organic papers
o1854
Pinkuset al. C16H22O2 doi:10.1107/S160053680601261X Acta Cryst.(2006). E62, o1854–o1855
Acta Crystallographica Section E
Structure Reports Online
ISSN 1600-5368
Dipropiodurene
A. G. Pinkus,* Kevin K.
Klausmeyer, Cody E. Carson and Herman C. Custard
Department of Chemistry and Biochemistry, Baylor University, One Bear Place No. 97348, Waco, TX 76798-7348, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 110 K
Mean(C–C) = 0.001 A˚ Rfactor = 0.038 wRfactor = 0.108
Data-to-parameter ratio = 20.2
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 15 March 2006 Accepted 6 April 2006
#2006 International Union of Crystallography All rights reserved
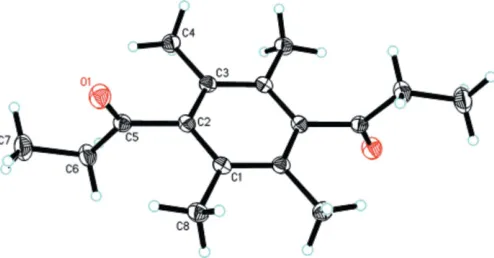
The conformer present in the crystal structure of the title compound [systematic name: 1,10-(2,3,5,6-tetramethyl-p
-phenylene)dipropan-1-one], C16H22O2, lies on an inversion
center and consequently has the two carbonyl groups in atrans
orientation relative to the central ring. In the crystalline state, the planes of the carbonyl and attached methylene groups are at a dihedral angle of 86.50 (11)relative to the central ring.
Comment
In the present crystallographic study of the title compound, (I), the two carbonyl groups and attached methylene C atoms are in a transorientation relative to the central ring (Fig. 1) due to the inversion center at the mid-point of the benzene ring. The planes of the carbonyl group and attached methylene C atom are at an angle of 86.50 (11) relative to the central
ring. This can be compared with a crystallographic study of 1,4-bis(4-chlorobenzoyl)-2,3,5,6-tetramethylbenzene by Ferguson et al. (1993), who found that the two p -chloro-benzoyl groups were also in atransorientation with a corre-sponding angle of 85.9. A crystallographic study (Bearet al.,
1973) of a compound with a large pivaloyl group between two methyl groups (2,4,6-trimethyl-3-pivaloylbenzoic acid) reported the mean planes through the keto group and the benzene ring to have a dihedral angle of 89.9. This compound was resolved but was found to undergo rapid racemization (Pinkus et al., 1968). The corresponding angle for a related hindered duryl ester, ethyl 2,3,5,6-tetramethylbenzoate (Pinkuset al., 2005), was found to be 79.9.
Selected geometric parameters are presented in Table 1. All other bonds and angles are within expected ranges.
Experimental
washings with water and final drying over anhydrous sodium sulfate. The solvent was removed by distillation and the product was recrystallized from benzene in 19% yield (9.3 g), m.p. 453.0–453.6 K [literature value 451 K (Baum & Meyer, 1895) 451 K]. Analysis calculated for C16H22O2: C 78.01, H 9.00%; found (Galbraith Labs,
Knoxville, Tennessee, USA): C 78.19, H, 9.07%. IR (dichloro-methane): 1703 cm1(C Ostr).
Crystal data
C16H22O2
Mr= 246.34
Orthorhombic,Pbca a= 8.6268 (14) A˚
b= 7.682 (3) A˚
c= 21.034 (4) A˚
V= 1393.9 (6) A˚3
Z= 4
Dx= 1.174 Mg m 3 MoKradiation
= 0.08 mm1
T= 110 (2) K Block, colorless 0.300.250.21 mm
Data collection
Bruker X8 APEX area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin= 0.974,Tmax= 0.987
12799 measured reflections 1715 independent reflections 1535 reflections withI> 2(I)
Rint= 0.034
max= 28.2
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.038
wR(F2) = 0.108
S= 1.06 1715 reflections 85 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0591P)2 + 0.3963P]
whereP= (Fo2+ 2Fc2)/3 (/)max= 0.001
max= 0.37 e A˚
3 min=0.16 e A˚
3
Table 1
Selected geometric parameters (A˚ ,).
