS. saprophyticus-specific PCR amplification primers. The PCR assay was specific forS. saprophyticuswhen tested with DNA from 49 gram-positive and 31 gram-negative bacterial species. This assay was also able to amplify efficiently DNA from all 60 strains ofS. saprophyticus from various origins tested. This assay was adapted for direct detection from urine samples. The sensitivity levels achieved with urine samples was 19 CFU with 30 cycles of amplification and 0.5 CFU with 40 cycles of amplification. This PCR assay for the specific detection ofS. saprophyticusis simple and rapid (approximately 90 min, including the time for urine specimen preparation).
Coagulase-negative staphylococci are commensal organisms of human skin flora but have become major etiological agents of nosocomial bacteremia and can colonize a variety of medical devices (18). These nosocomial infections are usually (⬎80%) caused byStaphylococcus epidermidis(18, 24). However, Staph-ylococcus saprophyticusis the second most frequently encoun-tered agent of acute urinary tract infections (UTIs) after Esch-erichia coli(10, 11).S. saprophyticusis often isolated from the urine of young, sexually active female outpatients presenting with symptoms of acute UTI (1, 15, 25) indistinguishable from the symptoms of UTIs caused byEscherichia coli. These coag-ulase-negative staphylococci are rarely found as a cause of UTIs in hospitalized patients or as a contaminant of urine cultures (15) and are characterized by the low bacterial counts (less than 105 CFU per ml) required to elicit a UTI (24).S.
saprophyticuscould be the cause of chronic bacterial prostatitis in men (4), and there is evidence that suggests that this staph-ylococcal species could be the etiological agent of sexually transmitted urethritis (9). The use of spermicide-coated con-doms has now been associated with an increased risk of UTIs caused byS. saprophyticus(6).
The classical phenotypic identification of staphylococci by Kloos and Schleifer (19) remains the “gold standard” for ref-erence laboratories, but it is too lengthy and cumbersome for routine use in hospital microbiology laboratories.S. saprophy-ticus is differentiated from other urinary coagulase-negative
staphylococci (i.e.,S. epidermidis) by its uniform resistance to novobiocin, aerobic growth requirements, urease production, and carbohydrate utilization (15). Several culture-based com-mercially available systems including API Staph strips and the RapiDEC system have been evaluated for the identification of
S. saprophyticus (12, 26). However, these systems require at least 20 h for staphylococcal species identification and occa-sionally misidentifyS. saprophyticus. A few DNA-based assays that target variable regions of the 16S rRNA gene of S. sap-rophyticushave been developed (7, 8). However, these assays have not been evaluated for direct detection ofS. saprophyticus
from clinical specimens.
Although S. saprophyticus is easy to cultivate, phenotypic analysis requires overnight growth of the microorganism. A rapid and sensitive DNA-based assay which is specific for S. saprophyticus and which is suitable for direct detection of the organism from urine specimens would allow a significant re-duction in the time required for the diagnosis ofS. saprophy-ticusinfections. In this study, we present the development of an S. saprophyticus-specific DNA-based assay. An arbitrarily primed PCR (AP-PCR) protocol was used to find a prominent fingerprinting product of 380 bp shared by a panel of clinical strains ofS. saprophyticusbut not encountered in other closely related bacterial species. This DNA fragment was sequenced and used to design a pair of PCR primers suitable for the specific and ubiquitous detection ofS. saprophyticus. This S. saprophyticus-specific PCR assay was adapted for direct detec-tion of the organism from urine specimens. This assay will be combined in multiplex with other PCR assays currently under development in our laboratory to allow the concomitant de-tection of other bacteria frequently associated with UTIs. * Corresponding author. Mailing address: Centre de Recherche en
Infectiologie, CHUQ (Pavillon CHUL), 2705 Boul. Laurier, Ste-Foy, Que´bec, Canada G1V 4G2. Phone: (418) 2705. Fax: (418) 654-2715. E-mail: [email protected].
3280
on May 15, 2020 by guest
http://jcm.asm.org/
MATERIALS AND METHODS
Bacterial strains.The bacterial isolates used in this study were selected from the culture collection of the Microbiology Laboratory of the Centre Hospitalier Universitaire de Que´bec (Pavillon Centre Hospitalier de l’Universite´ Laval [CHUL], Ste-Foy, Que´bec, Canada). Three S. saprophyticusstrains obtained from the American Type Culture Collection (ATCC; strains ATCC 15305, ATCC 35552, and ATCC 43867) were also used for this study. The strains were cultured on sheep blood agar or in brain heart infusion (BHI) medium. Bacterial cultures were stored frozen (⫺80°C) in BHI broth containing 10% glycerol.
