Copyright © 2002, American Society for Microbiology. All Rights Reserved.
Microevolution of the Direct Repeat Region of
Mycobacterium
tuberculosis
: Implications for Interpretation of
Spoligotyping Data
R. M. Warren,
1E. M. Streicher,
1S. L. Sampson,
1G. D. van der Spuy,
1M. Richardson,
1D. Nguyen,
2M. A. Behr,
2T. C. Victor,
1and P. D. van Helden
1*
MRC Centre for Molecular and Cellular Biology, Department of Medical Biochemistry, Faculty of Health
Sciences, Stellenbosch University, Tygerberg 7505, South Africa,1and Department of Medicine,
McGill University Health Centre, Montreal H3G 1A4, Canada2
Received 21 June 2002/Returned for modification 6 August 2002/Accepted 14 September 2002
The direct repeat (DR) region has been determined to be an important chromosomal domain for studying
the evolution ofMycobacterium tuberculosis. Despite this, very little is known about microevolutionary events
associated with clonal expansion and how such events influence the interpretation of both restriction fragment length polymorphism (RFLP) and spoligotype data. This study examined the structure of the DR region in
three independently evolving lineages ofM. tuberculosiswith a combination of DR-RFLP, spoligotyping, and
partial DNA sequencing. The results show that the duplication of direct variable repeat (DVR) sequences and
single-nucleotide polymorphisms is rare; conversely, the deletion of DVR sequences and IS6110-mediated
mutation is observed frequently. Deletion of either single or contiguous DVR sequences was observed. The deletion of adjacent DVR sequences occurred in a dependent manner rather than as an accumulation of
independent events. Insertion of IS6110 into either the direct repeat or spacer sequences influenced the
spoligotype pattern, resulting in apparent deletion of DVR sequences. Homologous recombination between
adjacent IS6110 elements led to extensive deletion in the DR region, again demonstrating a dependent
evolutionary mechanism. Different isolates from the same strain family and isolates from different strain families were observed to converge to the same spoligotype pattern. In conclusion, the binary data of the spoligotype are unable to provide sufficient information to accurately establish genotypic relationships between
certain clinical isolates ofM. tuberculosis. This has important implications for molecular epidemiologic strain
tracking and for the application of spoligotype data to phylogenetic analysis ofM. tuberculosisisolates.
Sequencing of theMycobacterium tuberculosis genome has identified a number of different repeat sequences (5). Perhaps the most intensively investigated repeat is the direct repeat (DR) (15). This region has a unique structure, comprising directly repeated sequences interspersed with unique variable sequences (15), which in combination have been termed the direct variable repeat (DVR) sequences (13). While the func-tion of this region of theM. tuberculosisgenome remains to be determined, its preservation throughout the evolutionary his-tory of the M. tuberculosiscomplex argues for a functionally important domain.
Repeated sequences such as the IS6110element and DR are the most commonly used targets for the molecular typing ofM.
tuberculosisisolates. IS6110is a transposable element that may
be repeated up to 25 times perM. tuberculosisgenome (31). Transposition is thought to be random, generating a diverse array of insertions throughout the chromosomes of different clinical isolates. The polymorphic nature of the IS6110 inser-tions and the DR region has been exploited to create relatively reproducible bacterial “DNA fingerprints,” which are used for epidemiologic tracking (1, 21, 29, 31, 37). To simplify the DR typing technique, a PCR-based method termed spoligotyping
was developed, which determines the presence or absence of 43 DVR sequences in the DR region (13, 16). This method is widely used for epidemiological investigations (6, 22, 23, 26) and lends itself readily to interlaboratory and international strain comparisons (17, 24).
A recent study extended the application of spoligotyping beyond epidemiologic tracking to investigate its potential util-ity for phylogenetic reconstruction of clinical isolates from different geographical regions (25). This study predicts the existence of at least seven distinct evolutionary groups, provid-ing a scenario regardprovid-ing the global spread ofM. tuberculosis. Although this study provided new insights into the macroevo-lution of the DR region, the microevomacroevo-lution of the DR region in well-defined evolutionary lineages remains largely unknown. Only one study has investigated the evolution of the DR region within a limited number of closely related low-IS6110 -copy-number strains (10). In that study, it was suggested that the DR region evolved primarily through homologous recombination. However, these low-copy-number strains were collected from vastly different geographical regions, and therefore the ob-served polymorphisms probably represent ancient evolutionary events.
More recently, it has been shown that the low-copy-number strains are evolutionarily distinct from the high-copy-number strains (12, 36). Hence, it is not possible to assume that the evolutionary mechanisms seen in the low-copy-number strains are the same as those occurring in high-copy-number strains.
* Corresponding author. Mailing address: MRC Centre for Molec-ular and CellMolec-ular Biology, Department of Medical Biochemistry, Stel-lenbosch University, P.O. Box 19063, Tygerberg 7505, South Africa. Phone: 27 21 9389401. Fax: 27 21 9389467. E-mail: [email protected].
4457
on May 15, 2020 by guest
http://jcm.asm.org/
Therefore, the mechanism and rate of DR region evolution in a large number of isolates representing defined evolutionary lineages of high-copy-number strains remain to be fully deter-mined. This knowledge will be important for the accurate in-ference of phylogenetic relationships.
