organic papers
o1200
Sonodaet al. C18H14N2O4 doi:10.1107/S1600536805009736 Acta Cryst.(2005). E61, o1200–o1202 Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
(
E
,
E
,
E
)-1,6-Bis(4-nitrophenyl)hexa-1,3,5-triene
Yoriko Sonoda,a* Yuji
Kawanishiaand Midori Gotob
a
Nanotechnology Research Institute, National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan, andbTechnical Centre,
National Institute of Advanced Industrial Science and Technology (AIST), 1-1-1 Higashi, Tsukuba, Ibaraki 305-8565, Japan
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 183 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.040
wRfactor = 0.126
Data-to-parameter ratio = 16.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
In the title compound, C18H14N2O4, the torsion angles of the
single bonds between the hexatriene chain and 4-nitrophenyl rings are 18.0 (2) and 0.7 (2). The molecules are linked
weakly via intermolecular C—H O hydrogen bonds to form sheets. These sheets are linked further by N O dipole interactions between the nitro groups and aromatic –
stacking interactions, to form a three-dimensional framework.
Comment
(E,E,E)-1,6-Diphenylhexa-1,3,5-triene [(E,E,E)-DPH] has been widely used as a fluorescence probe in photochemical and biological studies. Due to the extended -conjugation systems, DPH and its ring-substituted derivatives are also attractive in the field of applied physics, because of their potential use as materials for third-order nonlinear optics and optical power limiting, and as two-photon absorbing chromophores (Rodenberger et al., 1995; Spangler, 1999; Rumi et al., 2000; Geskin et al., 2003). We have previously reported the crystal structure of 1,6-bis(2,4-dichlorophenyl)-hexa-1,3,5-triene (Sonoda, Kawanishi & Goto, 2003). In this paper, we report the structure of the title compound, (I), which is a symmetrically substituted DPH having nitro groups on the benzene rings.
During our study of the solid-state fluorescence properties and photochemical behaviours of a series of ring-substituted DPHs, we found that (I) showed strongly red-shifted fluores-cence relative to that in solution (Sonoda, Kawanishi et al., 2003), and that it showed no photoreactivity in the solid state (Sonoda, Miyazawa et al., 2001). We undertook the crystal structure analysis of (I) in order to find an explanation for its red-shifted emission and photostability in the solid state. Our
[image:1.610.219.451.641.723.2]Received 14 March 2005 Accepted 29 March 2005 Online 31 March 2005
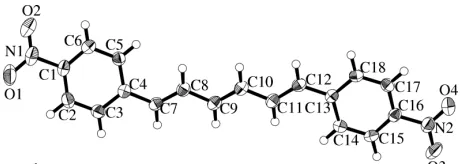
Figure 1
data can be compared with those for the (E,E,E)-isomers of DPH and 1,6-bis(2-methoxyphenyl)hexa-1,3,5-triene (Hall et al., 1989), 1,6-bis(4- and 1,6-bis(2-chlorophenyl)-3,4-dimethylhexa-1,3,5-triene (Stam & Riva di Sanseverino, 1966), and all-(E)-isomers of other diphenylpolyenes (Drenth & Wiebenga, 1954, 1955; Glaseret al., 2003).
The molecular structure of (I) is shown in Fig. 1 and selected geometric parameters are given in Table 1. In the hexatriene chain of (I), the torsion angles are almost 180,
indicating that the triene chain is planar. Distinct bond alter-nation is observed. Although the C—C single bonds [1.437 (2)–1.459 (2) A˚ ] are shorter than the standard value of 1.54 A˚ , they are significantly longer than the C C double bonds [1.340 (2)–1.342 (2) A˚ ]. Although C C bonds in conjugated olefins are expected to be longer than the standard value of 1.34 A˚ for isolated olefins, the C C bond lengths in (I) are all similar to the standard value for mono-olefins. This may be due to the thermal vibrations around the C—Ph and/or C—C bonds, as discussed previously for the structure of a 2,4-dichloro-derivative of DPH (Sonoda, Kawanishi & Goto, 2003). The C—C—C bond angles in the chain are all some-what larger than 120. In particular, the C4—C7—C8 and
C11—C12—C13 angles are 126.0 (1) and 127.4 (1),
respec-tively, in order to minimize the steric interaction between the H atoms of the chain and the benzene ring. The benzene rings are planar, and the nitro groups are coplanar with the rings.