Forty-nine gram-positive, 31 gram-negative, and 60 clinical isolates were used to establish the performance of theS. saprophyticusPCR assay. This battery of bacterial strains includes isolates obtained from both ATCC and the Microbiol-ogy Laboratory of CHUL.
Clinical specimens.A total of five culture-negative urine specimens received at the Microbiology Laboratory of CHUL, all collected from different patients, were used in this study. Urine samples (stored at 4°C) were tested by PCR less than 48 h after reception at the laboratory.
DNA isolation.Genomic DNA was purified with the G NOME kit (Bio 101, Inc., Vista, Calif.) according to the manufacturer’s instructions, except that the bacterial cells were initially resuspended in 250l of a lysis solution containing 200g of lysostaphin (Sigma Chemical Co., St. Louis, Mo.) per ml, 20 mM Tris, 2 mM EDTA, and 1.2% Triton X-100 and were incubated for 30 min at 37°C. Purified genomic DNA was diluted at a concentration of 1 ng/l in TE buffer (10 mM Tris [pH 8.0], 1 mM EDTA).
Urine specimens were prepared for PCR amplification by using the IDI DNA extraction kit (Infectio Diagnostic [IDI] Inc., Sainte-Foy, Que´bec, Canada) ac-cording to the manufacturer’s instructions.
AP-PCR amplification.Twenty 10-nucleotide primers (kit AD; Operon Tech-nologies Inc., Alameda, Calif.) were used for AP-PCR to search for a specific amplicon shared exclusively by the species of interest,S. saprophyticus(5, 27, 28). Amplifications were performed directly from 1l (0.1 ng/l) of purified genomic DNA from 5S. saprophyticusstrains and 27 other staphylococcal (non-S. sapro-phyticus) strains. The 25-l AP-PCR mixture contained 50M KCl, 10 mM Tris-HCl (pH 9.0), 0.1% Triton X-100, 2.5 mM MgCl2, 1 of the 20 10-nucleotide
primers at a concentration of 1.5M, 200M (each) the four deoxynucleoside triphosphates, and 0.5 U ofTaqDNA polymerase (Promega Corp., Madison, Wis.) combined with the TaqStart antibody (Clontech Laboratories Inc., Palo Alto, Calif.). The TaqStart antibody, which is a neutralizing monoclonal antibody ofTaqDNA polymerase, was added to all PCR mixtures to enhance the effi-ciency of the amplifications (17). The PCR mixtures were subjected to thermal cycling (3 min at 96°C and then 42 cycles of 1 min at 94°C for the denaturation step, 1 min at 31°C for the annealing step, and 2 min at 72°C for the extension step) with a PTC-200 thermal cycler (MJ Research Inc., Watertown, Mass.). A final extension step of 7 min at 72°C was performed to allow completion of amplicons. Random amplified polymorphic DNA (RAPD) fragment fingerprints were obtained by electrophoresis in 1.5% agarose gels containing 0.5g of ethidium bromide per ml in Tris-borate-EDTA buffer (89 mM Tris, 89 mM boric acid, 2 mM EDTA) at 4 V/cm for 90 min. The gels were visualized under 254-nm UV light. The sizes of the amplification products were estimated by comparison with a 50-bp-molecular-size standard ladder.
Subsequently, AP-PCR products of the predicted size were recovered from the gel by using the QIAquick gel extraction kit (QIAGEN Inc., Mississauga, On-tario, Canada). The purified DNA fragments were then cloned into the pCR 2.1 T/A cloning vector (Invitrogen Corp., Carlsbad, Calif.). Plasmids were isolated from transformedE. colistrains by using the QIAGEN plasmid mini kit (QIA-GEN Inc.). The presence of a DNA insert in the recombinant plasmids was confirmed by digesting the purified plasmid DNA withEcoRI (New England Biolabs Ltd., Mississauga, Ontario, Canada), which allowed the inserted frag-ment to be cut out. Both strands of the DNA inserts for each of the selected recombinant plasmids were sequenced with the PRISM Ready Reaction DyeDeoxy Terminator cycle sequencing kit with an Applied Biosystems 373A sequencer (Perkin-Elmer Corp., Foster City, Calif.). From the 380-bp sequence, we designed a pair of PCR primers suitable for the specific and ubiquitous detection ofS. saprophyticus. The selected primer pair was verified by using the primer analysis software Oligo, version 5.0 (National Bioscience, Plymouth, Minn.).
