Reverse Dot Blot Assay (Insertion Site Typing) for Precise
Detection of Sites of IS
6110
Insertion in the
Mycobacterium tuberculosis
Genome
LAUREN M. STEINLEINANDJACK T. CRAWFORD*
Division of AIDS, STD, and TB Laboratory Research, National Center for Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia 30333
Received 14 September 2000/Returned for modification 29 November 2000/Accepted 31 December 2000
We have developed an amplification-based reverse dot blot assay for the detection of specific sites of insertion of theMycobacterium tuberculosis insertion sequence IS6110. In this assay, a set of biotin-labeled amplicons representing the various copies of IS6110 and their flanking sequences is generated by linker-mediated PCR. The amplicons are then hybridized to immobilized oligonucleotide probes that are specific for known IS6110insertion sites. The method was evaluated using an array of oligonucleotide probes correspond-ing to IS6110 insertion sites from M. tuberculosisstrains CDC1551, Erdman, and H37Rv, and multidrug-resistant strain W. A set of 72 DNA samples from 60M. tuberculosis clinical isolates was analyzed for the presence or absence of these insertion sites, and the assay was found to be highly reproducible. This method of identifying insertion sites has been named “insite” and can be used for the genotyping ofM. tuberculosis
complex strains based on IS6110insertion site profiles.
It is well established that genotyping ofMycobacterium tu-berculosisisolates provides valuable support for the investiga-tion of suspected outbreaks, the detecinvestiga-tion of unsuspected transmission, the tracing of tuberculosis within a community, and the identification of possible sources of infection for newly diagnosed cases (1, 3, 9, 18). At the national or international level, fingerprinting allows strains from different geographic areas to be compared and the movement of individual strains to be tracked. IS6110 restriction fragment length polymor-phism (RFLP) fingerprinting is the most widely used method for the genotyping ofM. tuberculosisstrains (19) because of its reproducibility and discriminatory power (9). Unfortunately, this method requires high-quality genomic DNA, which is not only difficult to prepare but also requires culturing of the organism, resulting in a long turnaround time. In addition, fingerprint interpretation and matching can be complicated and require sophisticated computer software for large-scale analysis.
In contrast, nucleic acid amplification-based assays do not require culturing of the organisms, allowing the analysis of samples in real time. In many PCR-based typing assays, the target DNA of interest is amplified and labeled by PCR and the labeled products are hybridized to an array of immobilized diagnostic probes. This method has been successfully used for the detection of mutations in drug resistance genes of M. tuberculosis(21) and forMycobacteriumspecies identification (17). Spoligotyping (6, 8, 12, 20), a reverse dot blot assay that detects the presence of a series of unique spacers in the direct repeat (DR) locus, meets the need for a simple and rapid method by which to distinguishM. tuberculosiscomplex strains.
However, spoligotyping has significantly less discriminatory power than IS6110RFLP fingerprinting for strains that contain more than five copies of IS6110(9).
We report here a new method similar to spoligotyping that targets specific sites of insertion of IS6110. We refer to this insertion site mapping method as “insite” because it can be used to gain insight into the IS6110insertion sites of anM. tuberculosis complex strain. Here we demonstrate that insite can be used to detect known IS6110insertion sites to provide an IS6110-based fingerprint forM. tuberculosiscomplex strains.
MATERIALS AND METHODS
Strains and clinical samples.M. tuberculosisstrain H37Rv DNA and strain
Erdman DNA were provided by J. Belisle (Colorado State University). M. tuberculosisstrain CDC1551 and multidrug-resistant (MDR) strain W were from the Centers for Disease Control and Prevention mycobacteriology laboratory collection. A randomized, blinded set of 72 DNA samples from 60M. tuberculosis
clinical isolates prepared for an unrelated study of secondary typing was used to evaluate reproducibility. Genomic DNA was prepared using standard methods (19).
LMPCR amplification.Labeled target DNAs were prepared by a modification
of previously published mixed-linker PCR (7) and linker-mediated PCR (LMPCR) methods using asymmetric linkers (11, 15). The basis for specificity of the amplification is described in the original paper (7). A partially double-stranded linker was prepared by combining oligonucleotides linker 1 and linker 2 (Table 1) at a final concentration of 20M each in 1⫻PCR Buffer (Perkin-Elmer Applied Biosystems, Foster City, Calif.). The mixture was incubated at 80°C for 5 min and then slowly cooled to 25°C. The annealed linker was stored at⫺20°C. Genomic DNA (0.1 to 10 ng) was digested with 20 U ofHhaI in 20l of 1⫻Reaction Buffer 4 (NEB, Beverly, Mass.) for 3 h at 37°C. The linker (20 pmol) was ligated to 10l of digested DNA with 1 U of T4 DNA ligase (Gibco-BRL) in 20l of 1⫻Ligation Buffer overnight at 16°C. The ligation products were digested with 20 U ofHhaI for 30 min at 37°C to eliminate any religatedHhaI sites. Templates for the primary PCR amplification were pre-pared by diluting the sample 1:100 in 10 mM Tris-HCl, pH 8.3. The primary PCR amplification (50-l total volume) consisted of 1⫻PCR Buffer, 1 U of AmpliTaq DNA polymerase (Perkin-Elmer Applied Biosystems), deoxynucleoside triphos-phates (0.2 mM each), 5% dimethyl sulfoxide, primer IS-Primary (1M), linker primer (1M), and 5l of the diluted template. Amplification was performed in * Corresponding author. Mailing address: Mailstop F08, NCID/
DASTLR, CDC, 1600 Clifton Rd., Atlanta, GA 30333. Phone: (404) 639-1281. Fax: (404) 639-1287. E-mail: [email protected].