C5—O1 1.2124 (13) C2—C5 1.5137 (12)
C5—C6 1.5101 (14)
C6—C5—C2 116.46 (8)
All H atoms were included in calculated positions, with C—H = 0.98 and 0.99 A˚ , and withUiso(H) = 1.2Ueq(C).
Data collection:APEX2(Bruker, 2003); cell refinement:APEX2; data reduction:SAINT-Plus(Bruker, 2003); program(s) used to solve
structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure: SHELXL97 (Sheldrick, 1997); molecular graphics:
SHELXTL(Sheldrick, 2000); software used to prepare material for publication:SHELXTL.
The Bruker X8 APEX diffractometer was purchased with funds received from the National Science Foundation Major Research Instrumentation Program, grant No. CHE-0321214. KKK and AGP thank the Robert A. Welch Foundation for support (grant Nos. AA-1508 and AA-111, respectively).
References
Baum, F. & Meyer, V. (1895).Berichte,28, 3212–3215.
Bear, C. A., Macdonald, A. L. & Trotter, J. (1973).Acta Cryst.B29, 2617–2619. Bruker (2003).APEX2 (Version 1.0-27) and SAINT-Plus (Version 6.25).
Bruker AXS Inc., Madison, Wisconsin, USA.
Ferguson, G., Patterson, K. H. & Smith, D. M. (1993).Acta Cryst.C49, 190– 192.
Pinkus, A. G., Klausmeyer, K. K., Feazell, R. P. & Lin, E. C. H. Y. (2005).Acta Cryst.E61, o662–o663.
Pinkus, A. G., Riggs, J. I. & Broughton, S. I. (1968).J. Am. Chem. Soc.90, 5043–5044.
Sheldrick, G. M. (1996).SADABS. University of Gottingen, Germany. Sheldrick, G. M. (1997). SHELXS97 and SHELXL97. University of
Go¨ttingen, Germany.
[image:2.610.316.563.72.201.2]Sheldrick, G. M. (2000).SHELXTL. Version 6.10. Bruker AXS Inc., Madison, Wisconsin, USA.
Figure 1
A view of the molecular structure of (I), with the atom-numbering scheme. Displacement ellipsoids are drawn at the 50% probability level and H atoms are shown as small spheres of arbitrary radii. Unlabeled atoms are related to labeled atoms by the symmetry code (x+ 1,y,
supporting information
sup-1 Acta Cryst. (2006). E62, o1854–o1855
supporting information
Acta Cryst. (2006). E62, o1854–o1855 [https://doi.org/10.1107/S160053680601261X]
Dipropiodurene
A. G. Pinkus, Kevin K. Klausmeyer, Cody E. Carson and Herman C. Custard
1,1′-(2,3,5,6-tetramethyl-p-phenylene)dipropan-1-one or 2,3,5,6-tetramethyl-1,4-dipropiophenone
Crystal data
C16H22O2 Mr = 246.34
Orthorhombic, Pbca
Hall symbol: -P 2ac 2ab
a = 8.6268 (14) Å
b = 7.682 (3) Å
c = 21.034 (4) Å
V = 1393.9 (6) Å3 Z = 4
F(000) = 536
Dx = 1.174 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 6682 reflections
θ = 3.1–35.0°
µ = 0.08 mm−1 T = 110 K Block, colorless 0.30 × 0.25 × 0.21 mm
Data collection
Bruker SMART CCD area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Sheldrick, 1996)
Tmin = 0.974, Tmax = 0.987
12799 measured reflections 1715 independent reflections 1535 reflections with I > 2σ(I)
Rint = 0.034
θmax = 28.2°, θmin = 3.1°
h = −11→11
k = −9→10
l = −27→19
