has been used to screen for metabolic disorders, genetic diseases, and viral infection and for evolutionary studies of the genome. In this study, DHBV from serum and blood dried on filters was detected by PCR. A 0.1-l sample was sufficient for detection. The immobilization potential of filter papers for DHBV was examined, and the highest yield of PCR products was observed with Whatman paper. Dried serum was stable under different storage temperatures for 4 weeks, but the yields of PCR products decreased when the temper-ature was肁4°C. The optimal condition for storage wasⴚ70°C. A newly developed quantitative PCR based on monitoring the amplification by measuring the increase in fluorescence caused by the binding of SYBR green dye to double-stranded products was applied herein. DHBV genomic DNA cloned in a plasmid was used for the generation of standard DHBV DNA for quantitative PCR. It validated results from PCR in terms of the copy number of DHBV particles. The specificity of PCR was demonstrated by melting curve analysis, and the differentiation of two DHBV isolates amplified from dried serum was demonstrated based on their melting temperatures determined by GC contents and sequence. It was easier and simpler than other PCR-based DNA techniques. The use of serum dried on filters allows samples from distant field for which cold storage and transportation are a problem to be mailed to the diagnostic laboratory. Samples can be archived for compar-ison and used as a source of DNA for cloning and sequencing.
Duck hepatitis B virus(DHBV) is the first hepatitis B virus (HBV) identified from an avian host. The natural route of transmission is from the bloodstream of persistently infected laying ducks to the egg, resulting in congenital infection (18, 21). Congenitally infected ducks are at risk of developing hep-atoma or secondary amyloidosis due to chronic stimulation of the immune system (9, 10, 11). HBV is a worldwide human health problem, with carriers at risk for developing cirrhosis and hepatocarcinoma. The molecular biology of HBV is in-tensely researched for its medical importance and life cycle. It is dangerous to work with because of nosocomial infections through blood and body fluids from infected patients. There-fore, attention has been devoted to a safe model for HBV such as DHBV (23).
Specimens of serum or blood dried on filter paper have been used for serologic and screening tests for human immunode-ficiency virus (HIV), measles virus, human cytomegalovirus, phenylketonuria, and HBV surface antigens (7, 11, 15, 24, 27). This method provides a simple and powerful approach for large-scale screening and epidemiological studies. Dried sam-ples are light, are easy to store or mail, cannot be broken or spilled, and take up little space. When the PCR was used to amplify a defined DNA sequence, it offered a quick and
reli-able diagnosis and analysis of DNA isolated from serum dried on filter papers (3, 6). In this study, a sensitive PCR assay was applied to detect DHBV from dried serum or blood on filters. The immobilization potential of different filter papers and the temperature effect of storage were compared by using this system. The precise quantification of the DHBV level from each experiment was first accomplished with the use of the newly developed SYBR green quantitative PCR. In conclu-sion, this technique not only facilitated the field investigation of DHBV in flocks but also extended the usefulness of DHBV as a model of HBV.
MATERIALS AND METHODS
Virus loading onto filter papers.DHBV-infected serum (2.9⫻109copies of
DHBV genome/ml) was obtained from 1-week-old congenitally infected ducks. The filters and membranes investigated for the immobilization of serum samples were (i) Whatman paper no. 1, (ii) nitrocellulose membrane no. 71002 (Amer-sham), and (iii) HYBOND-M nylon membrane (Amersham). Samples of 50, 10,
1, and 0.1l of virus-infected serum were loaded onto each filter and dried at
room temperature. Whatman papers, each containing 10l of virus-infected
serum, were dried, sealed into the microtene bags, and stored at 37, 4,⫺20, and
⫺70°C for 4 weeks before use.
DHBV DNA isolation.Viral nucleic acid was prepared from serum or blood dried on filters by using a QiaAmp blood and tissue kit (Qiagen, Hilden, Ger-many) or phenol-chloroform extraction. For the QiaAmp kit, dried serum filters
were incubated with 200l of distilled water for 1 h at 37°C and then used for
the lysis reaction with chaotropic salts, detergents, and a final proteinase K concentration of 1 mg/ml. After the addition of 2-propanol to decrease solubility,
DNA was bound to spin columns and washed twice before elution in 20l of
distilled water. For the phenol-chloroform extraction, filter paper (Whatman
paper no. 1) with dried viral serum (1l) was incubated for 1 h at 37°C in a 400
* Corresponding author. Mailing address: Poultry Annex Building, Poultry Science Department, Auburn University, Auburn, AL 36849-5416. Phone: (334) 844-2642. Fax: (334) 844-2641. E-mail: jgiambro @acesag.auburn.edu.
