metal-organic papers
m80
Brendan Twamleyet al. [CrBr2(C4H8O)2] DOI: 101107/S1600536801001660 Acta Cryst.(2001). E57, m80±m81 Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
Solvated CrBr2(thf)2, a linear chain in the solid state
Brendan Twamley,aRalph
Zehnderband Pamela J.
Shapirob*
aUniversity Research Office, 109 Morrill Hall, University of Idaho, Moscow, ID 83844-3010, USA, andbDepartment of Chemistry, University of Idaho, Moscow, ID 83844-2343, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 203 K
Mean(C±C) = 0.005 AÊ
Rfactor = 0.027
wRfactor = 0.071
Data-to-parameter ratio = 15.8
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2001 International Union of Crystallography Printed in Great Britain ± all rights reserved
In the title compound, dibromobis(tetrahydrofuran-O )-chromium(II), [CrBr2(C4H8O)2], CrBr2 is solvated by two
tetrahydrofuran (thf) molecules, with a CrÐO distance of 2.072 (3) AÊ and a CrÐBr distance of 2.5825 (5) AÊ. The Cr atom lies on the center of inversion. The Cr atom is octahedrally coordinated, with the thf CrÐO bond orthogonal to the CrÐBr bond, and the vacant sites are occupied with a Cr Br intermolecular interaction of 2.9874 (5) AÊ. The latter interaction is almost orthogonal to the CrÐO vector. This extends the system into a linear chain along the [100] direction.
Comment
CrBr2(thf)2, (I), bears a close resemblance to the parent
compound CrBr2 (Tracy et al., 1962). Both show slightly
distorted octahedral coordination and both have similar in-plane CrÐBr bond distances, 2.5825 (5) AÊ in (I) and 2.545 (1) AÊ in CrBr2. The solvent molecule, thf in this case,
occupies an in-plane coordination site with a CrÐO distance of 2.072 (3) AÊ. The solvent molecule is coordinated ortho-gonally to the CrÐBr vector with an angle of 90.009 (8). The
thf molecule is in a half-chair conformation with a total puckering amplitude ofQT= 0.363 AÊ (Cremer & Pople, 1975).
The orientation of the thf molecule is in¯uenced by the intramolecular interactions C4ÐH4A Br1 and C1Ð H1A Br1i(see Table 1) which tilt the ring towards each Br
atom. The out-of-plane coordination site is occupied by an intermolecular Cr1 Br1ii interaction of 2.9874 (5) AÊ
[symmetry code: (ii) 1 +x, y, z]. The corresponding interaction in CrBr2has a Cr Br distance of 2.998 (1) AÊ. The distortion
from ideal octahedral geometry is also re¯ected in the Br1Ð Cr1ÐBr1iiangle of 94.65 (2). With the thf molecules
occu-pying the other coordination sites, (I) can only form a stag-gered chain instead of the planar sheets found in CrBr2. In this
case, the direction of propagation is along [100] (Fig. 1). Other solvent coordinated CrIIBr
2 species that are octahedrally
coordinated show a wide range of CrÐBr distances. The complex CrBr2(CH3CN)2 (Halepotoet al., 1990), which also
forms linear chains, has an in-plane CrÐBr distance of 2.545 AÊ and an out-of-plane distance of 2.976 (1) AÊ. However,
the molecular species trans-CrBr2(pyridine)4 (Holah et al.,
1998) has a CrÐBr bond distance of 2.998 (1) AÊ which indi-cates a very weakly associated Br atom.
Experimental
CrBr2(thf)2was synthesized as a side product during the attempted
synthesis of a chromocene bromide. Tetramethylethylenediyl(5
-bis-cyclopentadienyl)chromoceneII carbonyl (331 mg, 1.1 mmol) was
reacted with 1-bromo-2-methylpropane (155 mg, 1.1 mmol) in tetra-hydrofuran for 20 h at 378 K, with no color change. Dibromomethane (excess) was then added to increase the bromide concentration. Excess solvent and reagents were removed under reduced pressure and the green residue redissolved in tetrahydrofuran (20 ml) and ®ltered. The resulting solution was concentrated to promote crys-tallization and cooled in a 278 K refrigerator overnight. The super-natant was decanted yielding a green crystalline product. The supernatant was concentrated under reduced pressure to yield a ®ne pale green powder. Combined yield 68 mg, 19.1%, m.p. > 573 K.