The C5—C4—C7—C8 and C11—C12—C13—C14 torsion angles are 18.0 (2) and 0.7 (2), respectively. The
corre-sponding dihedral angles for the least-squares planes of the ring and the chain are 18.9 (1) and 2.4 (1), respectively. The
twist around the C4—C7 bond axis is probably due to inter-molecular interactions. The dihedral angles between the planes of the ring and the chain are 1.9 for unsubstituted
DPH, 15.6for its 2-methoxy-derivative (Hallet al., 1989) and
9.9 (1)for the 2,4-dichloro-derivative (Sonoda, Kawanishi &
Goto, 2003). It is interesting that the dihedral angle of
18.9 (1) for (I) is even larger than the values for the 2-methoxy- and 2,4-dichloro-derivatives, having a large steric hindrance between the olefinic H atoms and the 2-substituents of the rings.
Figs. 2 and 3 show the crystal structure of (I). Atoms O1, O2 and O3 of the nitro groups are in close contact with either a neighbouring aromatic H atom or with the olefinic H atom of a neighbouring molecule, to form weak C—H O hydrogen bonds (Table 2) of the type described by Desiraju & Steiner (1999). The molecules are linked weakly by these inter-molecular hydrogen bonds into sheets (Fig. 2). The shortest intermolecular distances between the H and O atoms and between the C and O atoms are H7 O3i = 2.44 A˚ and C7 O3i = 3.371 (2) A˚ , respectively, and the corresponding intermolecular angle is C7—H7 O3i= 166[symmetry code:
(i)x,y1 2,
1 2z].
The molecules are assembled along the a axis to form parallel plane-to-plane stacks. Attractive intermolecular interactions between the nitro O atoms and nitro N atoms of neighbouring molecules are observed. The sheets formed by the C—H O hydrogen bonds are linked by the inter-molecular N O dipole interactions and the aromatic –
stacking interactions into a three-dimensional framework (Fig. 3). The shortest intermolecular distance between the N and O atoms is N2 O3iv= 3.366 (2) A˚ [symmetry code: (iv) 1 +x,y,
z], which is comparable with the shortest C O distance of 3.371 (2) A˚ in the C—H O hydrogen bonds. The line of N2 O3iv makes an angle of ca 76 with the least-squares
plane defined by the nitrophenyl ring including atoms C13 and N2.
organic papers
Acta Cryst.(2005). E61, o1200–o1202 Sonodaet al. C
[image:2.610.344.538.71.322.2]18H14N2O4
o1201
Figure 2A packing diagram of (I), viewed along theaaxis. Intermolecular C— H O hydrogen bonds are represented by dashed lines. The shortest intermolecular distances between the H and O atoms and between the C and O atoms are H7 O3i = 2.44 A˚ and C7 O3i = 3.371 (2) A˚ , respectively, and the corresponding intermolecular angle is C7— H7 O3i= 166. [Symmetry codes: (i)x,y1
2, 1
2z, (ii)x2, 3 2
y,z1
2, (iii) 3x, 1y, 1z.]
Figure 3
[image:2.610.56.284.72.231.2]Experimental
Compound (I) was prepared according to the literature method of Spangleret al.(1989). Crystals of (I) were grown from an acetonitrile solution by slow evaporation (m.p. 513–514 K).