2584
on May 15, 2020 by guest
l of extraction buffer containing 0.01 M Tris, 0.01 M EDTA, 0.01 M NaCl, 0.5% (wt/vol) sodium dodecyl sulfate, and 50 mg of proteinase K/ml. Following incu-bation, DHBV DNA was phenol-chloroform extracted, ethanol precipitated, and
suspended in 10l of distilled water. Preparations served as the template for
both conventional and quantitative PCR.
PCR amplification.Viral DNA sequences were amplified by using the
oligo-nucleotide primers DCD 03 (5⬘-ACTAGAAAACCTCGTGGACT-3⬘, bp 241 to
260) and DCD 04 (5⬘-GGGAGAGGGGAGCCCGCACG-3⬘, bp 349 to 368). A
single PCR sample containing 10l of DNA was amplified in a 50-l reaction
mixture containing 10 mM Tris-HCl (pH 8.3), 2.0 mM MgCl2, each
deoxynucleo-side triphosphate at 0.2 mM, each primer at 0.5M, and 50 U ofTaqpolymerase
(Perkin-Elmer) per ml. PCRs were subjected to 30 cycles (denaturation, 30 s at 95°C; annealing, 30 s at 55°C; extension, 1 min at 72°C) and 1 final extension cycle at 72°C for 10 min. Primers mapped in the N-terminal third of the duck
HBVpolgene. Ten microliters of the reaction mixtures was analyzed on 3.0%
agarose/Tris-Cl–sodium acetate–EDTA (NuSieve, FMC) in the presence of 0.5
g of ethidium bromide per ml and photographed under UV illumination. The
results were compared to those for uninfected controls.
Construction of standard plasmids for SYBR green quantitative PCR.The plasmid (pDHBV) used as the standard DNA was constructed by ligation of a PCR fragment in pTrueBlue-VUII according to the instructions of the manu-facturer (Genomics One, Quebec, Canada). The cloned fragment comprised a 128-bp region of the DHBV DNA polymerase gene amplified by primers DCD
03 and DCD 04. Plasmids were linearized withNcoI (Roche) and purified over
a 0.8% agarose gel in 1⫻Cl–Tris sodium acetate–EDTA. The band of correct
size was excised and extracted by using a QIAquick gel extraction kit (Qiagen) according to the manufacturer’s instructions. The concentrations of linearized plasmid preparations were determined by measuring the optical density at 260 nm. Conversion to genome equivalents was done by using the following
equa-tions: an optical density at 260 nm of 1⫽50g/ml, and 1 bp⫽660 g/mol. This
resulted in a molecular mass for the plasmids (2,815 bp) of 1.82⫻106g/mol. For
the standard curve, a series of 10-fold-diluted plasmids pDHBV was included in each test.
SYBR green quantitative PCR.The SYBR green PCR kit (PE Applied
Bio-systems) was used with 50-l reaction mixtures in a model 7700 sequence
de-tector (PE Applied Biosystems). The PCR mixture contained 1⫻SYBR PCR
buffer, 2.0 mM MgCl2, 0.2 mM (each) dATP, dCTP, and dGTP, 0.5 mM dUTP,
0.1M primers DCD 03 and DCD 04, 0.5l of AmpErase uracil-N-glycosylase
(UNG) (1 U/l), and 0.25l of AmpliTaq Gold polymerase (1 U/l). After
initial incubation at 50°C for 2 min to activate the UNG and 95°C for 10 min to
activate theTaqpolymerase, thermal conditions followed 40 cycles of 95°C for
30 s, 63°C for 30 s, and 72°C for 1 min. For each run, quadruplicates of standard
plasmids pDHBV representing 104to 107copies of DHBV genome and DNA
samples isolated from 1l of dried viral serum in different filters were used. This
was done because all copy numbers of DHBV genome of tested samples fell in
the 104to 107copies of DHBV genomes in pilot experiments. A copy number of
DHBV genome was calculated according to the standard curve generated by the
real-time PCR software of the model 7700 sequence detector (PE Applied Biosystems).
Comparison of sensitivities and specificities for DHBV DNA detection of fresh serum, dried serum, and dried blood.Ten microliters of 30 blood or serum samples randomly obtained from the ducklings was directly used or spotted onto Whatman paper no. 1 and allowed to dry before DNA extraction. Viral nucleic acid was prepared by using a QiaAmp blood and tissue kit (Qiagen), dissolved in
10l of distilled water, and subjected to PCR. The sensitivities and specificities
of these assays were calculated.