Conventional PCR amplification.Amplifications were performed either from 1l of a purified genomic DNA preparation or from a standardized bacterial suspension whose turbidity was adjusted to equal that of a 0.5 McFarland stan-dard, which corresponds to approximately 1.5⫻108bacteria per ml. The 20-l
PCR mixture contained 0.4M (each) the twoS. saprophyticus-specific primers 5⬘-TCA AAA AGT TTT CTA AAA AAT TTA C-3⬘(annealing positions 169 to 193) and 5⬘-ACG GGC GTC CAC AAA ATC AAT AGG A-3⬘ (annealing positions 355 to 379), 200M (each) the four deoxyribonucleoside triphosphates (Pharmacia Biotech Inc., Baie d’Urfe´, Que´bec, Canada), 10M Tris-HCl (pH 9.0), 50M KCl, 0.1% Triton X-100, 2.5 mM MgCl2, 3.3g of bovine serum
albumin (Sigma-Aldrich Canada Ltd., Oakville, Ontario, Canada) per ml, 10 copies of linearized plasmid pSL1138, which served as a target for the internal control, and 0.5 U ofTaqDNA polymerase (Promega Corp.) combined with the TaqStart antibody (Clontech Laboratories Inc.) (16, 22). An internal control was integrated into every PCR mixture (16). Use of this control allowed verification
of the efficiency of the amplification and ensured that significant PCR inhibition was absent. The PCR mixtures were subjected to thermal cycling (3 min at 96°C and then 30 or 40 cycles of 1 s at 95°C for the denaturation step and 30 s at 55°C for the annealing-extension step with a PTC-200 thermal cycler). Analysis by agarose gel electrophoresis was performed as described previously (22).
The specificities of the DNA-based tests were verified by using a panel of clinical isolates consisting of 49 gram-positive and 31 gram-negative bacterial species (Table 1). The ubiquity (i.e., the ability to detect all strains ofS. sapro-phyticus) of the DNA-based tests was verified by using a panel of 60 clinical isolates identified asS. saprophyticusby using the MicroScan Autoscan-4 system equipped with the Positive BP Combo Panel Type 6 (Dade Diagnostics, Missis-sauga, Ontario, Canada).
For determination of the sensitivities of the 30- and 40-cycle PCR assays, cultures of three strains ofS. saprophyticus(strains ATCC 15305, ATCC 35552, and ATCC 43867) in the logarithmic phase of growth (optical density at 600 nm, ⬇0.7 to 0.8) were diluted in phosphate-buffered saline (PBS). Each dilution (1 l) was tested in PCR assays to determine the minimal number of CFU which could be detected. The number of CFU was estimated by standard plating procedures. A similar approach was applied to determine the minimal number of genome copies which could be detected.
To assess the sensitivity of the PCR assay for detection ofS. saprophyticus directly from urine specimens, five bacterium-free urine specimens were spiked with various amounts ofS. saprophyticuscells in the mid-logarithmic phase of growth in order to determine the minimal number of CFU which could be detected. The sensitivity was determined with 40 cycles of amplification.
Nucleotide sequence accession number.The nucleotide sequence of theS. saprophyticus-specific AP-PCR amplicon is available from GenBank as accession no. AF144088.
RESULTS
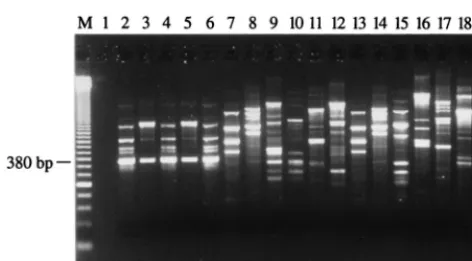
Isolation of an S. saprophyticus-specific DNA fragment by AP-PCR.The generation of RAPD fingerprints for 5S. sapro-phyticus strains (including the 3 ATCC strains) and 29 other staphylococcal species with the 20 different AP-PCR primers (10-mer) allowed determination of which primer produced am-plification patterns specific for the 5 S. saprophyticusstrains. Primer OPAD-16 (5⬘-AACGGGCGTC-3⬘) allowed isolation of a DNA fragment of 380 bp found in all RAPD patterns for the 5S. saprophyticusstrains tested but absent from the RAPD patterns for the other bacterial species tested (Fig. 1). Subse-quently, we confirmed that this 380-bp amplification product was also absent from a wider array of bacterial species consist-ing of 19 other genetically related gram-positive species (Table 1). ThisS. saprophyticus-specific amplification product was gel purified and then cloned into the T/A cloning vector pCR 2.1. Subsequently, the sequences of both strands of theS. sap-rophyticus 380-bp genomic DNA insert were determined for the five strains. We performed a multiple sequence alignment of these sequences and found homology of over 99%, indicat-ing that this genomic target is well conserved inS. saprophyti-cus and, consequently, is promising for diagnostic purposes. Searches for this sequence in various data banks did not reveal any significant homologies with known sequences. A pair of PCR primers for the specific detection ofS. saprophyticuswas derived from conserved regions of this DNA fragment with the help of the Oligo software.