871
on May 15, 2020 by guest
http://jcm.asm.org/
a GeneAmp PCR System 9700 (Perkin-Elmer Applied Biosystems) with an amplification profile that consisted of an initial denaturation step of 94°C for 1 min; 40 cycles of denaturation at 94°C for 30 s, annealing at 65°C for 30 s, and extension at 72°C for 1 min; and a final extension at 72°C for 1 min. Templates for the nested PCR (labeling reaction) were prepared by diluting the primary PCR products 1:100 in 10 mM Tris-HCl, pH 8.3. The nested PCR conditions were identical to the primary PCR conditions, except that the primer IS-Primary was replaced with the biotinylated primer IS-Nested. All primer sequences, including those used for the specific amplification of the IS6110flanking se-quences in strain 1551, are listed in Table 1.
Membrane preparation.Amino-linked oligonucleotides (Table 2) were
syn-thesized with a C6spacer by the Biotechnology Core Facility, Centers for Disease
Control and Prevention, and were diluted to the indicated concentration with 0.5 M NaHCO3. Membranes were prepared following previously published
proce-dures (12). A Biodyne C membrane (Pall Biosupport, Ann Arbor, Mich.) was activated by incubation in 16% (wt/vol) 1-ethyl-2-(3-dimethylaminopropyl)car-bodiimide (Sigma Chemical, St. Louis, Mo.) for 10 min at 25°C. Following a brief wash with deionized water, 150l of each diluted oligonucleotide was applied in a line by using a miniblotter system (MN45; Immunetics, Cambridge, Mass.). After a 5-min incubation at room temperature, the oligonucleotide solutions were removed from the membrane by aspiration. The membrane was inactivated by incubation in 100 mM NaOH for 9 min at room temperature, followed by a brief wash with 2⫻SSPE (0.36 M NaCl, 20 mM NaH2PO4, 2 mM EDTA, pH 7.7;
Gibco-BRL, Grand Island, N.Y.) and a 5-min incubation in 2⫻SSPE–0.1% sodium dodecyl sulfate (SDS) at 58°C. The membrane was incubated in 20 mM EDTA for 20 min at room temperature and stored at 4°C.
Insite assay.A 20-l volume of LMPCR product was diluted with 150l of 2⫻
SSPE–0.1% SDS and boiled for 10 min. The samples were then cooled on ice for 2 min, briefly centrifuged, and stored on ice until use. The insite membrane was incubated in 2⫻SSPE–0.1% SDS for 5 min at room temperature and then inserted into the miniblotter apparatus such that the lines of previously applied oligonucleotides were perpendicular to the sample lanes. Residual liquid was removed by vacuum aspiration. For each sample, 150l of diluted PCR-ampli-fied biotinylated product was added to one slot of the miniblotter. The entire apparatus was then incubated at 58°C for 1 h. Following hybridization, the samples were removed by vacuum aspiration for at least 1 min. The membrane was removed from the miniblotter and washed twice with 25 ml of 2⫻SSPE– 0.5% SDS for 10 min at 58°C. The membrane was transferred to a roller bottle and incubated with 3.75 U of streptavidin-POD (Boehringer Mannheim, India-napolis, Ind.) diluted in 30 ml of 2⫻SSPE–0.5% SDS for 1 h at 39°C. The membrane was washed twice with 25 ml of 2⫻SSPE–0.5% SDS for 10 min at 39°C and then twice with 100 ml of 2⫻SSPE for 5 min at room temperature. ECL chemiluminescence detection reagents (26 ml/membrane; Amersham Phar-macia Biotech, Piscataway, N.J.) were added, and the membrane was exposed to X-Omat AR film (Kodak) for 30 min to 16 h. For repeated use (up to 12 times), the membrane was stripped by a 1-h incubation in 1% SDS at 76°C. The mem-brane was incubated in 20 mM EDTA for 20 min at room temperature and stored at 4°C.
Identification of strain Erdman insertion sites.LMPCR products prepared
from strain Erdman genomic DNA were separated in a 2% agarose gel. Indi-vidual amplicons were isolated from the gel using the QIAquick Gel Extraction Kit (Qiagen, Valencia, Calif.) and inserted into the cloning vector pT7Blue using the Perfectly Blunt Cloning Kit (Novagen, Madison, Wis.). The inserts were sequenced using the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (Perkin-Elmer Applied Biosystems) and ABI 373 sequencer with XL upgrade.