Melting curve analysis and direct sequencing of PCR products.Melting curve analysis was performed to measure the specificity of the PCR product. It also was used to rapidly differentiate strains of DHBV based on melting temperature
(Tm). After PCR, samples were heated to 95°C for 30 s and 63°C for 20 s and
then heated to 95°C at a ramp rate of 0.2°C/s for 19 min 59 s. Results were analyzed with the melting curve analysis software of the model 7700 sequence
detector (PE Applied Biosystems). TheTmof PCR products was calculated at
the same time. PCR products were purified by using a Genelute PCR purification kit (Sigma) and then sequenced in both directions with primers DCD 03 and DCD 04 by using a PRISM ready reaction DyeDeoxy terminator cycle sequenc-ing kit as recommended by the manufacturer on a UA373 DNA sequencsequenc-ing system (PE Applied Biosystems).
RESULTS
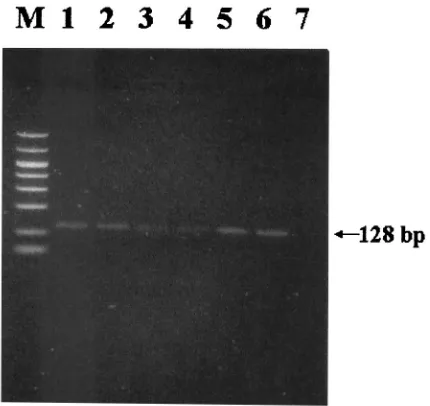
PCR detection of DHBV DNA from dried serum filter pa-pers.DHBV DNA was extracted and amplified by PCR from all filter papers. Yields of amplicons were proportional to the amount of sample (0.1 to 50l) loaded onto filters (Fig. 1). The highest and lowest yields of PCR products were observed on Whatman paper no. 1 and nylon membrane, respectively, as determined by the intensities of the electrophoretic bands. These revealed that DNBV DNA is stable in a dried condition and that Whatman paper no. 1 provides the most efficient immobilization.
Effect of storage temperature and extraction method of dried serum filter papers. Whatman paper was selected for experiments due to prior results. PCR products were observed after storage of serum dried on filters at 37, 4,⫺20, and⫺70°C for 4 weeks. The yields of PCR products from storage at⫺70 and⫺20°C were significantly higher than those from 37 and 4°C (Fig. 2). Results showed no differences between the yields of PCR products obtained with a silica gel-based column (Qia-FIG. 1. Detection of DHBV by PCR in dried serum samples spotted onto Whatman paper no. 1 (A), nylon membrane (B), and nitrocellulose membrane (C). Lanes M, 50- to 1,000-bp size markers; lanes 1, 50l of serum sample; lanes 2, 10l of serum sample; lanes 3, 1l of serum sample; lanes 4, 0.1l of serum sample; lanes 5, negative control.
on May 15, 2020 by guest
http://jcm.asm.org/
gen) and those obtained by using the phenol-chloroform tech-nique for viral DNA extraction (Fig. 2).
Comparison of sensitivities and specificities for DHBV DNA detection of fresh serum, dried serum, and dried blood.The results for the sensitivities and specificities for DHBV DNA detection of fresh serum, dried serum, and dried blood are shown in Table 1. Both sensitivity and specificity for fresh and dried serum were 100%. For dried blood, the sensitivity and specificity were 82 and 100%, respectively.
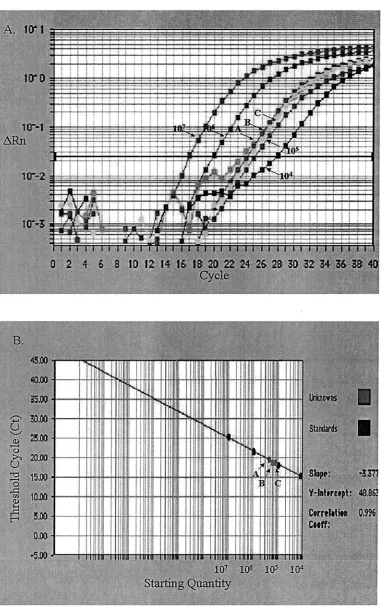
Quantification of DHBV DNA from dried serum filter pa-pers.One microliter of serum containing 3.0⫻105copies of
DHBV genome was dried on different filters and then recov-ered for quantification of DHBV. Results for Whatman paper no. 1, nitrocellulose membrane, and nylon membrane were 2.72⫻105⫾0.09⫻105, 2.23⫻105⫾1.3⫻105, and 1.68⫻
105⫾0.43⫻105copies of DHBV genome, respectively (P⬍
0.05) (Fig. 3). This showed that the dried serum from filters was detected with SYBR green quantitative PCR and offered quantitative proof of the immobilization potentials of different filters. To monitor the effect of storage temperature, 10-l
DISCUSSION
Human HBV has a worldwide distribution and poses a threat to public health. The molecular biology of HBV is sub-ject to intense research for its medical importance, but efforts are hampered by the lack of a good cell culture system and its contagious nature. DHBV is widely accepted as a surrogate for HBV due to their similarities (18, 21). Compared to the chim-panzee experimental model for HBV, this model has advan-tages in economics and handling (23).