Crystal data
[CrBr2(C4H8O)2]
Mr= 356.03
Triclinic,P1
a= 4.1043 (7) AÊ
b= 7.4530 (13) AÊ
c= 9.2875 (16) AÊ
= 87.777 (3)
= 78.619 (3)
= 87.281 (3)
V= 278.07 (8) AÊ3
Z= 1
Dx= 2.126 Mg mÿ3
MoKradiation Cell parameters from 839
re¯ections
= 2.2±27.4
= 8.18 mmÿ1
T= 203 (2) K Plate, pale yellow 0.250.200.15 mm
Data collection
Siemens SMART 1000 diffract-ometer
!scans
Absorption correction: empirical (SADABS; Sheldrick, 1999)
Tmin= 0.184,Tmax= 0.293
2917 measured re¯ections 962 independent re¯ections 897 re¯ections withI> 2(I)
Rint= 0.029
max= 25.0
h=ÿ4!4
k=ÿ8!8
l=ÿ11!11
50 frames remeasured standard re¯ections
every beginning and end re¯ec-tions
intensity decay: none
Re®nement
Re®nement onF2
R[F2> 2(F2)] = 0.027
wR(F2) = 0.071
S= 1.08 962 re¯ections 61 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0436P)2
+ 0.1523P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.58 e AÊÿ3
min=ÿ0.70 e AÊÿ3
Table 1
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
C4ÐH4A Br1 0.98 2.99 3.599 (4) 122 C1ÐH1A Br1i 0.98 2.99 3.607 (4) 122
Symmetry code: (i) 1ÿx;1ÿy;1ÿz.
Data collection:SMART(Bruker, 1997); cell re®nement:SMART; data reduction:SAINT-Plus(Bruker, 1999); program(s) used to solve
structure: SHELXTL (Sheldrick, 1998); program(s) used to re®ne structure:SHELXTL; molecular graphics:SHELXTL; software used to prepare material for publication:SHELXTL.
This work was supported by a grant from the National Science Foundation (CHE-9816730), the DOE EPSCoR Program (Notice 98-02) and the Petroleum Research Foun-dation (ACS-PRF No. 33831-AC). The Bruker (Siemens) SMART CCD diffraction facility was established at the University of Idaho with the assistance of the NSF-EPSCoR program under NSF OSR-9350539 and the M. J. Murdock Charitable Trust, Vancouver, Wa, USA.
References
Bruker (1997).SMART. Version 5.059. Bruker AXS Inc., Madison, Wisconsin, USA.
Bruker (1999).SAINT-Plus.Bruker AXS Inc., Madison, Wisconsin, USA. Cremer, D. & Pople, J. A. (1975).J. Am. Chem. Soc.97, 1354±1358. Halepoto, D. M., Larkworthy, L. F., Povey, D. C., Tucker, B. J. & Smith, G. W.
(1990).J. Chem. Soc. Dalton Trans.pp. 699±701.
Holah, D. G., Hughes, A. N., Lundeen, C. & Magnuson, V. R. (1998).
Polyhedron,17, 2101±2104.
Sheldrick, G. M. (1998).SHELXTL.Version 5.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Sheldrick G. M. (1999).SADABS. Version 2.01. Bruker AXS Inc., Madison, Wisconsin, USA.
Tracy, J. W., Gregory, N. W. & Lingafelter, E. C. (1962).Acta Cryst.15, 672±674.