Crystal data
C18H14N2O4
Mr= 322.31 Monoclinic, P21=c a= 3.8710 (4) A˚
b= 21.440 (2) A˚
c= 18.7091 (19) A˚
= 95.555 (2)
V= 1545.4 (3) A˚3
Z= 4
Dx= 1.385 Mg m 3
MoKradiation Cell parameters from 3502
reflections
= 2.2–28.1 = 0.10 mm1
T= 183 (2) K Needle, yellow 0.300.250.02 mm
Data collection
Bruker SMART CCD area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.909,Tmax= 0.998
9325 measured reflections
3490 independent reflections 2640 reflections withI> 2(I)
Rint= 0.026 max= 28.2
h=5!5
k=28!20
l=22!23
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.040
wR(F2) = 0.126
S= 1.06 3490 reflections 218 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0743P)2
+ 0.088P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.002
max= 0.24 e A˚ 3
min=0.20 e A˚ 3
Extinction correction:SHELXTL
(Sheldrick, 2000)
[image:3.610.314.565.110.186.2]Extinction coefficient: 0.0075 (15)
Table 1
Selected geometric parameters (A˚ ,).
C1—N1 1.463 (2)
C4—C7 1.459 (2)
C7—C8 1.340 (2)
C8—C9 1.437 (2)
C9—C10 1.342 (2)
C10—C11 1.437 (2)
C11—C12 1.341 (2)
C12—C13 1.457 (2)
C16—N2 1.463 (2)
N1—O2 1.225 (2)
N1—O1 1.226 (2)
N2—O3 1.223 (2)
N2—O4 1.224 (2)
C3—C4—C5 117.9 (1)
C5—C4—C7 122.7 (1)
C8—C7—C4 126.0 (1)
C11—C12—C13 127.4 (1)
C14—C13—C18 118.0 (1)
C14—C13—C12 123.0 (1)
O2—N1—O1 123.3 (1)
O1—N1—C1 118.2 (1)
O3—N2—O4 123.4 (1)
O4—N2—C16 118.3 (1)
C5—C4—C7—C8 18.0 (2)
C4—C7—C8—C9 178.2 (1)
C7—C8—C9—C10 177.5 (1)
C8—C9—C10—C11 179.0 (1)
C9—C10—C11—C12 179.9 (1)
C10—C11—C12—C13 178.6 (1)
C11—C12—C13—C14 0.7 (2)
C6—C1—N1—O2 1.4 (2)
C2—C1—N1—O1 1.0 (2)
C15—C16—N2—O3 2.7 (2)
C17—C16—N2—O4 3.2 (2)
Table 2
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
C7—H7 O3i
0.95 2.44 3.371 (2) 166
C14—H14 O2ii
0.95 2.57 3.366 (2) 141
C15—H15 O1ii
0.95 2.61 3.454 (2) 148
C2—H2 O1iii
0.95 2.71 3.364 (2) 127
Symmetry codes: (i) x;y1 2;zþ
1
2; (ii) x2;yþ 3 2;z
1 2; (iii)
xþ3;yþ1;zþ1.
H atoms were positioned geometrically and treated as riding atoms, with C—H distances of 0.95A˚ and withUiso(H) = 1.2Ueq(C).
Data collection:SMART(Bruker, 2001); cell refinement:SAINT (Bruker, 2001); data reduction:SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2000); program(s) used to refine structure: SHELXTL; molecular graphics: TEXSAN (Molecular Structure Corporation, 1999); software used to prepare material for publication:SHELXTL.
References
Bruker (2001).SMART(Version 5.625) andSAINT(Version 6.22). Bruker AXS Inc., Madison, Wisconsin, USA.
Desiraju, G. R. & Steiner, T. (1999).The Weak Hydrogen Bond in Structural Chemistry and Biology. New York: Oxford University Press.
Drenth, W. & Wiebenga, E. H. (1954).Recl Trav. Chim. Pays-Bas,73, 218–228. Drenth, W. & Wiebenga, E. H. (1955).Acta Cryst.8, 755–760.
Geskin, V. M., Lambert, C. & Bre´das, J.-L. (2003).J. Am. Chem. Soc.125, 15651–15658.
Glaser, R., Dendi, L. R., Knotts, N. & Barnes, C. L. (2003).Cryst. Growth Des.
3, 291–300.
Hall, T., Bachrach, S. M., Spangler, C. W., Sapochak, L. S., Lin, C. T., Guan, H. W. & Rogers, R. D. (1989).Acta Cryst.C45, 1541–1543.