RESULTS
Reverse dot blot hybridization assays involve the amplifica-tion and labeling of the DNA sequence of interest (the target DNA), followed by hybridization of the labeled amplicon to oligonucleotides (the probes) immobilized on a membrane. For example, in spoligotyping (6, 8, 12, 20), the individual spacers in the DR locus serve as target DNA and are amplified using biotin-labeled primers specific for the 36-bp DR element. The labeled target DNAs are hybridized to 43 immobilized oligonucleotide probes, each representing a unique spacer.M. tuberculosiscomplex strains can be typed based on the pres-ence or abspres-ence of spacers. We desired to develop a reverse dot blot method by which to detect specific IS6110 insertion sites. The method is based on the premise that, under the appropriate hybridization conditions, a probe representing the sequence at the junction of the IS6110 insertion site would hybridize specifically with the corresponding target DNA but would not hybridize with either IS6110or the flanking DNA alone. A set of labeled target DNAs representing the various copies of IS6110and their flanking sequences is prepared by LMPCR amplification (Fig. 1) using a biotinylated primer. The oligonucleotide probes correspond to known IS6110insertion sites and are complementary to the biotin-labeled strand gen-erated during LMPCR amplification of the target DNA. The target DNA is hybridized to the immobilized probes, and the presence or absence of a copy of IS6110in a particular inser-tion site in the genome can be detected.
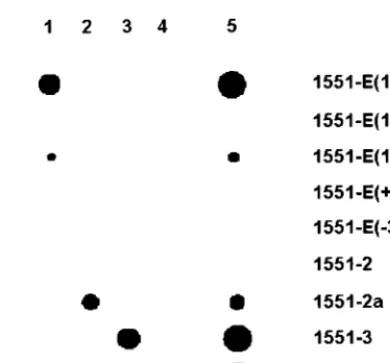
The potential use of this assay was investigated using the four IS6110insertion sites inM. tuberculosisstrain CDC1551. First, to investigate the optimal composition of the oligonucle-otide probe, several different probes for the detection of one of the insertions sites, 1551-E, were designed (Fig. 1). Probes 1551-E(10/10), 1551-E(12/8), and 1551-E(15/7) are 20- to 22-base oligonucleotide probes that contain slightly different flanking sequence and IS6110sequence lengths. Oligonucleo-tide probes 1551-1(⫹3) and 1551-1(⫺3) contain a three-base insertion and a three-base deletion, respectively, located be-tween the end of IS6110and the flanking sequence, simulating insertion of IS6110at three bases on either side of the actual 1551-E insertion site. Each probe (25 pmol) was chemically cross-linked to an activated Biodyne-C membrane. To gener-ate labeled target DNA for this copy of IS6110, a primer corresponding to a sequence 20 bases downstream of insertion site 1551-E and the biotinylated IS-Nested primer were used, producing an amplicon analogous to the fragment generated during LMPCR amplification. The target DNA bound to the five immobilized probes with varying efficiency (Fig. 2). Probe 1551-E(10/10) produced the strongest signal intensity, and probe 1551-E(12/8) produced the weakest. The target DNA did not hybridize to the probe containing either the three-base insertion or the three-base deletion. This suggested that 20- to 22-base oligonucleotides are of sufficient length for hybridiza-tion under the condihybridiza-tions used and that the probe must con-form closely to the IS6110insertion site in order to bind the target DNA.
[image:2.612.53.296.80.182.2]To test the specificity of the assay, probes for the three remaining insertion sites in strain CDC1551 (1551-2, 1551-3, and 1551-4, Table 2) were designed with 10 bases of the IS6110 sequence and 10 bases of the flanking sequence. Each of these TABLE 1. Sequences of oligonucleotides used in this study
Oligonucleotide Sequence
Linker 1...5⬘CCTGCAGGAGCTCGGATCCGACG Linker 2...5⬘TCGGATCC
IS-Primary ...5⬘CCAGTACTGCGGCGACGTCCCG IS-Nested...5⬘GCCGGTCGAACTCGAGGCTGCC Linker primer ...5⬘CCTGCAGGAGCTCGGATCCG 1551-E (PCR)...5⬘GCGCTCCTCGCGGATCACCTTGAAC 1551-2 (PCR)...5⬘GCGCCAATGAAGCCAGCAACGCCGT 1551-3 (PCR)...5⬘GCGCGTGTCCCGATGTTGAGGTGGT 1551-4 (PCR)...5⬘GCGCCGGATGATGGTGGTGCTGAAG
on May 15, 2020 by guest
http://jcm.asm.org/
probes (125 pmol), along with each of the five probes for 1551-E (25 pmol), was cross-linked to a membrane. Each of the four insertion sites was amplified individually using specific primers and the IS-Nested primer. Each target hybridized spe-cifically to the corresponding probe, although differences in signal intensity were again observed (Fig. 2). In particular, the signal for probe 1551-2 was barely detectable. Because the same amount of each target was used, this suggested that the probes were binding with different efficiencies. Analysis of the probe nucleotide sequences revealed that both probes 1551-2 and 1551-E(12/8) were capable of forming strong secondary structures whereas probes 1551-E(10/10) and 1551-E(15/7) were not. To test the hypothesis that secondary structures reduce the efficiency of target binding, a second probe,
1551-2a, was designed so that the hairpin structure was destroyed. As can be seen in Fig. 2, probe 1551-2a bound the target DNA significantly better than did probe 1551-2.