To further explore the evolution of the DR region, we have examined clinical isolates belonging to three strain families, grouped according to the similarity of the IS6110 banding pattern (36), as well as uniquely inherited polymorphisms (30, 33, 34). Isolates from each strain family were subjected to DR-restriction fragment length polymorphism (RFLP) analy-sis, spoligotyping, and partial DNA sequencing to identify the evolutionary mechanisms leading to the polymorphic variants observed in the DR and flanking regions. We demonstrate that the DR region evolves by deletion of DVR sequences and by IS6110-mediated mutation. IS6110 insertion strongly influ-ences the interpretation of spoligotype data, while mapping of the insertion points suggests the presence of preferential inte-gration sites. Recombination between adjacent IS6110 ele-ments or adjacent repeat sequences may lead to evolutionary convergence of DR-based genotypes. This has important im-plications for the interpretation of phylogenetic predictions.
MATERIALS AND METHODS
Study population.M. tuberculosisisolates were collected from patients visiting primary health care facilities in Cape Town, South Africa (2).
DNA fingerprinting.Each isolate was classified by DNA fingerprinting with the internationally standardized protocol (31). ThePvuII Southern blots were sequentially hybridized with the enhanced chemiluminescence (ECL)-labeled probes IS-3⬘(31), IS-5⬘(35), and DRr (15), each probe being stripped from the membrane by denaturation before the next probe was applied. The IS-3⬘, IS-5⬘, and DRr autoradiographs were scanned and normalized with GelCompar 4.0 to support accurate positioning of the respective hybridizing bands.
Strain family groupings were defined by isolates having an IS6110Dice simi-larity index of⬎65% (36), where the index is calculated as the sum of the total number of matching bands divided by the total number of bands in each isolate. The delineation of the strain family groups used in this study was supported by previously identified polymorphisms uniquely associated with each IS6110 -de-fined group (30, 33, 34). Each polymorphism is assumed to reflect a discrete evolutionary event, which is then inherited in subsequent generations. Therefore, a collection of isolates with an identical polymorphism is thought to represent clonal expansion from a common progenitor.
All isolates were classified according to polymorphisms inkatGandgyrA(27) with the dot blot hybridization method (36).
DR-RFLP analysis.In order to identify mutations leading to polymorphisms within the direct repeat locus and surrounding genome, the DRr DNA finger-prints of isolates grouped into strain families were analyzed as previously de-scribed (35). Briefly, thePvuII DRr DNA fingerprints were superimposed on the IS-3⬘and IS-5⬘DNA fingerprints to identify cohybridizing bands. Such cohybrid-ization was indicative of an IS6110insertion(s) within the direct repeat region. If cohybridization could not be demonstrated, this was indicative of the absence of an IS6110element in the DR region. A single IS6110insertion in the DR region was characterized by two DRr-hybridizing bands, while two IS6110insertions were identified by the presence of three DRr-hybridizing bands, since each IS6110insertion generates an additionalPvuII restriction site. Direct repeats of the IS6110elements were characterized by cohybridization of both the IS-5⬘and IS-3⬘probes with one of the DRr-containing fragments, while inverted repeats of the IS6110elements were characterized by cohybridization of the IS-3⬘probe with only one DRr-containing fragment, together with cohybridization of the IS-5⬘probe with the remaining two DRr fragments, or vice versa.
Spoligotyping.M. tuberculosisisolates were spoligotyped with the internation-ally standardized PCR protocol in combination with primers DRa (GGT TTT GGG TCT GAC GAC, 5⬘biotinylated) and DRb (CCG AGA GGG GAC GGA AAC) (13, 16). PCR amplifications were conducted in physically separated workstations to prevent contamination. Negative water controls were PCR am-plified and included on each blot to identify any possible amplicon contamina-tion. In addition, H37Rv DNA was amplified and included on each blot to
determine the reproducibility of the spoligotyping method. To determine whether the spoligotypes identified in this study were found in other geograph-ical regions, they were visually compared with the spoligotypes deposited in the worldwide database (25).
Sequencing.In order to determine the point of IS6110integration in the DR region or flanking regions, the chromosomal domain between the ancestral IS6110element (7) and the second IS6110element was PCR amplified with outward-reading primers complementary to the 3⬘(TTCAACCATCGCCGCC TCTACC) and 5⬘(GGTACCTCCTCGATGAACCAC) regions of the IS6110
element. Alternatively, the region between the inserted IS6110element and a flanking genomic sequence was PCR amplified with the above IS6110primers in combination with primers complementary to the 3⬘-flanking region (GCCGAA GTCACGGCAGACTG) or 5⬘-flanking region (CCTTGCTGTCCCGCCAA TAC or CAGCGCAGAGGAGTTTGTG) .