Molecular Structure Corporation (1999).TEXSAN. Version 1.11. MSC, 9009 New Trails Drive, The Woodlands, TX 77381-5209, USA.
Rodenberger, D. C., Heflin, J. R. & Garito, A. F. (1995).Phys. Rev. A,51, 3234–3245.
Rumi, M., Ehrlich, J. E., Heikal, A. A., Perry, J. W., Barlow, S., Hu, Z., McCord-Maughon, D., Parker, T. C., Ro¨ckel, H., Thayumanavan, S., Marder, S. R., Beljonne, D. & Bre´das, J.-L. (2000).J. Am. Chem. Soc.122, 9500–9510. Sheldrick, G. M. (1996).SADABS. University of Go¨ttingen, Germany. Sheldrick, G. M. (2000).SHELXTL. Version 6.12. Bruker AXS, Madison,
Wisconsin, USA.
Sonoda, Y., Kawanishi, Y. & Goto, M. (2003).Acta Cryst.C59, o311–o313. Sonoda, Y., Kawanishi, Y., Ikeda, T., Goto, M., Hayashi, S., Yoshida, Y.,
Tanigaki, N. & Yase, K. (2003).J. Phys. Chem. B,107, 3376–3383. Sonoda, Y., Miyazawa, A., Hayashi, S. & Sakuragi, M. (2001).Chem. Lett.pp.
410–411.
Spangler, C. W. (1999).J. Mater. Chem.9, 2013–2020.
Spangler, C. W., McCoy, R. K., Dembek, A. A., Sapochak, L. S. & Gates, B. D. (1989).J. Chem. Soc. Perkin Trans. 1.pp. 151–154.
Stam, C. H. & Riva di Sanseverino, L. (1966).Acta Cryst.21, 132–138.
organic papers
o1202
Sonodaet al. C [image:3.610.45.296.452.628.2]supporting information
sup-1 Acta Cryst. (2005). E61, o1200–o1202
supporting information
Acta Cryst. (2005). E61, o1200–o1202 [https://doi.org/10.1107/S1600536805009736]
(
E
,
E
,
E
)-1,6-Bis(4-nitrophenyl)hexa-1,3,5-triene
Yoriko Sonoda, Yuji Kawanishi and Midori Goto
(E,E,E)-1,6-Bis(4-nitrophenyl)hexa-1,3,5-triene
Crystal data
C18H14N2O4
Mr = 322.31 Monoclinic, P21/c
a = 3.8710 (4) Å
b = 21.440 (2) Å
c = 18.7091 (19) Å
β = 95.555 (2)°
V = 1545.4 (3) Å3
Z = 4
F(000) = 672
Dx = 1.385 Mg m−3
Melting point: 513-514 K K Mo Kα radiation, λ = 0.71073 Å Cell parameters from 3502 reflections
θ = 2.2–28.1°
µ = 0.10 mm−1
T = 183 K Needle, yellow 0.30 × 0.25 × 0.02 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: rotating anode Graphite monochromator
Detector resolution: 8.366 pixels mm-1
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.909, Tmax = 0.998
9325 measured reflections 3490 independent reflections 2640 reflections with I > 2σ(I)
Rint = 0.026
θmax = 28.2°, θmin = 1.5°
h = −5→5
k = −28→20
l = −22→23
Refinement