PCR assays.Specificity tests performed with the panel of gram-positive and gram-negative bacterial species (Table 1) with 30 and 40 cycles of amplification showed that the selected PCR primer pair amplified only DNA from S. saprophyticus
strains. In order to ensure that the negative PCR results ob-tained with the bacterial species other than the target species were not attributable to PCR inhibitors or to the inadequacy of the PCR assay, all reactions included an internal control si-multaneously amplified. This control was always efficiently am-plified when no target DNA was present, thereby showing the absence of PCR inhibitors. It is important that the S. sapro-phyticus-specific PCR assay did not yield any specific amplifi-cation product with 27 staphylococcal species other than S. saprophyticus(Table 1). No false-positive results with the set of
on May 15, 2020 by guest
http://jcm.asm.org/
49 gram-positive bacteria comprising 30 staphylococcal species and 19 other genetically related gram-positive species was ob-served, indicating that the targeted genomic sequences are unique toS. saprophyticus. Increasing the number of amplifi-cation cycles from 30 to 40 did not appear to affect the spec-ificity of theS. saprophyticus-specific PCR assay because all staphylococcal species other than S. saprophyticusas well as closely related species (Table 1) could not be amplified by the 40-cycle PCR assay (data not shown).
The S. saprophyticus-specific PCR assay was further vali-dated by testing DNA from 60 clinical isolates ofS. saprophy-ticusfrom the region of Quebec City, Quebec, Canada. These ubiquity tests showed that DNAs from all isolates were effi-ciently amplified by this PCR assay, thereby showing a perfect correlation with standard bacterial identification methods.
DNAs from all reference strains tested were also shown to be efficiently amplified, thereby demonstrating a 100% ubiquity.
We determined the sensitivity of the 30-cycle PCR assay by using genomic DNA purified from the threeS. saprophyticus
strains from ATCC. These results indicated a detection limit of 100 copies of the S. saprophyticus genome for the three S. saprophyticusstrains. In order to enhance the sensitivity of the assay, we increased the number of cycles. For PCR assays with 40 cycles, the sensitivity was increased to about six copies of the
S. saprophyticusgenome, while the length of time for comple-tion of the assay was increased by approximately 10 min.
Sensitivity assays were also performed to determine the min-imal number ofS. saprophyticuscells which can be detected in urine specimens spiked with various amounts of cells in the mid-logarithmic phase of growth (Table 2). The detection lim-TABLE 1. Bacterial strains used to test specificity ofS. saprophyticus-specific PCR assay
Gram-positive bacteria (n⫽49) Gram-negative bacteria (n⫽31)
Staphylococcus arlettaeATCC 43957 Acinetobacter baumanniiATCC 19606
Staphylococcus aureussubsp.anaerobiusATCC 35844 Bacteroides fragilisATCC 25285
Staphylococcus aureussubsp.aureusATCC 43300 Bordetella pertussisATCC 9797
Staphylococcus auricularisATCC 33803 Bulkholderia cepaciaATCC 25416
Staphylococcus capitissubsp.capitisATCC 27840 Citrobacter diversusATCC 27028
Staphylococcus capraeATCC 35538 Citrobacter freundiiATCC 8090
Staphylococcus carnosusATCC 51365 Enterobacter aerogenesATCC 13048