The PCR products were then cloned into the vector pGEMTeasy (Promega) according to the manufacturer’s instructions, and the inserts were sequenced with the T7 and SP6 primers. These sequences were deposited in GenBank with the following accession numbers: isolate 108 (AY099013); isolate 118 (AY052534, AY052535); isolate 125 (AY052536, AY052537); isolate 176 (AF390058, AF390059, AF390060, AF390061, AF390062, AF390063); isolate 211 (AY099015, AY099018, AY099019, AY099021, AY099023); isolate 235 (AY053426, AY053427, AY053428); isolate 278 (AF504308, AF504309); iso-late 286 (AY053429, AY053430, AY053431); isoiso-late 305 (AF504310, AF504311); isolate 348 (AF390057, AF390056, AF390055); isolate 392 (AF390069, AF390070, AF390071); isolate 397 (AF390064, AF390065, AF390066, AF390067,AF390068); isolate 453 (AY053432, AY053433, AY053434, AY099016); isolate 480 (AY053435, AY053436, AY053437, AY053438, AY053439); isolate 602 (AF421346); isolate 677 (AY099014, AY099020); isolate 704 (AF390047, AF390048, AF390049, AF390050); isolate 780 (AF390039S1, AF390039S2); isolate 907 (AY099008, AY099011, AY099012); isolate 973 (AF390051, AF390052, AF390053, AF390054); isolate 1124 (AF421344, AF421345); isolate 1227 (AF390041, AF390042, AF390043, AF390044, AF390045, AF390046); isolate 1633 (AY099009 AY099010); iso-late 1662 (AY099022); isoiso-late 1764 (AY099017, AY099024); and isoiso-late 1985 (AF411183, AF411182).
The BlastN (http://www.ncbi. nlm.nih.gov) algorithm was used to identify the positions of IS6110insertions and to localize DVR deletions and mutations in relation to the genome sequence of H37Rv (cosmid MTCY16B7, accession number Z81331) (5).
RESULTS
A total of 379 high-IS6110-copy-number strains (⬎5 IS6110
insertions) were identified in the study setting during the pe-riod from mid-1992 to the end of 1998. Each high-copy-num-ber strain has a distinct IS6110banding pattern. One hundred and seventy-six of these strains were grouped into three strain families (F11, F28, and Beijing) (Fig. 1), according to a Dice similarity index of⬎65% (36). These groupings were support-ed by chromosomal polymorphisms unique to each IS6110 -defined group. Isolates grouped as strain family F11 showed a C-to-T transition at position 491 of the rrs gene (34), while isolates grouped as strain family F28 showed a G-to-A transi-tion at positransi-tion 619 in the Rv3566c gene (30). The Beijing family isolates were characterized by the absence of the ISL540 sequence (33).
Strain family F11 was represented by 97 different IS6110
banding patterns, family F28 was represented by 41 patterns; and the Beijing family was represented by 38 patterns (Table 1). These strain families represent the three largest groupings found in the study community (36).katG and gyrA polymor-phism data (27) showed that strain families F11 and F28 were part of genetic group 2 (Table 1), although phylogenetic anal-ysis has inferred that these strain families evolved indepen-dently and that they do not share a recent progenitor (36). The Beijing family was part of genetic group 1 (27) (Table 1).
Spoligotyping (13, 16) of the DR regions was used to deter-mine the presence or absence of the 43 DVR sequences (Fig.
on May 15, 2020 by guest
http://jcm.asm.org/
2). Strain families F11 and F28 each showed a total of 14 different spoligotypes, while the Beijing family showed only a single spoligotype (Table 1 and Fig. 2).
The spoligotype pattern evolved primarily by the deletion of
[image:3.603.103.486.67.515.2]single DVRs, although, in 11 instances, contiguous DVRs were deleted (Fig. 2). The absence of intermediate DR structures suggests that the deletion of adjacent DVR sequences oc-curred as single deletion events rather than as a series of
FIG. 1. Dendrogram showing the relative IS6110band sizes of isolates belonging to strain families F11, F28, and Beijing.M. tuberculosisisolates were genotypically classified by RFLP analysis with the internationally standardized method together with the IS6110probe (31). Autoradiographs were normalized by GelCompar 4.1 software, and cluster analysis was done with the unweighted pair group method with arithmetic mean and Dice coefficient (14). Strain family groupings were defined by isolates having an IS6110Dice similarity index of⬎65% (36).
TABLE 1. Polymorphic nature of DR regions from three independently evolving strain families Strain
family No. of ISvariants6110 Geneticgroupa worldwide databaseSpoligotype in b spoligotypesNo. of No. of DR-RFLPvariants Combined no. ofDR variants Ratio of DR variantsto IS6110variants
F11 97 2 S103 (33) 14 18 21 0.217
F28 41 2 S19 (34) 14 12 16 0.390
Beijing 38 1 S1 (1) 1 2 2 0.053
aGenetic group classified according to mutations in thekatGandgyrAgenes (27).
bThe first entry is the spoligotype classification from reference 22; the spoligotype classification from reference 25 is shown in parentheses.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.603.44.544.651.708.2]FIG.
2.
Spoligotype
patterns
of
three
independently
evolving
strain
families.
Each
isolate
was
spoligotyped
with
the
internationally
standardize
d
method
(13,
16).
The
DVRs
are
numbered
according
to
the
variable
sequences
immobilized
on
the
membrane.
Open
squares
indicate
the
absence
of
a
speci
fic
DVR,
and
solid
squares
indicate
the
presence
of
a
speci
fic
DVR.
Shaded
squares
indicate
DVR
sequences
not
detected
by
spoligotyping
but
shown
to
be
present
by
DNA
sequencing.