Refinement on F2 Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.040
wR(F2) = 0.126
S = 1.06 3490 reflections 218 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0743P)2 + 0.088P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.002 Δρmax = 0.24 e Å−3 Δρmin = −0.20 e Å−3
supporting information
sup-2 Acta Cryst. (2005). E61, o1200–o1202
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C1 1.3424 (3) 0.59348 (6) 0.59861 (8) 0.0368 (3)
C2 1.2112 (4) 0.57741 (7) 0.52994 (8) 0.0411 (3)
H2 1.2433 0.5365 0.5122 0.049*
C3 1.0328 (4) 0.62165 (6) 0.48752 (8) 0.0383 (3)
H3 0.9380 0.6107 0.4404 0.046*
C4 0.9889 (3) 0.68226 (6) 0.51257 (7) 0.0312 (3)
C5 1.1238 (3) 0.69624 (6) 0.58283 (7) 0.0351 (3)
H5 1.0933 0.7370 0.6010 0.042*
C6 1.2997 (3) 0.65251 (6) 0.62640 (7) 0.0376 (3)
H6 1.3889 0.6626 0.6741 0.045*
C7 0.8069 (3) 0.72819 (6) 0.46529 (7) 0.0342 (3)
H7 0.6665 0.7127 0.4247 0.041*
C8 0.8196 (3) 0.79021 (6) 0.47386 (7) 0.0351 (3)
H8 0.9540 0.8061 0.5151 0.042*
C9 0.6449 (3) 0.83450 (7) 0.42533 (7) 0.0366 (3)
H9 0.5023 0.8192 0.3850 0.044*
C10 0.6736 (3) 0.89643 (6) 0.43431 (7) 0.0364 (3)
H10 0.8143 0.9109 0.4754 0.044*
C11 0.5088 (3) 0.94263 (6) 0.38688 (7) 0.0356 (3)
H11 0.3669 0.9289 0.3456 0.043*
C12 0.5458 (3) 1.00413 (6) 0.39816 (7) 0.0348 (3)
H12 0.6851 1.0162 0.4405 0.042*
C13 0.3969 (3) 1.05453 (6) 0.35289 (7) 0.0309 (3)
C14 0.1893 (3) 1.04421 (6) 0.28848 (7) 0.0335 (3)
H14 0.1388 1.0026 0.2733 0.040*
C15 0.0565 (3) 1.09322 (6) 0.24656 (7) 0.0346 (3)
H15 −0.0838 1.0857 0.2029 0.042*
C16 0.1322 (3) 1.15352 (6) 0.26948 (7) 0.0330 (3)
C17 0.3357 (3) 1.16595 (6) 0.33257 (7) 0.0346 (3)
H17 0.3859 1.2077 0.3472 0.042*
C18 0.4648 (3) 1.11635 (6) 0.37400 (7) 0.0338 (3)
H18 0.6029 1.1243 0.4178 0.041*
N1 1.5364 (3) 0.54706 (6) 0.64365 (8) 0.0488 (3)
N2 −0.0165 (3) 1.20575 (6) 0.22655 (6) 0.0422 (3)
supporting information
sup-3 Acta Cryst. (2005). E61, o1200–o1202
O2 1.6594 (3) 0.56252 (6) 0.70385 (7) 0.0640 (4)
O3 −0.2134 (3) 1.19414 (6) 0.17311 (6) 0.0576 (3)
O4 0.0580 (4) 1.25883 (5) 0.24665 (7) 0.0678 (4)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C1 0.0332 (6) 0.0355 (7) 0.0426 (8) 0.0036 (5) 0.0078 (6) 0.0109 (6)
C2 0.0471 (8) 0.0300 (7) 0.0470 (8) 0.0017 (6) 0.0094 (6) 0.0010 (6)