Staphylococcus chromogenesATCC 43764 Enterobacter cloacaeATCC 13047
Staphylococcus capraeATCC 35538 Escherichia coliATCC 25922
Staphylococcus cohniisubsp.urealyticumATCC 20260 Gardnerella vaginalisATCC 14018
Staphylococcus delphiniATCC 49171 Haemophilus ducreyiATCC 33940
Staphylococcus epidermidisATCC 14990 Haemophilus influenzaeATCC 9007
Staphylococcus equorumATCC 43958 Hafnia alveiATCC 13337
Staphylococcus felisATCC 49168 Kingella indologenesATCC 25869
Staphylococcus gallinarumATCC 35539 Klebsiella oxytocaATCC 13182
Staphylococcus haemolyticusATCC 29970 Klebsiella pneumoniaeATCC 13883
Staphylococcus hominisATCC 27844 Moraxella catarrhalisATCC 25240
Staphylococcus hyicusATCC 11249 Morganella morganiiATCC 25830
Staphylococcus intermediusATCC 29663 Neisseria gonorrhoeaeATCC 35201
Staphylococcus kloosiATCC 43959 Neisseria meningitidisATCC 13077
Staphylococcus lentusATCC 29070 Pasteurella aerogenesATCC 27883
Staphylococcus lugdunensisATCC 43809 Proteus mirabilisATCC 25933
Staphylococcus saprophyticusATCC 15305 Proteus vulgarisATCC 13315
Staphylococcus saprophyticusATCC 35552 Providencia rettgeriATCC 9250
Staphylococcus saprophyticusATCC 43867 Pseudomonas aeruginosaATCC 27853
Staphylococcus schleiferisubsp.coagulansATCC 49545 Salmonella typhimuriumATCC 14028
Staphylococcus sciurisubsp.sciuriATCC 29060 Serratia marcescensATCC 8100
Staphylococcus simulansATCC 27848 Shigella flexneriATCC 12022
Staphylococcus warneriATCC 27836 Shigella sonneiATCC 29930
Staphylococcus xylosusATCC 29971 Stenotrophomonas maltophiliaATCC 13843
Bacillus subtilisATCC 27370 Yersinia enterocoliticaATCC 9610
Enterococcus aviumATCC 14025
Enterococcus duransATCC 19432
Enterococcus faecalisATCC 29212
Enterococcus faeciumATCC 19434
Enterococcus flavescensATCC 49996
Enterococcus gallinarumATCC 49573
Lactococcus lactisATCC 11454
Lactobacillus acidophilusATCC 4356
Listeria monocytogenesATCC 15313
Macrococcus caseolyticusATCC 13548
Mobiluncus curtissiATCC 35242
Streptococcus agalactiaeATCC 27591
Streptococcus anginosusATCC 33397
Streptococcus bovisATCC 33317
Streptococcus dysgalactiaeATCC 43078
Streptococcus pneumoniaeATCC 27336
Streptococcus pyogenesATCC 19615
Streptococcus salivariusATCC 7073
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.612.56.555.82.548.2]its in terms of the numbers of CFU determined with the IDI DNA extraction kit with five different urine specimens spiked with various amounts ofS. saprophyticuscells were in the range of 300 to 1,400 CFU/ml of urine for the 40-cycle PCR. Fur-thermore, there was no significant PCR inhibition because the internal control was always efficiently amplified. For compari-son, we have determined the sensitivity levels achieved with the same spiked urine specimens added directly to the PCR mix-ture without pretreatment. We found much lower detection limits (i.e., in the range of 700,000 to 800,000 CFU/ml of urine). Moreover, there was partial inhibition of the PCR on the basis of the amplification of the internal control. The sen-sitivity levels achieved withS. saprophyticuscells diluted in PBS were 500 CFU/ml with the IDI extraction kit, as opposed to 650,000 CFU/ml for samples added directly to the PCR mix-ture without pretreatment (Table 2). As expected, no signifi-cant PCR inhibition was observed for any experiment with PBS.