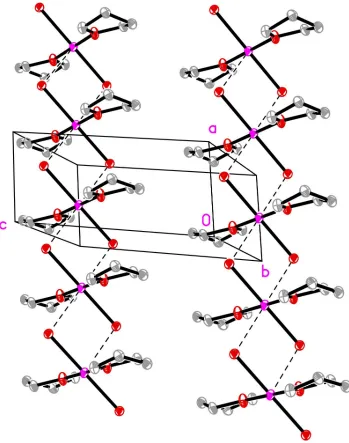
Figure 1
supporting information
sup-1
Acta Cryst. (2001). E57, m80–m81supporting information
Acta Cryst. (2001). E57, m80–m81 [doi:10.1107/S1600536801001660]
Solvated CrBr
2(thf)
2, a linear chain in the solid state
Brendan Twamley, Ralph Zehnder and Pamela J. Shapiro
S1. Comment
(THF)2CrBr2, (I), bears a close resemblance to the parent compound CrBr2 (Tracy et al., 1962). Both show slightly
distorted octahedral coordination and both have similar in-plane Cr—Br bond distances, 2.5825 (5) Å in (I) and 2.545 (1)
Å in CrBr2. The solvent molecule, THF in this case, occupies an in-plane coordination site with a Cr—O distance of
2.072 (3) Å. The solvent molecule is coordinated orthogonally to the Cr—Br vector with an angle of 90.009 (8)°. The
THF molecule is in a half-chair conformation with a total puckering amplitude of QT = 0.363 Å (Cremer & Pople, 1975).
The orientation of the THF molecule is influenced by the intramolecular interactions C4—H4a···Br1 and C1—H1a···Br1i
(see Table 1) which tilt the ring towards each Br atom. The out-of-plane coordination site is occupied with an
intermolecular Cr1···Br1ii interaction of 2.9874 (5) Å [symmetry code: (ii) 1 + x, y, z]. The corresponding interaction in
CrBr2 has a Cr···Br distance of 2.998 (1) Å. The distortion from ideal octahedral geometry is also reflected in the Br1—
Cr1—Br1ii angle of 94.65 (2)°. With the THF molecules occupying the other coordination sites, (I) can only form a
staggered chain instead of the planar sheets found in CrBr2. In this case, the direction of propagation is along [100] (Fig.
1). Other solvent coordinated CrIIBr
2 species that are octahedrally coordinated show a wide range of Cr—Br distances.
The complex CrBr2(CH3CN)2 (Halepoto et al., 1990), which also forms linear chains, has an in-plane Cr—Br distance of
2.545 Å and an out-of-plane distance of 2.976 (1) Å. However, the molecular species trans-CrBr2(pyridine)4 (Holah et al.,
1998) has a Cr—Br bond distance of 2.998 (1) Å which indicates a very weakly associated Br atom.
S2. Experimental
(THF)2CrBr2 was synthesized as a side product during the attempted synthesis of a chromocene bromide.
Tetramethyl-ethylenediyl(η5-biscyclopentadienyl)chromoceneII carbonyl (331 mg, 1.1 mmol) was reacted with
1-bromo-2-methyl-propane (155 mg, 1.1 mmol) in THF for 20 h at 378 K, with no color change. Dibromomethane (excess) was then added
to increase the bromide concentration. Excess solvent and reagents were removed under reduced pressure and the green
residue redissolved in THF (20 ml) and filtered. The resulting solution was concentrated to promote crystallization and
cooled in a 278 K refrigerator overnight. The supernatant was decanted yielding a green crystalline product. The
supernatant was concentrated under reduced pressure to yield a fine pale green powder. Combined yield 68 mg, 19.1%,
Figure 1
Packing diagram of (I) showing the staggered propagation along the [100] direction. H atoms have been omitted for
clarity.