C3 0.0423 (7) 0.0356 (7) 0.0368 (7) −0.0023 (6) 0.0033 (6) −0.0020 (6)
C4 0.0262 (6) 0.0329 (7) 0.0347 (7) −0.0017 (5) 0.0038 (5) 0.0021 (5)
C5 0.0339 (6) 0.0331 (7) 0.0378 (7) 0.0021 (5) 0.0004 (5) −0.0014 (5)
C6 0.0362 (7) 0.0410 (8) 0.0349 (7) 0.0011 (6) 0.0006 (5) 0.0031 (6)
C7 0.0296 (6) 0.0392 (8) 0.0331 (7) −0.0001 (5) −0.0009 (5) 0.0003 (5)
C8 0.0298 (6) 0.0382 (7) 0.0366 (7) −0.0002 (5) 0.0002 (5) 0.0031 (6)
C9 0.0317 (6) 0.0418 (8) 0.0360 (7) 0.0021 (6) 0.0011 (5) 0.0044 (6)
C10 0.0324 (6) 0.0392 (8) 0.0373 (7) 0.0012 (5) 0.0015 (5) 0.0051 (6)
C11 0.0316 (6) 0.0402 (8) 0.0349 (7) 0.0008 (6) 0.0024 (5) 0.0036 (6)
C12 0.0302 (6) 0.0399 (8) 0.0336 (7) −0.0014 (5) −0.0002 (5) 0.0021 (5)
C13 0.0254 (5) 0.0366 (7) 0.0310 (6) 0.0007 (5) 0.0038 (5) 0.0018 (5)
C14 0.0333 (6) 0.0344 (7) 0.0326 (7) −0.0011 (5) 0.0027 (5) −0.0032 (5)
C15 0.0331 (6) 0.0440 (8) 0.0262 (6) 0.0002 (6) 0.0006 (5) −0.0018 (5)
C16 0.0319 (6) 0.0370 (7) 0.0308 (6) 0.0043 (5) 0.0057 (5) 0.0048 (5)
C17 0.0329 (6) 0.0331 (7) 0.0378 (7) −0.0025 (5) 0.0038 (5) −0.0017 (5)
C18 0.0306 (6) 0.0380 (7) 0.0319 (6) −0.0020 (5) −0.0011 (5) −0.0018 (5)
N1 0.0472 (7) 0.0461 (8) 0.0546 (9) 0.0099 (6) 0.0122 (6) 0.0195 (6)
N2 0.0456 (7) 0.0441 (7) 0.0374 (7) 0.0088 (5) 0.0070 (5) 0.0073 (5)
O1 0.1054 (11) 0.0459 (7) 0.0723 (9) 0.0311 (7) 0.0135 (8) 0.0148 (6)
O2 0.0669 (8) 0.0649 (8) 0.0577 (8) 0.0122 (6) −0.0070 (6) 0.0217 (6)
O3 0.0610 (7) 0.0662 (8) 0.0425 (6) 0.0087 (6) −0.0101 (5) 0.0134 (5)
O4 0.0983 (10) 0.0381 (7) 0.0641 (8) 0.0119 (6) −0.0067 (7) 0.0049 (5)
Geometric parameters (Å, º)
C1—C2 1.379 (2) C11—C12 1.341 (2)
C1—C6 1.384 (2) C11—H11 0.950
C1—N1 1.463 (2) C12—C13 1.457 (2)
C2—C3 1.379 (2) C12—H12 0.950
C2—H2 0.950 C13—C14 1.400 (2)
C3—C4 1.398 (2) C13—C18 1.401 (2)
C3—H3 0.950 C14—C15 1.381 (2)
C4—C5 1.399 (2) C14—H14 0.950
C4—C7 1.459 (2) C15—C16 1.384 (2)
C5—C6 1.378 (2) C15—H15 0.950
C5—H5 0.950 C16—C17 1.381 (2)
C6—H6 0.950 C16—N2 1.463 (2)
C7—C8 1.340 (2) C17—C18 1.381 (2)
supporting information
sup-4 Acta Cryst. (2005). E61, o1200–o1202
C8—C9 1.437 (2) C18—H18 0.950
C8—H8 0.950 N1—O2 1.225 (2)
C9—C10 1.342 (2) N1—O1 1.226 (2)
C9—H9 0.950 N2—O3 1.223 (2)
C10—C11 1.437 (2) N2—O4 1.224 (2)
C10—H10 0.950
C2—C1—C6 122.1 (1) C12—C11—C10 123.1 (1)