DISCUSSION
S. saprophyticusis relatively easy to culture and identify by phenotypic methods. However, there is a need for the devel-opment of rapid and sensitive DNA-based assays which are suitable for the direct detection ofS. saprophyticusfrom clin-ical specimens, especially urine specimens, to improve the ra-pidity and the accuracy of the diagnosis of S. saprophyticus
infections. At present, resistance to novobiocin is still used in most laboratories to presumptively identify S. saprophyticus, but other coagulase-negative species, includingS. epidermidis, are occasionally resistant to novobiocin. We have previously demonstrated the usefulness of nucleic acid amplification by PCR for detection and identification ofS. aureus,S. epidermi-dis, and their clinically relevant antibiotic resistance genes (21– 23). In the present study, we have developed a rapid PCR-based assay suitable for specific detection ofS. saprophyticusin urine specimens. Initially, a set of 20 10-mer AP-PCR primers was tested with five different strains ofS. saprophyticusin order to find a prominent shared amplicon. By this strategy, we were able to obtain such an amplicon of 380 bp that was consistently
found in allS. saprophyticusstrains tested but that was absent from other staphylococcal species and genetically related gram-positive bacteria. ThisS. saprophyticus-specific AP-PCR amplicon was sequenced and used to derive optimal PCR primers for the detection ofS. saprophyticus. TheS. saprophyti-cus-specific PCR assay developed in this study was specific because it did not amplify DNAs from a variety of gram-positive and gram-negative bacterial species including 26 staphylococcal species other than S. saprophyticus. Further-more, this assay was shown to be 100% ubiquitous on the basis of testing of 60S. saprophyticusclinical isolates from various patients, of which 92% were etiologic agents of UTIs. All 60 of these clinical isolates from CHUL as well as the three strains from ATCC were initially reconfirmed to beS. saprophyticus
with the MicroScan Autoscan-4 system, thereby showing a perfect correlation with the identification obtained by theS. saprophyticus-specific PCR assay. Therefore, theS. saprophyti-cusgenomic target of unknown coding potential selected for the PCR assay appears to be present in allS. saprophyticus
strains and also appears to be well conserved in this species at the nucleotide level but either absent from or distinct in other closely related bacterial species including other staphylococcal species. The PCR assay, which was performed directly from standardized bacterial suspensions or urine specimens spiked with a known number ofS. saprophyticus cells, was designed and optimized to be simple and performed in approximately an hour and a half.
Others have developedS. saprophyticus-specific PCR ampli-fication assays targeting variable regions V3 and V6 of the 16S rRNA gene (7, 8). However, these assays have not been ap-plied for direct detection ofS. saprophyticusfrom clinical spec-imens. In our study, we have used a different approach to developS. saprophyticus-specific DNA-based diagnostic tests. Our goal was to develop a simple and rapid PCR assay which is specific and ubiquitous forS. saprophyticusand which can be applied to detection directly from bacterial cultures or urine specimens in about 1 h. The 30-cycle PCR protocol showed sensitivity levels of about 100 copies of theS. saprophyticus
genome. This sensitivity level is sufficient for culture confirma-tion assays from urine specimens or blood cultures. Increased levels of sensitivity of the PCR are required for detection ofS. saprophyticusdirectly from urine specimens, in which the num-ber of target cells can be much lower. The 40-cycle PCR pro-tocol, which showed sensitivity levels of about six copies of the
[image:4.612.55.291.68.198.2]S. saprophyticus genome per PCR mixture or 300 to 1,400 CFU/ml of urine, appears to be suitable for that purpose. Such a high sensitivity level will be particularly critical in patients with UTIs with lowS. saprophyticuscell counts (i.e., less than 104CFU/ml), found in approximately one-third of women with
TABLE 2. Sensitivity levels achieved by the 40-cycle PCR assay with PBS or urine specimens spiked with various amount
ofS. saprophyticusATCC 15305 cells
Sample preparation
Detection limit CFU/PCR
mixture CFU/ml of PBSor urine
Spiked PBS
IDI DNA extraction kit 0.5 500
No treatment 650 650,000
Spiked urine samplesa
IDI DNA extraction kit 0.3–1.4 300–1,400
No treatment 700–800 700,000–800,000
aThe given ranges of detection limits are for five urine samples.
FIG. 1. AP-PCR amplification with the OPAD-16 primer performed with 100 pg of purified genomic DNA from reference and clinical strains ofS. sap-rophyticus, various staphylococcal species, and gram-positive bacteria genetically related toS. saprophyticus. The content of each lane is as follows: 2,S. sapro-phyticusATCC 15305; 3,S. saprophyticusATCC 33552; 4,S. saprophyticusATCC 43867; 5,S. saprophyticusCssa-18; 6,S. saprophyticusSsa-165; 7,S. aureusATCC 43300; 8,S. capitissubsp.capitisATCC 27840; 9,S. epidermidisATCC 14990; 10, S. haemolyticusATCC 29970; 11,S. hominisATCC 27844; 12,S. simulansATCC 27848; 13,S. warneriATCC 27836; 14,Bacillus subtilisATCC 27370; 15, Entero-coccus faecalisATCC 29212; 16,Lactobacillus acidophilusATCC 4356; 17, Lis-teria monocytogenesATCC 15313; 18,Streptococcus pneumoniaeATCC 27336; 1 and 19, controls to which no DNA was added; M, 50-bp ladder (molecular size standard).
on May 15, 2020 by guest
http://jcm.asm.org/
acute lower UTIs caused byS. saprophyticus(14, 20). Similarly, acute pyelonephritis caused by UTIs have been reported in association with low bacterial counts in voided urine (13).