The
ancestral
IS
6110
insertion
is
located
in
DVR
24
at
position
15600
in
the
H37Rv
genome
sequence
(cosmid
MTCY16B7)
(5).
Each
DR-RFLP
genotype
is
indicated
by
a
number.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:4.603.65.482.128.681.2]sequential deletion events. The deletion of the DVR se-quences appeared to be random, although there was a slight preference for the deletion of DVR sequences in the region 5⬘
to the ancestral IS6110 element (between DVRs 1 and 24) (Fig. 2).
Many of the isolates in each IS6110strain family were char-acterized by specific DVR deletions (Fig. 2), suggesting that these isolates have a common ancestor. Identical spoligotypes were identified in isolates in the worldwide spoligotype data-base (22, 25) (Table 1), suggesting that the strain families in our study could be globally distributed or that the spoligotypes observed in these databases arose convergently in different strains.
DR-RFLP analysis enabled an overview of the structure of the DR region and adjacent flanking regions. Strain family F11 strains showed 18 DR-RFLP variants, while family F28 showed 12 DR-RFLP variants. In contrast, the Beijing family showed only two DR-RFLP variants (Table 1). Comparison between the DR-RFLP data and the spoligotype data demonstrated that certain evolutionary mechanisms were not detected by the spoligotyping method and that the regions flanking the DR region were also subject to evolution (Fig. 3). In combination, the DR-RFLP and spoligotype data demonstrate that this chromosomal region has undergone substantial evolutionary change, although at a rate lower than that for IS6110(see ratio of DR variants to IS6110variants, Table 1).
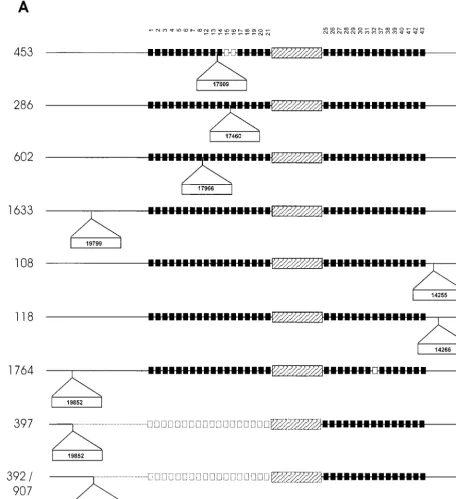
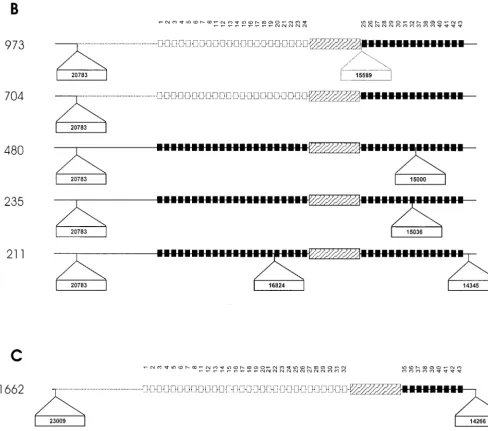
To identify the mechanisms leading to the different poly-morphic variants, chromosomal domains adjacent to the an-cestral IS6110insertion element as well as domains adjacent to additional IS6110elements were cloned and sequenced. Com-parison of the DNA sequences with the DNA sequence of the DR region of H37Rv (cosmid MTCY16B7) suggests that the duplication of DVR sequences was rare, as only one event was identified (isolate 453 contains a duplication of DVRs 17 and 18). Single-nucleotide polymorphisms were identified in six isolates, occurring in both the direct repeat and variable se-quences. Sequence analysis showed that in seven cases, the DR region had been disrupted by a second IS6110insertion (Fig. 3). In strain family F11, the additional DR IS6110insertions occurred between DVR 8 and DVR 21 (Fig. 3A, isolates 286, 453, and 602), while in family F28, the additional DR IS6110
insertions occurred between DVR 25 and DVR 37 (Fig. 3B, isolates 235, 480, and 973).
These IS6110insertions play an important role in the for-mulation of the spoligotype pattern. In two instances, the IS6110element was inserted in the variable spacer sequence, preventing hybridization of the amplified region to the immo-bilized complementary sequence (Fig. 2, IS6110 insertion in DVR 21 [F11 isolates] and isolate 235). In four instances, the IS6110element was asymmetrically inserted in the direct re-peat sequence, inhibiting PCR amplification of the adjacent variable sequence (Fig. 2, isolates 286, 480, 602, and 973) (11). The asymmetrical insertion of the IS6110element in the DR region of isolate 286 (Fig. 3A) led to the apparent evolution of a spoligotype pattern that was identical to the spoligotype seen in isolate 453 (Fig. 2).