dibromobis(tetrahydrofuran-O)chromium(II)
Crystal data
[CrBr2(C4H8O)2]
Mr = 356.03
Triclinic, P1
a = 4.1043 (7) Å
b = 7.4530 (13) Å
c = 9.2875 (16) Å
α = 87.777 (3)°
β = 78.619 (3)°
γ = 87.281 (3)°
V = 278.07 (8) Å3
Z = 1
F(000) = 174
Dx = 2.126 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 839 reflections
supporting information
sup-3
Acta Cryst. (2001). E57, m80–m81µ = 8.18 mm−1
T = 203 K
Plate, pale yellow 0.25 × 0.20 × 0.15 mm
Data collection
Siemens SMART 1000 diffractometer
Radiation source: normal-focus sealed tube Graphite monochromator
Detector resolution: 8.3 pixels mm-1
ω scans
Absorption correction: empirical (using intensity measurements)
(SADABS; Sheldrick, 1999)
Tmin = 0.184, Tmax = 0.293 2917 measured reflections 962 independent reflections 897 reflections with I > 2σ(I)
Rint = 0.029
θmax = 25.0°, θmin = 2.2°
h = −4→4
k = −8→8
l = −11→11
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.027
wR(F2) = 0.071
S = 1.08 962 reflections 61 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0436P)2 + 0.1523P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001 Δρmax = 0.58 e Å−3 Δρmin = −0.70 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 0.10559 (9) 0.70378 (5) 0.37413 (4) 0.02022 (17)
Cr1 0.5000 0.5000 0.5000 0.0180 (2)
O1 0.4032 (7) 0.6622 (3) 0.6810 (3) 0.0245 (6)
C1 0.2743 (10) 0.5980 (5) 0.8298 (4) 0.0242 (9)
H1A 0.3974 0.4886 0.8536 0.029*
H1B 0.0382 0.5724 0.8425 0.029*
C3 0.2812 (11) 0.9167 (5) 0.8283 (4) 0.0244 (9)
H3A 0.0454 0.9507 0.8342 0.029*
H3B 0.3948 1.0190 0.8558 0.029*
C2 0.3237 (10) 0.7515 (5) 0.9266 (4) 0.0240 (9)
H2A 0.5459 0.7433 0.9508 0.029*
H2B 0.1558 0.7528 1.0177 0.029*
H4A 0.3266 0.9144 0.6022 0.028*
H4B 0.6753 0.8848 0.6524 0.028*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.0210 (2) 0.0198 (2) 0.0199 (3) 0.00074 (15) −0.00461 (16) 0.00071 (15)
Cr1 0.0230 (5) 0.0167 (4) 0.0144 (5) 0.0034 (3) −0.0046 (4) −0.0026 (3)
O1 0.0390 (16) 0.0173 (13) 0.0152 (14) −0.0007 (12) −0.0004 (12) −0.0019 (11)
C1 0.029 (2) 0.022 (2) 0.018 (2) 0.0006 (16) 0.0015 (17) −0.0007 (16)
C3 0.027 (2) 0.024 (2) 0.021 (2) 0.0004 (16) −0.0020 (17) −0.0058 (16)
C2 0.028 (2) 0.027 (2) 0.017 (2) 0.0021 (17) −0.0026 (17) −0.0052 (17)
C4 0.032 (2) 0.0162 (19) 0.021 (2) −0.0041 (16) −0.0036 (17) −0.0010 (15)
Geometric parameters (Å, º)
Br1—Cr1 2.5825 (5) O1—C4 1.458 (4)
Cr1—O1i 2.072 (3) C1—C2 1.527 (6)
Cr1—O1 2.072 (3) C3—C4 1.512 (5)
Cr1—Br1i 2.5825 (5) C3—C2 1.528 (6)
O1—C1 1.450 (5)
O1i—Cr1—O1 180.00 (9) C1—O1—Cr1 124.1 (2)
O1i—Cr1—Br1i 90.09 (8) C4—O1—Cr1 125.1 (2)
O1—Cr1—Br1i 89.91 (8) O1—C1—C2 104.7 (3)
O1i—Cr1—Br1 89.91 (8) C4—C3—C2 103.6 (3)
O1—Cr1—Br1 90.09 (8) C1—C2—C3 102.1 (3)
Br1i—Cr1—Br1 180.0 O1—C4—C3 105.2 (3)
C1—O1—C4 110.7 (3)
Symmetry code: (i) −x+1, −y+1, −z+1.
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C4—H4A···Br1 0.98 2.99 3.599 (4) 122
C1—H1A···Br1i 0.98 2.99 3.607 (4) 122