The sensitivity assays performed withS. saprophyticuscells diluted in PBS or urine samples demonstrated the efficacy of the IDI DNA extraction kit for (i) control of the PCR inhibi-tors present in urine samples and (ii) lysis ofS. saprophyticus
cells. On the basis of the minimum number of CFU detected in PBS, this extraction kit allows a cell lysis which is about 1,300 times more efficient than that from the direct addition of di-luted cells to the PCR mixture without pretreatment. Further-more, the sensitivity levels achieved with spiked urine samples and cells diluted in PBS prepared with the IDI DNA extraction kit were both very similar, thereby suggesting that it eliminated PCR inhibition completely. For comparison, there was partial to complete PCR inhibition with spiked urine samples added directly to the PCR mixture without pretreatment.
Preliminary studies performed with the IDI DNA extraction kit, which requires about 10 min for urine sample preparation, indicate that it is also suitable for efficient recovery of DNA from gram-negative bacilli includingE. coli, enterococci, and streptococci, which are also frequently encountered in urine specimens. However, this application needs to be confirmed in a clinical study to validate the procedure for the diagnosis of UTIs. We have previously developed PCR assays for the spe-cific detection of other staphylococcal species as well as asso-ciated antibiotic resistance genes (21–23). TheS. saprophyticus
PCR assay reported in this study will be combined in multiplex with these PCR assays as well as with others which are under development, especially for the identification and detection from urine specimens ofE. coliand other bacteria frequently associated with UTIs. A direct impact of such diagnostic tests is that they should allow the faster establishment of effective antibiotic therapy and a reduction of empirical treatments with broad-spectrum antibiotics which are associated with high costs and toxicity (2, 3). The consequent reduction of antibiotic use should reduce the emergence of resistance.
ACKNOWLEDGMENTS
We thank Louise Coˆte´, who is the director of the Microbiology Laboratory of CHUL, for free access to the laboratory and for pro-viding theS. saprophyticusclinical isolates. We thank Maurice Boissi-not for critical comments regarding the manuscript.
Francis Martineau has a scholarship from the Fonds de la Recher-che en Sante´ du Que´bec. Marc Ouellette is a Medical Research Coun-cil Scientist. This research project was supported by grant PA-15586 from the Medical Research Council of Canada and by IDI.
REFERENCES
1.Abrahamsson, K., S. Hansson, U. Jodal, and K. Lincoln.1993. Staphylococ-cus saprophytiStaphylococ-cusurinary tract infections in children. Eur. J. Pediatr.152:69– 71.
2.Bergeron, M., and M. Ouellette. 1998. Preventing antibiotic resistance through rapid genotypic identification of bacteria and of their antibiotic resistance genes in the clinical microbiology laboratory. J. Clin. Microbiol.
36:2169–2172.
3.Bergeron, M. G., and M. Ouellette.1995. Diagnosing bacterial infectious diseases in one hour: an essential upcoming revolution. Infection23:69–72. 4.Bergman, B., H. Wedren, and S. E. Holm.1989.Staphylococcus saprophyticus
in males with symptoms of chronic prostatitis. Urology5:241–245. 5.Fani, R., G. Damiani, Di Serio, E. Gallori, A. Grifoni, and M. Bazzicalupo.
1993. Use of random amplified polymorphic DNA (RAPD) for generating specific DNA probes for microorganisms. Mol. Ecol.2:243–250.
6.Fihn, S. D., E. J. Boyko, C. L. Chen, E. H. Normand, P. Yarbro, and D. Scholes.1998. Use of spermicide-coated condoms and other risk factors for urinary tract infection caused byStaphylococcus saprophyticus. Arch. Intern. Med.158:281–287.
7.Gaszewska-Mastalarz, A., J. Zakrzewska-Czerwinska, and M. Mordarski.
1997. Rapid detection ofStaphylococcus saprophyticususing primer specific PCR. Acta Biol. Hung.48:319–322.
8.Gaszewska-Mastalarz, A., J. Zakrzewska-Czerwinska, and M. Mordarski.
1995. Rapid identification ofStaphylococcus saprophyticususing an oligonu-cleotide probe complementary to 16S rRNA. System. Appl. Microbiol.18:
123–126.