The close proximity of the IS6110insertions to each other suggests the presence of two different preferential IS6110 in-tegration loci in the DR region as well as preferential integra-tion loci in both the 3⬘- and 5⬘-flanking regions (Fig. 3). The
IS6110insertion in the 5⬘-flanking regions appears to mediate recombination with an IS6110 insertion in the DR region, leading to partial deletion of the DR region (Fig. 3, isolates 392, 397, 704, 907, and 973) (Sampson et al., submitted for publication). This is supported by the absence of the 3-bp duplication at the terminus of the IS6110element (4, 8). In these strains, the terminal 3 bp correspond to the 3-bp direct repeat of the ancestral IS6110element and of the 5⬘-flanking IS6110insertion. These large deletion events affect the spoli-gotype pattern, resulting in the convergent evolution of the spoligotype in isolates from different evolutionary lineages (Fig. 2, 3A, and 3B, compare isolates 704 with 392, 397, and 907).
Convergence of the spoligotype was also observed in isolates within a single evolutionary lineage (Fig. 2, compare isolates 392 and 397). In these isolates, recombination occurred be-tween the ancestral IS6110element and a 5⬘-flanking IS6110
element located in different chromosomal positions in the dif-ferent strains (Fig. 3A). In addition, convergence via recombi-nation is predicted to have occurred as parallel events between the ancestral and the same 5⬘-flanking IS6110insertion in two different isolates (392 and 907) (Fig. 2 and 3A). These isolates are positioned on different branches within the same evolu-tionary lineage (Fig. 1).
DISCUSSION
The DR region has become an important genomic locus which can be used to study the evolutionary history of clinical isolates ofM. tuberculosis. Phylogenetic analysis has provided new insights into the macroevolution of distinct clades and their global dissemination (25). However, the factors influenc-ing microevolution are not fully understood, and therefore their influence on phylogenetic predictions remains to be de-termined. In this study we have analyzed the structure of the DR region in three independently evolving strain families (36). The large number of observed DR variants suggests that the DR region is evolving more rapidly than was previously pre-dicted (10), although this rate is significantly slower than that of IS6110, as demonstrated by the ratio of DR variants to IS6110variants. Within the study population, the frequency of change appears to be strain family specific, which may suggest that the mechanisms leading to change are dependent either on the structure of the DR region or on the overall mutation rate of the strain family. This is particularly evident in the Beijing strain family, where the DR region appears to be in a (evolutionarily) fixed state (32). Only three spoligotype vari-ants have been previously documented in the Beijing family (3, 19).
Analysis of the data presented in this study supports sugges-tions that the DR region evolves by at least four different mechanisms (10, 32); IS6110-mediated mutation, homologous recombination between repeat sequences leading to DVR de-letion, strand slippage during replication leading to duplication of DVR sequences, and point mutation. In agreement with a previous report (32), the duplication of DVR sequences and point mutations was found to be rare. Conversely, the fre-quency of DVR deletions was high. From this study, it is evident that the deletion of specific DVR sequences generated spoligotype “signatures” which appear to be unique to a strain
on May 15, 2020 by guest
http://jcm.asm.org/
family (Fig. 2). Comparison between the spoligotype patterns identified in this study and those deposited in the worldwide database (22, 25) identified spoligotype patterns that were ei-ther identical or highly similar. This suggests that the strain families are widely disseminated, but the correlation between spoligotype signature and strain family needs further
investi-gation, as it is possible that the spoligotypes deposited in the different databases have arisen convergently in different strains. Furthermore, it is not known how the accumulation of mutations in the DR region will disturb such signatures.
[image:6.603.63.519.73.572.2]The evolution of the spoligotype pattern appears to occur via the deletion of single DVRs or contiguous DVR sequences.
FIG. 3. Schematic diagram showing points of IS6110insertion in the DR region and flanking regions of isolates from three independently evolving strain families. (A) DR region ofM. tuberculosisisolates belonging to strain family F11. (B) DR region ofM. tuberculosisisolates belonging to strain family F28. (C) DR region ofM. tuberculosisisolates belonging to the Beijing family. Solid squares depict the presence of specific DVRs in the DR region, while open squares depict the deletion of specific DVRs. The DVRs are numbered according to the variable sequences immobilized on the membrane. The ancestral IS6110inserted in the DR region is depicted by the hatched rectangular box, while newly inserted IS6110elements are depicted as outset open rectangular boxes (position of insertion is indicated in the rectangle). Dotted blocks and lines represent deleted elements and flanking regions, respectively.
on May 15, 2020 by guest
http://jcm.asm.org/
In this study, isolates representing intermediate numbers of DVR deletions were not observed, suggesting that the deletion of contiguous DVR sequences does not occur via a process of sequential deletion but rather via a single deletion event span-ning a number of DVRs. This mechanism has been previously proposed to explain the deletion of contiguous DVR se-quences (32) or DVR sese-quences in association with an (inter-nal) IS6110element (10). This implies that certain deletions of contiguous DVR sequences will occur in a dependent manner, which may restrict the use of spoligotype data to accurately predict evolutionary relationships. Most of the algorithms used to calculate phylogenetic trees are based on the assumption that the markers evolve independently (28). Furthermore, the sampling methods used to gain statistical support for inferred phylogenies are dependent on the markers’ evolving indepen-dently.