[image:3.612.52.549.82.511.2]To determine if the assay could detect all four insertion sites that had been amplified simultaneously, the membrane con-taining the nine probes described above was incubated with LMPCR products from CDC1551 (Fig. 2). Each insertion site could be detected successfully but not equally. During LMPCR amplification, individual fragments may not be amplified with equal efficiency, and in the final set of amplicons, smaller fragments are often represented far more than are larger frag-ments. The unequal proportions of each target in the LMPCR amplification products can be seen after agarose gel electro-phoresis analysis (data not shown) and in the variation of TABLE 2. Insertion site oligonucleotide probes used in this study
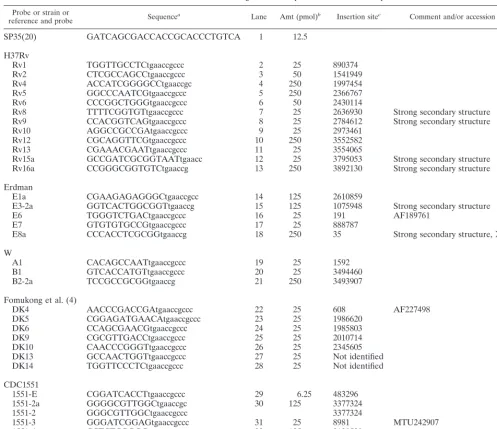
Probe or strain or
reference and probe Sequencea Lane Amt (pmol)b Insertion sitec Comment and/or accession no.
SP35(20) GATCAGCGACCACCGCACCCTGTCA 1 12.5
H37Rv
Rv1 TGGTTGCCTCtgaaccgccc 2 25 890374
Rv2 CTCGCCAGCCtgaaccgccc 3 50 1541949
Rv4 ACCATCGGGGCCtgaaccgc 4 250 1997454
Rv5 GGCCCAATCGtgaaccgccc 5 250 2366767
Rv6 CCCGGCTGGGtgaaccgccc 6 50 2430114
Rv8 TTTTCGGTGTtgaaccgccc 7 25 2636930 Strong secondary structure
Rv9 CCACGGTCAGtgaaccgccc 8 25 2784612 Strong secondary structure
Rv10 AGGCCGCCGAtgaaccgccc 9 25 2973461
Rv12 CGCAGGTTCGtgaaccgccc 10 250 3552582
Rv13 CGAAACGAATtgaaccgccc 11 25 3554065
Rv15a GCCGATCGCGGTAATtgaacc 12 25 3795053 Strong secondary structure
Rv16a CCGGGCGGTGTCtgaaccg 13 250 3892130 Strong secondary structure
Erdman
E1a CGAAGAGAGGGCtgaaccgcc 14 125 2610859
E3-2a GGTCACTGGCGGTtgaaccg 15 125 1075948 Strong secondary structure
E6 TGGGTCTGACtgaaccgccc 16 25 191 AF189761
E7 GTGTGTGCCGtgaaccgccc 17 25 888787
E8a CCCACCTCGCGGtgaaccg 18 250 35 Strong secondary structure, X95799
W
A1 CACAGCCAATtgaaccgccc 19 25 1592
B1 GTCACCATGTtgaaccgccc 20 25 3494460
B2-2a TCCGCCGCGGtgaaccg 21 250 3493907
Fomukong et al. (4)
DK4 AACCCGACCGAtgaaccgccc 22 25 608 AF227498
DK5 CGGAGATGAACAtgaaccgccc 23 25 1986620
DK6 CCAGCGAACGtgaaccgccc 24 25 1985803
DK9 CGCGTTGACCtgaaccgccc 25 25 2010714
DK10 CAACCCGGGTtgaaccgccc 26 25 2345605
DK13 GCCAACTGGTtgaaccgccc 27 25 Not identified
DK14 TGGTTCCCTCtgaaccgccc 28 25 Not identified
CDC1551
1551-E CGGATCACCTtgaaccgccc 29 6.25 483296
1551-2a GGGGCGTTGGCtgaaccgc 30 125 3377324
1551-2 GGGCGTTGGCtgaaccgccc 3377324
1551-3 GGGATCGGAGtgaaccgccc 31 25 8981 MTU242907
1551-4 CCTCTCGGGGtgaaccgccc 32 125 3120520
SP35 GATCAGCGACCACCGCACCCTGTCA 33 12.5
aIS6110sequence is in lowercase, and flanking sequence is in capital letters. bPicomoles of oligonucleotide probe loaded onto membrane.
cAll insertion sites refer to positions inM. tuberculosisstrain H37Rv (GenBank accession no. NC_000962) unless otherwise indicated by an alternative accession number.
on May 15, 2020 by guest
http://jcm.asm.org/
signal intensity after hybridization with the insertion site probes (Fig. 2).
Next, the insite assay was expanded to include selected in-sertion sites for strains H37Rv and Erdman and MDR strain W (Table 2). Oligonucleotide probes designed to detect insertion sites for 13 of the 16 copies of IS6110in the H37Rv genome (accession number AL123456) were added to the membrane. Rv1 and Rv14 are identical, and one probe (Rv1) detects both. A probe meant to detect one of the remaining insertion sites was already included; Rv11 is identical to 1551-4. Probes for the final two copies of IS6110were not included for technical reasons. Rv-IS61103 was not present in our stock of H37Rv, as determined by PCR amplification of this region (data not shown). Rv-IS61107 is inserted two bases upstream of anHhaI site, resulting in an only five-base flanking sequence in the linker-mediated amplicon of this copy, which is insufficient for specific hybridization. Also included on the insite membrane were oligonucleotides designed to detect three of the IS6110 insertion sites present in MDR strain W (11, 14), seven of the eight IS6110insertion sites in strain Erdman as sequenced in this study (E1, E3, E6 to -8, DK5, and 1551-1), and seven previously published IS6110insertion sites (DK4 to -6, 9, 10, 13, and 14; reference 5). The quantity of oligonucleotide probe immobilized on the insite membrane was determined empiri-cally to obtain signals of similar intensity for each insertion site. Amounts loaded ranged from 6.25 to 250 pmol (Table 2).