9.Goldenring, J. M.1986. Urinary tract infection withStaphylococcus sapro-phyticus. J. Adolesc. Health Care6:417–418.
10. Gupta, K., D. Scholes, and W. E. Stamm.1999. Increasing prevalence of antimicrobial resistance among uropathogens causing acute uncomplicated cystitis in women. JAMA281:736–738.
11. Henry, D., W. Ellison, J. Sullivan, D. L. Mansfield, D. J. Magner, M. B. Dorr, and G. H. Talbot for The Sparfloxacin Multi Center UUTI Study Group.
1998. Treatment of community-acquired acute uncomplicated urinary tract infection with sparfloxacin versus ofloxacin. Antimicrob. Agents Chemother.
42:2262–2266.
12. Janda, W. M., K. Ristow, and D. Novak.1994. Evaluation of RapiDEC Staph for identification ofStaphylococcus aureus,Staphylococcus epidermidis, and Staphylococcus saprophyticus. J. Clin. Microbiol.32:2056–2059.
13. Johnson, J. R., and W. E. Stamm.1987. Diagnosis and treatment of acute urinary tract infections. Infect. Dis. Clin. N. Am.1:773–791.
14. Johnson, J. R., and W. E. Stamm.1989. Urinary tract infections in women: diagnosis and treatment. Ann. Intern. Med.111:906–917.
15. Jordan, P. A., A. Iravani, G. A. Richard, and H. Baer.1980. Urinary tract infection caused byStaphylococcus saprophyticus. J. Infect. Dis.142:510–515. 16. Ke, D., F. J. Picard, F. Martineau, C. Me´nard, P. H. Roy, M. Ouellette, and M. G. Bergeron.1999. Development of a PCR assay for rapid detection of enterococci. J. Clin. Microbiol.37:3497–3503.
17. Kellogg, D. E., I. Rybalkin, N. Chen, N. Mukhamedova, T. Vlasik, P. D. Siebert, and A. Chenchick.1994.TaqStart Antibody: “hot start” PCR facil-itated by a neutralizing monoclonal antibody directed againstTaqDNA polymerase. BioTechniques16:1134–1137.
18. Kloos, W. E., and T. L. Bannerman.1994. Update on clinical significance of coagulase-negative staphylococci. Clin. Microbiol. Rev.7:117–140. 19. Kloos, W. E., and K. H. Schleifer. 1975. Simplified scheme for routine
identification of humanStaphylococcusspecies. J. Clin. Microbiol.1:82–88. 20. Kunin, C. M., L. Van Arsdale White, and T. Hua Hua.1993. A reassessment of the importance of “low-count” bacteriuria in young women with acute urinary symptoms. Ann. Intern. Med.119:454–460.
21. Martineau, F., F. J. Picard, N. Lansac, C. Me´nard, P. H. Roy, M. Ouellette, and M. G. Bergeron.2000. Correlation between the resistance genotype determined by multiplex PCR assays and the antibiotic susceptibility patterns inStaphylococcus aureusandStaphylococcus epidermidis. Antimicrob. Agents Chemother.44:231–238.
22. Martineau, F., F. J. Picard, P. H. Roy, M. Ouellette, and M. G. Bergeron.
1998. Species-specific and ubiquitous DNA-based assays for rapid identifi-cation ofStaphylococcus aureus. J. Clin. Microbiol.36:618–623.
23. Martineau, F., F. J. Picard, P. H. Roy, M. Ouellette, and M. G. Bergeron.
1996. Species-specific and ubiquitous DNA-based assays for rapid identifi-cation ofStaphylococcus epidermidis. J. Clin. Microbiol.34:2888–2893. 24. Rupp, M. E., and G. L. Archer.1994. Coagulase-negative staphylococci:
pathogens associated with medical progress. Clin. Infect. Dis.19:231–245. 25. Svanborg, C., and G. Godaly. 1997. Bacterial virulence in urinary tract
infections. Infect. Dis. Clin. N. Am.11:513–529.
26. von Baum, H., F. R. Klemme, H. K. Geiss, and H. G. Sonntag.1998. Comparative evaluation of a commercial system for identification of gram-positive cocci. Eur. J. Clin. Microbiol. Infect. Dis.17:849–852.
27. Welsh, J., and M. McClelland.1990. Fingerprinting genomes using PCR with arbitrary primers. Nucleic Acids Res.18:7213–7218.
28. Williams, J. G. K., A. R. Kubelik, K. J. Livak, J. A. Rafalski, and S. V. Tingey.1990. DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Res.18:6531–6535.