In contrast to previous studies, we show that IS6110- medi-ated mutation plays a significant role in the evolution of the spoligotype pattern. Mapping of the IS6110 insertion points
demonstrates the existence of preferential IS6110integration loci (9, 11, 15, 20). Two of these loci were located within the DR region, while a further two preferential integration loci were located in the 3⬘- and 5⬘-flanking regions. The preferen-tial integration loci in the DR region appear to be strain family specific, suggesting that the structure of the DR region may influence subsequent evolutionary events. The insertion of IS6110into repeat sequences in the DR region resulted in the apparent deletion of DVR sequences according to the spoli-gotype data (11, 19). However, DNA sequencing of these re-gions showed that the DVR sequences were present and that PCR amplification was inhibited by the insertion of the IS6110
element in the priming region (18).
[image:7.603.48.537.71.502.2]An alternative mechanism was also identified by which the IS6110element was inserted into the variable spacer sequence, preventing hybridization. The identification of these additional evolutionary mechanisms implies that the spoligotype pattern reflects an overrepresentation of the frequency of homologous recombination events occurring within the DR region. The
FIG. 3—Continued.
on May 15, 2020 by guest
http://jcm.asm.org/
inability of the spoligotype pattern to differentiate between the evolutionary mechanisms leading to the apparent loss of DVR sequences has important consequences for tracking of strains in epidemiological studies. Clustering algorithms will group such isolates as identical even though they have evolved inde-pendently by different mechanisms. This will influence the ac-curacy of both epidemiologic and phylogenetic studies based only on spoligotype data.
Homologous recombination between adjacent IS6110 ele-ments (4, 8) could explain the deletion of large portions of the DR region and 5⬘-flanking region (Sampson et al., submitted), thereby strongly influencing the spoligotype pattern. This evo-lutionary mechanism generated identical spoligotype patterns in different strain families, demonstrating convergent evolution of the spoligotype pattern. This also has important implications for phylogenetic predictions, as isolates will be seen as geno-typically identical even though they have evolved indepen-dently and belong to different evolutionary lineages. The true extent of convergence of spoligotype patterns or subsets of the spoligotype pattern is unknown and will depend on the fre-quency at which specific DVRs or contiguous DVRs are de-leted by homologous recombination. The significance of such events on phylogenetic reconstructions will vary depending on the number of DVRs deleted as well as the complexity of the underlying spoligotype. The lower the complexity, the greater the influence of convergent events on the phylogenetic recon-struction, while the greater the number of contiguous DVRs deleted, the greater the chance of convergence.
From this and other studies (10, 32), it is clear that the evolutionary process generates a substantial number of variant DR structures. However, this study implies that the binary data of the spoligotype are unable to provide sufficient information to accurately establish the evolutionary relationship between certain clinical isolates ofM. tuberculosis. To gain further in-sights into the evolutionary history ofM. tuberculosisisolates, we recommend the use of other genotyping methods, in asso-ciation with spoligotyping, to enhance the accuracy of geno-typic classification.
ACKNOWLEDGMENTS
This work was made possible by grants from the GlaxoSmithKline Action TB Initiative, the Sequella Global Tuberculosis Foundation, IAEA (projects SAF6/003 and CRP 9925), the Harry Crossely Foun-dation, and the National Research Foundation (THRP).
E. Engelke, S. Carlini, and M. De Kock are thanked for technical assistance.
REFERENCES
1. Alland, D., G. E. Kalkut, A. R. Moss, R. A. McAdam, J. A. Hahn, W. Bosworth, E. Drucker, and B. R. Bloom.1994. Transmission of tuberculosis in New York City. An analysis by DNA fingerprinting and conventional epidemiologic methods. N. Engl. J. Med.330:1710–1716.
2. Beyers, N., R. P. Gie, H. L. Zietsman, M. Kunneke, J. Hauman, M. Tatley, and P. R. Donald.1996. The use of a geographical information system (GIS) to evaluate the distribution of tuberculosis in a high-incidence community. S. Afr. Med. J.86:40–41, 44.
3. Bifani, P., B. Mathema, M. Campo, S. Moghazeh, B. Nivin, E. Shashkina, J. Driscoll, S. S. Munsiff, R. Frothingham, and B. N. Kreiswirth.2001. Mo-lecular identification of streptomycin monoresistantMycobacterium tubercu-losisrelated to multidrug-resistant W strain. Emerg. Infect. Dis.7:842–848. 4. Brosch, R., W. J. Philipp, E. Stavropoulos, M. J. Colston, S. T. Cole, and S. V. Gordon.1999. Genomic analysis reveals variation between Mycobacte-rium tuberculosisH37Rv and the attenuatedM. tuberculosisH37Ra strain. Infect. Immun.67:5768–5774.
5. Cole, S. T., R. Brosch, J. Parkhill, T. Garnier, C. Churcher, D. Harris, S. V.
Gordon, K. Eiglmeier, S. Gas, C. E. Barry III, F. Tekaia, K. Badcock, D. Basham, D. Brown, T. Chillingworth, R. Connor, R. Davies, K. Devlin, T. Feltwell, S. Gentles, N. Hamlin, S. Holroyd, T. Hornsby, K. Jagels, B. G. Barrell, et al.1998. Deciphering the biology ofMycobacterium tuberculosis
from the complete genome sequence. Nature393:537–544.