The results of the hybridization of LMPCR products of strains 1551, Erdman, H37Rv, and W can be seen in Fig. 3. With one exception, each labeled product tested hybridized specifically with oligonucleotide probes containing the respec-tive insertion site and gave the expected insertion site, or insite, profile. The LMPCR product from strain H37Rv consistently hybridized with probe DK4 (Fig. 3, lane 3), which differs from Rv10 in three positions. The 3⬘14 bases in Rv10 and DK4 are
perfectly matched and the calculated melting temperature (Tm) of this 14-mer oligonucleotide is 62.5°C, above the
hy-bridization temperature used. One insertion site, Rv12, was not detected by the probes used in this assay, and two addi-tional insertion sites, MDR strain W B2 and Rv16, were de-tected only weakly. The presence of Rv12 and Rv16 in the H37Rv strain used was confirmed by PCR using site-specific primers, and the insertion sites in these PCR fragments could easily be detected after hybridization with the probes (data not shown), suggesting that the inability to detect the insertion site after hybridization with the LMPCR products was due to in-sufficient labeled target in the set of amplicons.
An oligonucleotide probe meant to detect spacer 35 in the DR locus (20) was included on the insite membrane. Note that the spacer numbers used here refer to the recent study on the evolution of the DR locus by van Embden et al. (20) and not to the spacer numbers originally used in the description of the spoligotyping method (8); new spacer 35 is previous spacer 24. MostM. tuberculosis strains, including strains CDC1551 and H37Rv, have an IS6110insertion in the DR preceding spacer 35, resulting in the amplification of this spacer during LMPCR amplification for insite analysis. As can be seen in Fig. 3, insite samples prepared for these strains hybridized to spacer 35. Strains Erdman and W also have copies of IS6110in the DR locus. Strain Erdman has an asymmetric insertion in the DR preceding the second copy of spacer 35, followed by spacers 42 and 51, resulting in the hybridization of insite samples to both insite probe E6 (DR locus insertion) and spacer 35. Spacer 35 is not present in the DR locus of strain W (20).
[image:4.612.333.528.66.248.2]To test the reproducibility of the assay, LMPCR products from a randomized set of 72 DNA samples (Table 3) from 60 M. tuberculosisisolates were hybridized to the membrane con-taining the 32 insertion site probes (Fig. 4). The set included duplicate samples from 12 isolates with IS6110copy numbers
FIG. 1. General strategy of the insite assay. IS6110-specific LMPCR amplification is used to generate a set of labeled amplicons representing the copies of IS6110and their flanking sequences. The biotinylated IS-Nested primer is indicated. Oligonucleotide probes are designed to consist of both the right end of IS6110and the flanking sequence and are complementary to the biotinylated strand generated during LMPCR amplification. Probes 1 to 5 were designed to detect the first insertion site inM. tuberculosis strain 1551-E. The IS6110
sequence is indicated by lowercase letters, the flanking sequence is indicated by capital letters, and the nucleotides involved in predicted secondary structures are underlined.
FIG. 2. Specificity of strain CDC1551 insertion site probes. Oligo-nucleotide probes designed to detect the four insertion sites inM. tuberculosisstrain CDC1551 were hybridized with biotinylated PCR products. Lanes: 1, PCR-amplified IS6110flank E; 2, PCR-amplified IS6110flank 2; 3, PCR-amplified IS6110 flank 3; 4, PCR-amplified IS6110 flank 4; 5, LMPCR products for M. tuberculosis strain CDC1551.
on May 15, 2020 by guest
http://jcm.asm.org/
ranging from 0 to 20 and 48 isolates from tuberculosis inves-tigations in Arkansas and Tennessee. Although the insertion site profiles of the 12 duplicate samples were not particularly informative, they were reproducible.
Thirty isolates from Arkansas have been grouped into six clusters. The 11 isolates in clusters A to C were from patients with epidemiologic links and match by IS6110 RFLP finger-prints and spoligotype patterns (data not shown). All of the samples within each cluster had identical insite patterns. Clus-ter A (IS6110copy no. 5) was characterized as having the four insertion sites in strain CDC1551. For cluster B (IS6110copy no., 10), only insertion site 1551-4 was detected, and for cluster C (IS6110copy no. 12), insertion sites Rv6 and 1551-4 were detected. Spacer 35 was detected for cluster B but not for cluster C, which agrees with its respective spoligotype pattern (data not shown).