Collection of serum on filter paper has been used to screen for metabolic disorders such as phenylketonuria and sickle cell disease (27). It enables large-scale, worldwide screening of viruses, evolutionary studies, and searches for mutations (22, 26). It allows samples from distant field or developing coun-tries, in which cold storage and transportation are problems, to be concentrated into one laboratory (12). Samples can be mailed after collection, stored, and archived for comparison or used as a source of DNA for cloning and sequencing (19). Studies have shown the efficacy of collecting blood or serum on filter paper followed by PCR analysis for screening and sur-veillance of perinatal transmission of HIV and human cyto-megalovirus and global distribution of malaria genotypes (5, 24). When PCR was applied for viral detection in infected ducks, dried or fresh serum gave fewer false negatives than did dried blood. This is due to a more complete removal of eryth-rocyte contaminants from serum preparations during drying and contaminants such as the porphyrin moiety of heme, which is known to be a strong inhibitor of PCR (17). This inhibition effect of dried blood on filters caused a decrease in sensitivity (82%). Although microwaving dried blood samples on filters prior to PCR was reported to enhance the sensitivity for de-tection of HBV, blood still had larger amounts of potential erythrocyte contaminants than did serum (14). Viral DNA was extracted and amplified by PCR from fresh serum or dried serum on filters. Due to the high levels of DHBV in the serum of infected ducks, 0.1l was sufficient for detection of virus on the filter paper. The highest yield of PCR products was ob-served when DHBV-infected serum was loaded into Whatman paper, because of its higher absorption and rehydration poten-tials to serum. Other advantages include its availability and price, which is lower than that of other filters.
No significant difference in DNA extraction yield was ob-FIG. 2. Detection of DHBV by PCR in dried serum samples (1l)
[image:3.587.58.273.74.277.2]spotted into Whatman paper no. 1. In lanes 1 to 4, a study of storage temperature is shown. Lane M, 50- to 1,000-bp size markers; lane 1, ⫺70°C; lane 2,⫺20°C; lane 3, 4°C; lane 4, 37°C. In lanes 5 to 7, a study of extraction method is shown. Lane 5, Qiagen column; lane 6, phenol-chloroform extraction; lane 7, negative control.
TABLE 1. Comparison of fresh serum, dried serum, and dried blood for detection of DHBV DNAa
Assay
No. of samples with indicated result
Sensitivity
(%) Specificity(%)
True
positive negativeTrue positiveFalse negativeFalse
Fresh serum 22 8 0 0 100 100
Dried serum 22 8 0 0 100 100
Dried blood 18 8 0 4 82 100
aSensitivity and specificity were calculated as (TP)100/(TP ⫹ FP) and
(TN)100/(TN⫹FP), respectively (TP, true positive; FP, false positive; TN, true
negative).
on May 15, 2020 by guest
[image:3.587.41.284.633.703.2]FIG. 3. Quantitative PCR quantification of DHBV in dried serum sample (1 l) spotted onto Whatman paper no. 1 (arrow A), nylon membrane (arrow B), and nitrocellulose membrane (arrow C). (A) Amplification plot of extracted DNA and the standard DHBV plasmids starting from 104to 107molecules. The cycle number was plotted versus the change in fluorescent intensity. (B) Standard curve generated from the
amplification plot. The starting copy number was plotted versus the threshold cycle.
on May 15, 2020 by guest
http://jcm.asm.org/
served between silica gel-based columns (Qiagen) and phenol-chloroform. It was more convenient to use Qiagen columns in large-scale screening. Similar results were observed with other viruses (1, 26). Serum dried on filters was stable under different temperatures for 4 weeks, but the yields of PCR products decreased when the temperature was肁4°C. A temperature of
⫺70°C was optimal for storage. These findings are consistent
with those of studies with HIV and hemorrhagic enteritis virus (6, 26). The rate of partial degradation of dried DNA on filters or the presence of PCR inhibitors may increase proportion-ately with storage temperature. Also, difficulty in removing viral DNA from filters increases with prolonged storage time and higher temperature (12).