6. Cronin, W. A., J. E. Golub, L. S. Magder, N. G. Baruch, M. J. Lathan, L. N. Mukasa, N. Hooper, J. H. Razeq, D. Mulcahy, W. H. Benjamin, and W. R. Bishai.2001. Epidemiologic usefulness of spoligotyping for secondary typing ofMycobacterium tuberculosis isolates with low-copy-numbers of IS6110. J. Clin. Microbiol.39:3709–3711.
7. Dale, J. W.1995. Mobile genetic elements in mycobacteria. Eur. Respir. J. Suppl.20:633s-648s.
8. Fang, Z., C. Doig, D. T. Kenna, N. Smittipat, P. Palittapongarnpim, B. Watt, and K. J. Forbes.1999. IS6110-mediated deletions of wild-type chromo-somes ofMycobacterium tuberculosis. J. Bacteriol.181:1014–1020. 9. Fang, Z., and K. J. Forbes.1997. A Mycobacterium tuberculosis IS6110
preferential locus (ipl) for insertion into the genome. J. Clin. Microbiol.35:
479–481.
10. Fang, Z., N. Morrison, B. Watt, C. Doig, and K. J. Forbes.1998. IS6110
transposition and evolutionary scenario of the direct repeat locus in a group of closely relatedMycobacterium tuberculosisstrains. J. Bacteriol.180:2102– 2109.
11. Filliol, I., C. Sola, and N. Rastogi.2000. Detection of a previously unampli-fied spacer within the DR locus ofMycobacterium tuberculosis: epidemiolog-ical implications. J. Clin. Microbiol.38:1231–1234.
12. Fomukong, N., M. Beggs, H. el Hajj, G. Templeton, K. Eisenach, and M. D. Cave.1997. Differences in the prevalence of IS6110insertion sites in Myco-bacterium tuberculosisstrains: low and high-copy-number of IS6110. Tuber. Lung Dis.78:109–116.
13. Groenen, P. M., A. E. Bunschoten, D. van Soolingen, and J. D. van Embden.
1993. Nature of DNA polymorphism in the direct repeat cluster of Myco-bacterium tuberculosis; application for strain differentiation by a novel typing method. Mol. Microbiol.10:1057–1065.
14. Hermans, P. W., F. Messadi, H. Guebrexabher, D. van Soolingen, P. E. de Haas, H. Heersma, H. de Neeling, A. Ayoub, F. Portaels, and D. Frommel.
1995. Analysis of the population structure ofMycobacterium tuberculosisin Ethiopia, Tunisia, and the Netherlands: usefulness of DNA typing for global tuberculosis epidemiology. J. Infect. Dis.171:1504–1513.
15. Hermans, P. W., D. van Soolingen, E. M. Bik, P. E. de Haas, J. W. Dale, and J. D. van Embden.1991. Insertion element IS987fromMycobacterium bovis
BCG is located in a hot-spot integration region for insertion elements in
Mycobacterium tuberculosiscomplex strains. Infect. Immun.59:2695–2705. 16. Kamerbeek, J., L. Schouls, A. Kolk, M. van Agterveld, D. van Soolingen, S.
Kuijper, A. Bunschoten, H. Molhuizen, R. Shaw, M. Goyal, and J. Van Embden.1997. Simultaneous detection and strain differentiation of Myco-bacterium tuberculosisfor diagnosis and epidemiology. J. Clin. Microbiol.
35:907–914.
17. Kremer, K., D. van Soolingen, R. Frothingham, W. H. Haas, P. W. Hermans, C. Martin, P. Palittapongarnpim, B. B. Plikaytis, L. W. Riley, M. A. Yakrus, J. M. Musser, and J. D. van Embden.1999. Comparison of methods based on different molecular epidemiological markers for typing ofMycobacterium tuberculosiscomplex strains: interlaboratory study of discriminatory power and reproducibility. J. Clin. Microbiol.37:2607–2618.
18. Legrand, E., I. Filliol, C. Sola, and N. Rastogi.2001. Use of spoligotyping to study the evolution of the direct repeat locus by IS6110transposition in
Mycobacterium tuberculosis. J. Clin. Microbiol.39:1595–1599.
19. Mokrousov, I., O. Narvskaya, E. Limeschenko, T. Otten, and B. Vyshnevskiy.
2002. Novel IS6110insertion sites in the direct repeat locus of Mycobacte-rium tuberculosisclinical strains from the St. Petersburg area of Russia and evolutionary and epidemiological considerations. J. Clin. Microbiol. 40:
1504–1507.
20. Sampson, S. L., R. M. Warren, M. Richardson, G. D. van der Spuy, and P. D. van Helden.1999. Disruption of coding regions by IS6110insertion in My-cobacterium tuberculosis. Tuber. Lung Dis.79:349–359.
21. Small, P. M., P. C. Hopewell, S. P. Singh, A. Paz, J. Parsonnet, D. C. Ruston, G. F. Schecter, C. L. Daley, and G. K. Schoolnik.1994. The epidemiology of tuberculosis in San Francisco. A population-based study with conventional and molecular methods. N. Engl. J. Med.330:1703–1709.
22. Soini, H., X. Pan, A. Amin, E. A. Graviss, A. Siddiqui, and J. M. Musser.
2000. Characterization ofMycobacterium tuberculosisisolates from patients in Houston, Texas, by spoligotyping. J. Clin. Microbiol.38:669–676. 23. Soini, H., X. Pan, L. Teeter, J. M. Musser, and E. A. Graviss.2001.