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.038 wR(F2) = 0.108 S = 1.06 1715 reflections 85 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0591P)2 + 0.3963P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.37 e Å−3
Δρmin = −0.16 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
C2 0.47454 (10) 0.04316 (11) 0.56283 (4) 0.0168 (2)
C3 0.34707 (10) 0.02262 (12) 0.52226 (4) 0.0170 (2)
C1 0.62736 (11) 0.02124 (12) 0.54173 (4) 0.0171 (2)
C5 0.44501 (10) 0.08889 (12) 0.63178 (4) 0.0183 (2)
C4 0.18314 (11) 0.04672 (13) 0.54611 (4) 0.0213 (2)
H4A 0.1855 0.0812 0.5910 0.032*
H4B 0.1261 −0.0629 0.5417 0.032*
H4C 0.1316 0.1375 0.5211 0.032*
C6 0.41512 (13) −0.06156 (13) 0.67633 (4) 0.0240 (2)
H6A 0.5107 −0.1323 0.6796 0.029*
H6B 0.3333 −0.1363 0.6578 0.029*
C7 0.36562 (15) −0.00711 (17) 0.74265 (5) 0.0343 (3)
H7A 0.4465 0.0655 0.7617 0.051*
H7B 0.3496 −0.1110 0.7689 0.051*
H7C 0.2688 0.0592 0.7401 0.051*
O1 0.44569 (9) 0.23882 (10) 0.64977 (3) 0.0274 (2)
C8 0.76383 (11) 0.04693 (13) 0.58593 (4) 0.0217 (2)
H8A 0.8230 −0.0617 0.5889 0.033*
H8B 0.7262 0.0797 0.6282 0.033*
H8C 0.8307 0.1395 0.5693 0.033*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
C2 0.0185 (4) 0.0160 (4) 0.0159 (4) −0.0002 (3) −0.0006 (3) 0.0017 (3)
C3 0.0161 (4) 0.0167 (4) 0.0183 (4) 0.0001 (3) −0.0003 (3) 0.0020 (3)
C1 0.0173 (4) 0.0169 (4) 0.0170 (4) −0.0003 (3) −0.0024 (3) 0.0021 (3)
C5 0.0159 (4) 0.0220 (5) 0.0172 (4) 0.0004 (3) −0.0015 (3) 0.0002 (3)
C4 0.0168 (4) 0.0255 (5) 0.0214 (4) 0.0002 (4) 0.0001 (3) −0.0003 (3)
C6 0.0301 (5) 0.0244 (5) 0.0175 (4) −0.0011 (4) 0.0020 (4) 0.0021 (3)
C7 0.0463 (7) 0.0376 (6) 0.0189 (5) 0.0031 (5) 0.0064 (4) 0.0024 (4)
O1 0.0357 (4) 0.0226 (4) 0.0238 (4) −0.0004 (3) 0.0001 (3) −0.0032 (3)
C8 0.0179 (4) 0.0269 (5) 0.0204 (4) −0.0001 (4) −0.0043 (3) 0.0002 (4)
Geometric parameters (Å, º)
C2—C3 1.4009 (12) C4—H4C 0.9800
C2—C1 1.4012 (13) C6—C7 1.5177 (14)
supporting information
sup-3 Acta Cryst. (2006). E62, o1854–o1855
C5—C6 1.5101 (14) C7—H7A 0.9800
C3—C1i 1.4049 (13) C7—H7B 0.9800
C3—C4 1.5119 (13) C7—H7C 0.9800
C1—C8 1.5130 (12) C8—H8A 0.9800
C4—H4A 0.9800 C8—H8B 0.9800
C4—H4B 0.9800 C8—H8C 0.9800
C3—C2—C1 122.14 (8) C5—C6—C7 114.05 (9)
C3—C2—C5 118.54 (8) C5—C6—H6A 108.7
C1—C2—C5 119.32 (8) C7—C6—H6A 108.7
C2—C3—C1i 119.17 (8) C5—C6—H6B 108.7
C2—C3—C4 121.22 (8) C7—C6—H6B 108.7
C1i—C3—C4 119.61 (8) H6A—C6—H6B 107.6
C2—C1—C3i 118.68 (8) C6—C7—H7A 109.5
C2—C1—C8 121.46 (8) C6—C7—H7B 109.5
C3i—C1—C8 119.85 (8) H7A—C7—H7B 109.5
O1—C5—C6 122.29 (9) C6—C7—H7C 109.5
O1—C5—C2 121.25 (8) H7A—C7—H7C 109.5
C6—C5—C2 116.46 (8) H7B—C7—H7C 109.5
C3—C4—H4A 109.5 C1—C8—H8A 109.5
C3—C4—H4B 109.5 C1—C8—H8B 109.5
H4A—C4—H4B 109.5 H8A—C8—H8B 109.5
C3—C4—H4C 109.5 C1—C8—H8C 109.5
H4A—C4—H4C 109.5 H8A—C8—H8C 109.5
H4B—C4—H4C 109.5 H8B—C8—H8C 109.5
C1—C2—C5—O1 86.50 (11)