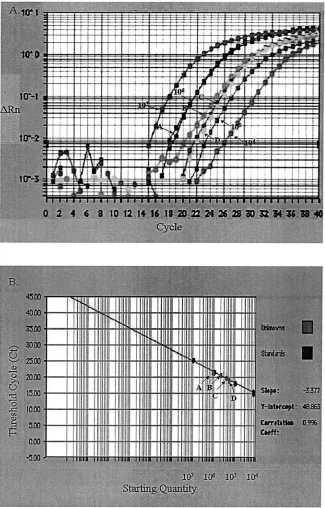
[image:5.587.132.457.76.584.2]To precisely estimate the amounts of virus in samples, a FIG. 4. Quantitative PCR quantification of DHBV in dried serum sample (10l) spotted into Whatman paper no. 1 stored at⫺70°C (arrow A),⫺20°C (arrow B), 4°C (arrow C), and 37°C (arrow D). (A) Amplification plot of extracted DNA and the standard DHBV plasmids starting from 104to 107molecules. The cycle number was plotted versus the change in fluorescent intensity. (B) Standard curve generated from the
amplification plot. The starting copy number was plotted versus the threshold cycle.
on May 15, 2020 by guest
quantitative PCR was applied herein. The detection and quan-tification of target DNA take place simultaneously. Amplifica-tion is performed in a closed tube and requires no post-PCR electrophoresis. Therefore, contamination with amplicons can be avoided (22). This assay is more reproducible and conve-nient than other quantitative PCR tests. Competitive quanti-tative PCR for DHBV DNA requires an exogenous competitor for each target as a control which needs to be constructed and characterized (2). Fluorogenic quantitative PCR requires an efficient probe with double-labeled fluorescent reporter dyes to be incorporated in each reaction (4, 8, 28). Our system has advantages in sensitivity over the fluorogenic probe system, because multiple dyes can bind to a single amplified molecule. We cloned a plasmid containing viral genome for the genera-tion of standard DHBV for quantitative PCR. The detecgenera-tion limit of this assay was 10 copies of DHBV DNA (data not shown), and it further validated results from PCR experiments in terms of the copy number of DHBV particles.
Steps were taken to avoid false positives in quantitative PCR, because SYBR green dye can bind to any double-stranded DNA, even nonspecific amplicons. A “hot start” Am-pliTaq Gold polymerase, which requires incubation at 95°C to activate just before the first denaturation step of thermocy-cling, was applied. The use of UNG and the incorporation of dUTP in place of dTTP can prevent carryover contamination by PCR product, because illegitimate amplifications that incor-porate dUTP can be cleaved by UNG. Any nonspecific ampli-con would be eliminated before the amplification, with no subsequent contamination detected on further runs (20). The specificity of PCR was verified by the performance of melting curve analysis for the PCR product, which depends on its GC contents, length, and sequence (25, 29). Two batches of sera dried on filters containing different DHBV isolates (California and Indiana isolates) were amplified and examined by melting curve analysis. The melting temperatures of PCR products from California and Indiana isolates were 80.6 and 81.2°C, respectively. The difference between the melting temperatures was attributed to the nucleotide sequence variations in these
isolates (sequence homology was 98.7%). Therefore, the dif-ferentiation of virus strains by melting curve analysis was suc-cessful. It was more applicable (with a difference of only four bases in this case) and faster (20 min) than PCR-restriction fragment length polymorphism or PCR–single-strand confor-mation polymorphism (13, 15, 16). No amplification from primer dimer or other sources was present in the controls. Sequence analysis further confirmed the specificity of PCR amplifications.
Although dried serum cannot provide live virus that can be grown and studied in culture, the loss of viral infectivity has the advantage of safety during shipping. This technique can be further refined and optimized for different applications, includ-ing the study of genetic diseases, forensics, the human genome, and persistent infections in humans and animals.
ACKNOWLEDGMENTS
We are grateful to Hongzhuan Wu for technical assistance. This work was supported partially by a grant from Southern Micro-biological Associates of Birmingham, Ala.