C2—C1—N1 119.3 (1) C12—C11—H11 118.4
C6—C1—N1 118.6 (1) C10—C11—H11 118.4
C3—C2—C1 118.9 (1) C11—C12—C13 127.4 (1)
C3—C2—H2 120.5 C11—C12—H12 116.3
C1—C2—H2 120.5 C13—C12—H12 116.3
C2—C3—C4 121.2 (1) C14—C13—C18 118.0 (1)
C2—C3—H3 119.4 C14—C13—C12 123.0 (1)
C4—C3—H3 119.4 C18—C13—C12 119.0 (1)
C3—C4—C5 117.9 (1) C15—C14—C13 121.3 (1)
C3—C4—C7 119.5 (1) C15—C14—H14 119.3
C5—C4—C7 122.7 (1) C13—C14—H14 119.3
C6—C5—C4 121.9 (1) C14—C15—C16 118.6 (1)
C6—C5—H5 119.1 C14—C15—H15 120.7
C4—C5—H5 119.1 C16—C15—H15 120.7
C5—C6—C1 118.0 (1) C17—C16—C15 122.1 (1)
C5—C6—H6 121.0 C17—C16—N2 118.9 (1)
C1—C6—H6 121.0 C15—C16—N2 119.0 (1)
C8—C7—C4 126.0 (1) C16—C17—C18 118.5 (1)
C8—C7—H7 117.0 C16—C17—H17 120.8
C4—C7—H7 117.0 C18—C17—H17 120.8
C7—C8—C9 124.8 (1) C17—C18—C13 121.5 (1)
C7—C8—H8 117.6 C17—C18—H18 119.2
C9—C8—H8 117.6 C13—C18—H18 119.2
C10—C9—C8 123.2 (1) O2—N1—O1 123.3 (1)
C10—C9—H9 118.4 O2—N1—C1 118.5 (1)
C8—C9—H9 118.4 O1—N1—C1 118.2 (1)
C9—C10—C11 125.3 (1) O3—N2—O4 123.4 (1)
C9—C10—H10 117.3 O3—N2—C16 118.3 (1)
C11—C10—H10 117.3 O4—N2—C16 118.3 (1)
C6—C1—C2—C3 0.2 (2) C18—C13—C14—C15 0.4 (2)
N1—C1—C2—C3 −179.2 (1) C12—C13—C14—C15 −179.2 (1)
C1—C2—C3—C4 1.1 (2) C13—C14—C15—C16 −0.1 (2)
C2—C3—C4—C5 −1.7 (2) C14—C15—C16—C17 0.1 (2)
C2—C3—C4—C7 178.3 (1) C14—C15—C16—N2 −178.1 (1)
C3—C4—C5—C6 1.0 (2) C15—C16—C17—C18 −0.4 (2)
C7—C4—C5—C6 −179.0 (1) N2—C16—C17—C18 177.8 (1)
C4—C5—C6—C1 0.3 (2) C16—C17—C18—C13 0.8 (2)
C2—C1—C6—C5 −0.9 (2) C14—C13—C18—C17 −0.7 (2)
supporting information
sup-5 Acta Cryst. (2005). E61, o1200–o1202
C3—C4—C7—C8 −162.0 (1) C2—C1—N1—O2 178.1 (1)
C5—C4—C7—C8 18.0 (2) C6—C1—N1—O2 −1.4 (2)
C4—C7—C8—C9 178.2 (1) C2—C1—N1—O1 −1.0 (2)
C7—C8—C9—C10 −177.5 (1) C6—C1—N1—O1 179.6 (1)
C8—C9—C10—C11 179.0 (1) C17—C16—N2—O3 −175.6 (1)
C9—C10—C11—C12 −179.9 (1) C15—C16—N2—O3 2.7 (2)
C10—C11—C12—C13 178.6 (1) C17—C16—N2—O4 3.2 (2)
C11—C12—C13—C14 −0.7 (2) C15—C16—N2—O4 −178.6 (1)
C11—C12—C13—C18 179.8 (1)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C7—H7···O3i 0.95 2.44 3.371 (2) 166
C14—H14···O2ii 0.95 2.57 3.366 (2) 141
C15—H15···O1ii 0.95 2.61 3.454 (2) 148
C2—H2···O1iii 0.95 2.71 3.364 (2) 127
C5—H5···O4iv 0.95 2.97 3.470 (2) 115
C6—H6···O4iv 0.95 2.92 3.437 (2) 115