The 19 isolates in clusters D to F are from patients with no
[image:5.612.51.550.472.703.2]epidemiologic links, but the isolates match by IS6110 RFLP. Cluster D contains two isolates with a 13-band IS6110RFLP fingerprint and identical spoligotype patterns; the insite pro-files (Rv1, Rv2, Rv5, Rv8, Rv9, Rv10, Rv13, 1551-3, 1551-4) of the two isolates were similar, with the addition of the E7 insertion being identified for one of the samples. With a single exception, cluster E was characterized as containing only the 1551-E and 1551-4 insertion sites. The insite profile of sample 4 (E1, E3, E6, DK5, 1551-E) was similar to that of strain Erdman; the spoligotype pattern of this strain also differed from others in the cluster (data not shown), suggesting labo-ratory error in the handling of this culture. Cluster F also contained a single sample whose insite profile differed from the other five in the cluster. Although the cluster was characterized as containing the four insertion sites in strain 1551, strain 28 contained only the A1 insertion site, which is typically found in Beijing family strains (10). This result is in agreement with its FIG. 3. Evaluation of insertion site probes using strains with defined insertion sites. LMPCR products were hybridized to the immobilized oligonucleotide probes (Oligos) listed in Table 2. Lanes: 1, MDRM. tuberculosisstrain W; 2, strain H37Rv; 3, strain Erdman; 4, strain CDC1551.
TABLE 3. Description of randomized set of DNA samples
Samples No. of IS6110copies Sample no.
Duplicate pairs no.
1 0 11, 55
2 2 16, 34
3 4 9, 66
4 6 32, 37
5 8 14, 39
6 10 57, 63
7 11 35, 38
8 12 10, 12
9 14 33, 64
10 16 15, 58
11 18 36, 56
12 20 13, 72
Tuberculosis outbreak investigation clusters
A (n⫽2) 5 1, 59
B (n⫽6) 10 2, 24, 47, 60, 62, 70
C (n⫽3) 12 23, 25, 48
D (n⫽2) 13 5, 51
E (n⫽11) 3 3, 4,a22, 26, 27, 44, 45, 46, 49, 50, 65
F (n⫽6) 4 20, 21, 28,b43, 61, 71
1551 (n⫽12) 4 6, 7, 18, 19, 29, 41, 42, 52, 53, 67,c68, 69
1551 RFLP 4 8,c17,c30, 31,c40,c54c
aSpoligotype pattern differs from others in cluster. bSpoligotype pattern differs from others in cluster. cSamples are mickle lysates.
on May 15, 2020 by guest
http://jcm.asm.org/
FIG. 4. Insite assay of a randomized, blinded set ofM. tuberculosis DNA samples from clinical isolates as described in Table 3. The oligonucleotide probes (oligos) are as ordered in Table 2.
on May 15, 2020 by guest
http://jcm.asm.org/
nine-spot spoligotype pattern (data not shown) characteristic of Beijing family strains (16), again suggesting laboratory error. Twelve DNA samples were from isolates associated with a tuberculosis outbreak in Tennessee; all of the isolates have matching IS6110RFLP fingerprints and spoligotype patterns and are from patients with epidemiologic links. These isolates have been identified asM. tuberculosisstrain CDC1551, which can also be identified by the 1551 PCR assay (14). Five addi-tional isolates have an IS6110RFLP fingerprint identical to that of strain 1551 but are unrelated to the outbreak, have two different spoligotype patterns, and are negative in the 1551 PCR assay (data not shown). A final isolate is characterized as having a positive 1551 diagnostic PCR result but has a different four-band IS6110 RFLP pattern and a different spoligotype pattern. All 18 of the isolates were shown to contain the four strain 1551 insertion sites. LMPCR products for the last five samples and one of the CDC1551 outbreak samples were pre-pared from a glass bead-mickle apparatus crude lysate of each, which demonstrates that it is not necessary to prepare purified genomic DNA samples for the insite assay.
DISCUSSION
We developed a method, designated insite, which can be used to detect IS6110insertion sites. The method relies on the hybridization of biotin-labeled target DNA, generated by IS6110-specific LMPCR amplification, to oligonucleotide probes representing known sites of insertion immobilized on a membrane. The assay can easily be completed in 3 days, and because the method is amplification based, it does not require large-scale purification of high-quality genomic DNA. A single colony suspension is a sufficient template for LMPCR ampli-fication (2). The specificity of the assay was demonstrated by both the specific hybridization of individual PCR products and the hybridization patterns of LMPCR amplification products from strains with well-defined insertion sites. The reproduc-ibility of the assay was demonstrated by the identical insite profiles generated for 12 matched pairs of strains and for samples from tuberculosis outbreak investigation clusters.
Some insertion sites were detected more reliably than oth-ers, and a combination of factors are likely to be responsible for this effect. Amplicons representing each copy of IS6110are not represented equally in the hybridizing DNA sample be-cause all targets are not amplified with equal efficiency using the LMPCR method. In particular, larger amplicons are not amplified as efficiently as smaller ones. The composition of the oligonucleotide probe also influences hybridization efficiency. Oligonucleotide probes that contain strong secondary struc-ture hybridize with the DNA sample with reduced efficiency under our assay conditions. These difficulties may be overcome by decreasing the length of the IS6110sequence (which has a very high GC content) in the probe to reduce the secondary structure and by increasing the amount of oligonucleotide im-mobilized on the membrane. For probes with highTmvalues
(⬎70°C), reducing the length of the IS6110sequence should also increase the specificity. The hybridization of Rv-IS611010 target DNA to probe DK4 highlights the importance of con-sideringTmvalues during oligonucleotide probe design.