REFERENCES
1.Abe, K., and N. Konomi.1998. Hepatitis C virus RNA in dried serum spotted
onto filter paper is stable at room temperature. J. Clin. Microbiol.36:3070–
3072.
2.Addison, W. R., W. W. S. Wong, K. P. Fischer, and D. L. Tyrrell.2000. A quantitative competitive PCR assay for the covalently closed circular form of
duck hepatitis B virus. Antivir. Res.48:27–37.
3.Beck, I. A., K. D. Drennan, A. J. Melvin, K. M. Mohan, A. M. Herz, J. Alarcon, J. Piscoya, C. Velazquez, and L. M. Frenkel.2001. Simple, sensitive, and specific detection of human immunodeficiency virus type 1 subtype B DNA in dried blood samples for diagnosis in infants in the field. J. Clin.
Microbiol.39:29–33.
4.Bhudevi, B., and D. Weinstock.2001. Fluorogenic RT-PCR assay (TaqMan) for detection and classification of bovine viral diarrhea virus. Vet. Microbiol.
83:1–10.
5.Cassol, S., T. Salas, M. Arella, P. Neumann, M. T. Schechter, and M. V. O’Shaughnessy.1991. Use of dried blood spot specimens in the detection of human immunodeficiency virus type 1 by the polymerase chain reaction.
J. Clin. Microbiol.29:667–671.
6.Cassol, S., T. Salas, M. J. Gill, M. Montpetit, J. Rudnik, C. T. Sy, and M. V. O’Shaughnessy.1992. Stability of dried blood spot specimens for detection of human immunodeficiency virus DNA by polymerase chain reaction.
J. Clin. Microbiol.30:3039–3042.
7.Cassol, S. A., N. Lapointe, T. Salas, C. Hankins, M. Arella, M. Fauvel, G. Delage, M. Boucher, J. Samson, J. Charest, M. L. Montpetit, and M. V. O’Shaughnessy.1992. Diagnosis of vertical HIV-1 transmission using the polymerase chain reaction and dried blood spot specimens. J. Acquir.
Im-mune Defic. Syndr.5:113–119.
8.Chen, R. W., H. Piiparinen, M. Seppanen, P. Koskela, S. Sarna, and M. Lappalainen.2001. Real-time PCR for detection and quantification of
hep-atitis B virus DNA. J. Med. Virol.65:250–256.
9.Cova, L., A. Duflot, M. Prave, and C. Trepo.1993. Duck hepatitis B virus
infection, alfatoxin B1 and liver cancer in ducks. Arch. Virol.8:81–87.
10.Duflot, A., R. Mehrota, S. Yu, L. Barraud, C. Trepo, and L. Cova.1995. Spectrum of liver disease and duck hepatitis B virus infection in a large series
of Chinese ducks with hepatocellular carcinoma. Hepatology21:1483–1491.
11.Farzadegan, H., K. H. Noori, and F. Ala.1978. Detection of hepatitis B surface antigen in blood and blood products dried on filter paper. Lancet
i:362–363.
12.Fiscus, S. A., D. Brambilla, L. Grosso, J. Shock, and M. Cronin.1998. Quantitation of human immunodeficiency virus type 1 RNA in plasma by
using blood dried on filter paper. J. Clin. Microbiol.36:258–260.
13.Fujiwara, K., O. Yokosuka, T. Ehata, F. Imazeki, and H. Saisho.2000.
PCR-SSCP analysis of 5⬘-nontranslated region of hepatitis A viral RNA:
comparison with clinicopathological features of hepatitis A. Dig. Dis. Sci.
45:2422–2427.
14.Gupta, B. P., N. Jayasuryan, and S. Jameel. 1992. Direct detection of hepatitis B virus from dried blood spots by polymerase chain reaction
am-plification. J. Clin. Microbiol.30:1913–1916.
15.Guthrie, R., and A. Susi.1963. A simple method for detecting
phenylketo-nuria in large populations of newborn infants. Pediatrics32:338–343.
16.Han, M. G., and S. J. Kim.2001. Analysis of Korean strains of infectious laryngotracheitis virus by nucleotide sequences and restriction fragment
[image:6.587.47.281.74.223.2]length polymorphism. Vet. Microbiol.83:321–331.
FIG. 5. Melting curve analysis of PCR products obtained following 40 cycles of amplification. Melting peaks were produced by plotting the change in the relative intensity of fluorescence for each sample over temperature. Note that theTms of the PCR products of DHBV Indi-ana and California isolates were 81.2 and 80.6°C, respectively. No peaks of amplification products from negative control groups (arrow C) were observed.