Trans-mission dynamics and molecular characterization ofMycobacterium tubercu-losisisolates with low-copy-numbers of IS6110. J. Clin. Microbiol.39:217– 221.
24. Sola, C., A. Devallois, L. Horgen, J. Maisetti, I. Filliol, E. Legrand, and N. Rastogi.1999. Tuberculosis in the Caribbean: with spacer oligonucleotide typing to understand strain origin and transmission. Emerg. Infect. Dis.
5:404–414.
25. Sola, C., I. Filliol, M. C. Gutierrez, I. Mokrousov, V. Vincent, and N. Rastogi.
2001. Spoligotype database ofMycobacterium tuberculosis: biogeographic
on May 15, 2020 by guest
http://jcm.asm.org/
distribution of shared types and epidemiologic and phylogenetic perspec-tives. Emerg. Infect. Dis.7:390–396.
26. Sola, C., L. Horgen, J. Maisetti, A. Devallois, K. S. Goh, and N. Rastogi.
1998. Spoligotyping followed by double-repetitive-element PCR as rapid alternative to IS6110fingerprinting for epidemiological studies of tubercu-losis. J. Clin. Microbiol.36:1122–1124.
27. Sreevatsan, S., X. Pan, K. E. Stockbauer, N. D. Connell, B. N. Kreiswirth, T. S. Whittam, and J. M. Musser.1997. Restricted structural gene polymor-phism in theMycobacterium tuberculosiscomplex indicates evolutionarily recent global dissemination. Proc. Natl. Acad. Sci. USA94:9869–9874. 28. Swofford, D. L., G. J. Olsen, P. J. Waddell, and D. M. Hillis.1996.
Phylo-genetic inference, p. 407–514.InD. M. Hillis, C. Moritz, and B. K. Mable (ed.), Molecular systematics. Sinauer Associates, Boston, Mass.
29. Torrea, G., G. Levee, P. Grimont, C. Martin, S. Chanteau, and B. Gicquel.
1995. Chromosomal DNA fingerprinting analysis with the insertion sequence IS6110and the repetitive element DR as strain-specific markers for epide-miological study of tuberculosis in French Polynesia. J. Clin. Microbiol.
33:1899–1904.
30. Upton, A. M., A. Mushtaq, T. C. Victor, S. L. Sampson, J. Sandy, D. M. Smith, P. V. van Helden, and E. Sim.2001. Arylamine N-acetyltransferase of
Mycobacterium tuberculosisis a polymorphic enzyme and a site of isoniazid metabolism. Mol. Microbiol.42:309–317.
31. van Embden, J. D., M. D. Cave, J. T. Crawford, J. W. Dale, K. D. Eisenach, B. Gicquel, P. Hermans, C. Martin, R. McAdam, and T. M. Shinnick.1993. Strain identification ofMycobacterium tuberculosisby DNA fingerprinting: recommendations for a standardized methodology. J. Clin. Microbiol.31:
406–409.
32. van Embden, J. D., T. van Gorkom, K. Kremer, R. Jansen, B. A. Der Zeijst,
and L. M. Schouls.2000. Genetic variation and evolutionary origin of the direct repeat locus ofMycobacterium tuberculosiscomplex bacteria. J. Bac-teriol.182:2393–2401.
33. van Rie, A., R. M. Warren, N. Beyers, R. P. Gie, C. N. Classen, M. Rich-ardson, S. L. Sampson, T. C. Victor, and P. D. van Helden.1999. Transmis-sion of a multidrug-resistantMycobacterium tuberculosisstrain resembling “strain W” among noninstitutionalized, human immunodeficiency virus-se-ronegative patients. J. Infect. Dis.180:1608–1615.
34. Victor, T. C., A. van Rie, A. M. Jordaan, M. Richardson, G. D. Der Spuy, N. Beyers, P. D. van Helden, and R. Warren.2001. Sequence polymorphism in therrsgene ofMycobacterium tuberculosisis deeply rooted within an evolu-tionary clade and is not associated with streptomycin resistance. J. Clin. Microbiol.39:4184–4186.
35. Warren, R. M., M. Richardson, S. L. Sampson, G. D. van der Spuy, W. Bourn, J. H. Hauman, H. Heersma, W. Hide, N. Beyers, and P. D. van Helden..2001. Molecular evolution ofMycobacterium tuberculosis: phyloge-netic reconstruction of clonal expansion. Tuberculosis (Edinburgh)81:291– 302.
36. Warren, R. M., S. L. Sampson, M. Richardson, G. D. van der Spuy, C. J. Lombard, T. C. Victor, and P. D. van Helden.2000. Mapping of IS6110 -flanking regions in clinical isolates ofM. tuberculosisdemonstrates genome plasticity. Mol. Microbiol.37:1405–1416.
37. Yang, Z. H., P. E. de Haas, D. van Soolingen, J. D. van Embden, and A. B. Andersen.1994. Restriction fragment length polymorphismMycobacterium tuberculosisstrains isolated from Greenland during 1992: evidence of tuber-culosis transmission between Greenland and Denmark. J. Clin. Microbiol.
32:3018–3025.