The majority of the IS6110insertion sites in this study were chosen simply because they are in strains with previously
se-quenced insertion sites. The four strains used in the develop-ment of this method contain a wide variation in IS6110copy number (4, 8, 16, or 22 copies) and share relatively few inser-tion sites. It was not expected that the inserinser-tion sites chosen for method development would be of great use in discriminating between strains ofM. tuberculosis. Although the insertion site profiles of the blinded set of 60 isolates showed the reproduc-ibility of the assay, limited discrimination of strains was ob-served. It should also be noted that three of the insertion sites most often detected in these 60 isolates are also found in most low-copy-number strains. Fomukong et al. (5) showed that of 42 strains with four or five copies of IS6110(all with different RFLP patterns), 88% had the 1551-1 (DK1) insertion, 76% had the 1551-3 (DK3) insertion, and 97% had the 1551-4 (DK2) insertion. Because over half of our set of 60 isolates have fewer than five copies of IS6110, it is not surprising that only a limited number of insertion sites were repeatedly de-tected.
By using a carefully selected, expanded set of insertion sites, the insite method should be useful forM. tuberculosisoutbreak investigations. Specimens with clearly different insite patterns could be assumed to be different, while specimens with the same insite pattern could be further analyzed using a second fingerprinting method. This “carefully selected set of insertion sites” will have to be determined empirically by screening a large number of isolates using the insite method itself along with sequencing of LMPCR products. The three probes for strain W were included in this study as examples of carefully selected insertion sites. As described by Kurepina et al. (10), the evolution of MDR strain W can be followed using IS6110 transposition events. The A1 insertion has only been identified in group 1 isolates, the B2 insertion can be found in a subset of group 1 isolates (e.g., strain W, strain N), and the B1 insertion, which presumably occurred after the acquisition of the MDR phenotype, can only be found in MDR strain W. Kremer et al. (9) showed that genotyping can be used successfully to define genotype families within the M. tuberculosis complex family. The insite approach will also be useful for defining large fam-ilies of strains. A small set of conserved insertion sites will likely define each family, with additional key sites defining major branches. The insite assay will recognize these insertion sites even when mutations have altered the size of the PvuII fragments for the conserved sites and the overall IS6110 pat-terns have diverged significantly.
In most instances, this method will provide less discrimina-tion than that seen with tradidiscrimina-tional IS6110RFLP fingerprint-ing because only known insertion sites can be detected. An important exception to this will be the detection of IS6110in hot spots—very small regions of the genome in which a large number of IS6110 insertion sites have been observed. Al-though these produce very small variations in fragment length that are often indistinguishable using RFLP typing methods, the insite assay should be able to easily discriminate between them. For example, one of the hot spots for IS6110in theM. tuberculosis chromosome is the ipl locus (4). Three probes (Rv1, E6, and E7) specific for IS6110 insertion sites in this locus were among the 32 probes utilized in this study, and all were highly specific for their respective insertion site se-quences.
Although the insite method is limited to known insertion
on May 15, 2020 by guest
http://jcm.asm.org/
sites, it is a significant improvement over currently used meth-ods for determining the presence or absence of IS6110in a specific position within the genome. In contrast to either prob-ing IS6110RFLP membranes with individual probes or ampli-fying a flanking sequence using specific primers, the insite method using a line blot apparatus can simultaneously screen up to 45 different samples for 45 different insertion sites. Use of the new microarray technologies should allow the screening of almost an unlimited number of probes. In conclusion, we have developed a method, insite, which is capable of the spe-cific detection of IS6110insertion sites at the nucleotide level. The application of the assay forM. tuberculosis strain differ-entiation will require additional knowledge regarding the dis-tribution of IS6110insertion sites in genotype families.
REFERENCES
1.Bauer, J., A. Kok-Jensen, P. Faurschou, J. Thuesen, E. Taudorf, and A. B.
Andersen.2000. A prospective evaluation of the clinical value of nation-wide
DNA fingerprinting of tuberculosis isolates in Denmark. Int. J. Tuberc. Lung Dis.4:295–299.
2.Bonora, S., M. C. Gutierrez, G. Di Perri, F. Brunello, B. Allegranzi, M.
Li-gozzi, R. Fontana, E. Concia, and V. Vincent.1999. Comparative evaluation
of ligation-mediated PCR and spoligotyping as screening methods for geno-typing ofMycobacterium tuberculosisstrains. J. Clin. Microbiol.37:3118–3123.
3.de la Salmoniere, Y.-O. G., H. M. Li, G. Torrea, A. Bunschoten, J. van Embden,
and B. Giquel.1997. Evaluation of spoligotyping in a study of the transmission
ofMycobacterium tuberculosis. J. Clin. Microbiol.35:2210–2214.
4.Fang, Z., and K. J. Forbes.1997. A Mycobacterium tuberculosis IS6110
preferential locus (ipl) for insertion into the genome. J. Clin. Microbiol.
35:479–481.
5.Fomukong, N., M. Beggs, H. El Hajj, G. Templeton, K. Eisenach, and M. D.
Cave.1998. Differences in the prevalence of IS6110insertion sites in
Myco-bacterium tuberculosisstrains: low and high copy number of IS6110. Tuber. Lung Dis.78:109–116.
6.Goyal, M., N. A. Saunders, J. D. A. van Embden, B. D. Young, and R. J Shaw.
1997. Differentiation ofMycobacterium tuberculosisisolates by spoligotyping and IS6110restriction fragment length polymorphism. J. Clin. Microbiol.
35:647–651.
7.Haas, W. H., W. R. Butler, C. L. Woodley, and J. T. Crawford.1993.
Mixed-linker polymerase chain reaction: a new method for rapid fingerprinting of isolates of theMycobacterium tuberculosiscomplex. J. Clin. Microbiol.31:
1293–1298.
8.Kamerbeek, J., L. Schouls, A. Kolk, M. van Agterveld, D. van Soolingen,
S. Kuijper, A. Bunschoten, H. Molhuizen, R. Shaw, M. Goyal, and J.
van Embden.1997. Simultaneous detection and strain differentiation of
Myco-baterium tuberculosisfor diagnosis and epidemiology. J. Clin. Microbiol.35:907– 914.
9.Kremer, K., D. van Soolingen, R. Frothingham, W. H. Haas, P. W. M.
Hermans, C. Martin, P. Palittapongarnpim, B. B. Plikaytis, L. W. Riley,
M. A. Yarkus, J. M. Musser, and J. D. A. van Embden.1999. Comparison of
methods based on different molecular epidemiological markers for typing of
Mycobacterium tuberculosiscomplex strains: interlaboratory study of discrim-inatory power and reproducibility. J. Clin. Microbiol.37:2607–2618.
10. Kurepina, N. E., S. Sreevatsan, B. B. Plikaytis, P. J. Bifani, N. D. Connell,
R. J. Donnelly, D. van Sooligen, J. M. Musser, and B. N. Kreiswirth.1998.
Characterization of the phylogenetic distribution and chromosomal insertion sites of five IS6110elements inMycobacterium tuberculosis: non-random integration in thednaA-dnaN region. Tuber. Lung Dis.79:31–42.
11. Mazurek, G. H., V. Reddy, B. J. Marston, W. H. Haas, and J. T. Crawford
1996. DNA fingerprinting by infrequent-restriction-site amplification. J. Clin. Microbiol.34:2386–2390.
12. Molhuizen, H. O. F., A. E. Bunschoten, L. M. Schouls, and J. D. A. van Embden.
1998. Rapid detection and simultaneous strain differentiation of Mycobacte-rium tuberculosiscomplex bacteria by spoligotyping. Methods Mol. Biol.
101:381–394.
13. Plikaytis, B. B., J. L. Marden, J. T. Crawford, C. L. Woodley, W. R. Butler,
and T. M. Shinnick.1994. Multiplex PCR assay specific for the
multidrug-resistant strain W ofMycobacterium tuberculosis. J. Clin. Microbiol.32:1542– 1546.
14. Plikaytis, B. B., N. Kurepina, C. L. Woodley, R. Fleischmann, B. Kreiswirth,
and T. M. Shinnick.1999. Multiplex PCR assay to aid in the identification of
the highly transmissibleMycobacterium tuberculosisstrain CDC1551. Tuber. Lung. Dis.79:273–278.
15. Prod’hom, G., C. Guilhot, M. C. Gutierrez, A. Varnerot, B. Giquel, and V.
Vincent.1997. Rapid discrimination ofMycobacterium tuberculosiscomplex
strains by ligation-mediated PCR fingerprint analysis. J. Clin. Microbiol.
35:3331–3334.
16. Qian, L., J. D. A. van Embden, A. G. M. van der Zanden, E. F. Weltevreden,
H. Duanmu, and J. T. Douglas.1999. Retrospective analysis of the Beijing
family ofMycobacterium tuberculosisin preserved lung tissues. J. Clin. Mi-crobiol.37:471–474.
17. Troesch, A., H. Nguyen, C. G. Miyada, S. Desvarenne, T. R. Gingeras, P. M.
Kaplan, P. Cros, and C. Mabilat.1999.Mycobacteriumspecies identification
and rifampin resistance testing with high-density DNA probe arrays. J. Clin. Microbiol.37:49–55.
18. van Embden, J., and D. van Soolingen.2000. Molecular epidemiology of
tuberculosis: coming of age. Int. J. Tuberc. Lung Dis.4:285–286.
19. van Embden, J. D. A., M. D. Cave, J. T. Crawford, J. W. Dale, K. D.
Eisenach, B. Giquel, P. Hermans, C. Martin, R. McAdam, T. M. Shinnick,
and P. M. Small.1993. Strain identification ofMycobacterium tuberculosisby
DNA fingerprinting: recommendations for a standardized methodology. J. Clin. Microbiol.31:406–409.
20. van Embden, J. D. A., T. van Gorkom, K. Kremer, R. Jansen, B. A. M.
van der Zeijst,and L. M. Schouls.2000. Genetic variation and evolutionary
origin of the direct repeat locus ofMycobacterium tuberculosiscomplex bacteria. J. Bacteriol.182:2393–2401.
21. Victor, T. C., A. M. Jordaan, A. van Rie, G. D. van der Spuy, M. Richardson,
P. D. van Helden, and R. Warren.1999. Detection of mutations in drug
resistance genes ofMycobacterium tuberculosisby a dot-blot hybridization strategy. Tuberc. Lung Dis.79:343–348.