JOURNAL OFVIROLOGY, Nov. 1996, p. 8019–8028 Vol. 70, No. 11 0022-538X/96/$04.0010
Copyrightq1996, American Society for Microbiology
Postbinding Events Mediated by Human Immunodeficiency
Virus Type 1 Are Sensitive to Modifications in the
D4-Transmembrane Linker Region of CD4
SUSAN MOIR,1,2JOSE´ E PERREAULT,1†ANDLOUISE POULIN1,2*
Centre de Recherche en Infectiologie du Centre Hospitalier Universitaire de Que´bec, Pavillon CHUL, Que´bec, Canada
G1V 4G2,1and De´partement de Microbiologie, Universite´ Laval, Que´bec, Canada G1K 7P42
Received 10 April 1996/Accepted 30 July 1996
Evidence from both structural and functional studies of the CD4 molecule suggests that several domains, including the transmembrane (TM) domain and the adjoining extracellular region (D4-TM linker), contribute to the post-gp120-binding events leading to human immunodeficiency virus-mediated membrane fusion. To investigate such a role in syncytium formation and cell-free infectivity, we generated several deletion and substitution mutations in the TM and D4-TM linker regions of the CD4 molecule. We found that while the TM domain of CD4 was dispensable for cell-cell and virus-cell interactions, modifications in the D4-TM linker led to perturbations in both processes. Deletion of the five amino acid residues linking D4 to the TM domain resulted in a delayed and reduced capacity to form syncytia, whereas replacement of the residues with the heterologous sequence from the CD8 molecule restored the kinetic profile to wild-type CD4 levels. On the other hand, both mutants of the CD4 D4-TM linker demonstrated delayed cell-free human immunodeficiency virus type 1 infectivity profiles. The defective fusion capacity may be linked to structural perturbations identified with anti-CD4 monoclonal antibodies in the D1-D2 interface and D3 domain of the deletion mutant yet absent in D1 and D4. While all cells were found to bind comparable levels of gp120, both D4-TM linker mutants appeared to induce a decrease in the V3 loop exposure of bound gp120. This underexposure may explain the delays in cell-free infectivities observed for both of these mutants. Together, these findings confirm a role for regions of the CD4 molecule located outside D1 in post-gp120-binding events and suggest that the D4-TM interface contributes to the conformational changes that direct the fusion process.
The CD4 molecule serves as the major portal of entry for human immunodeficiency virus type 1 (HIV-1). The high-af-finity binding of the viral external envelope glycoprotein gp120 to its host cell receptor, CD4 (41, 62), initiates a complex sequence of events that culminates in the fusion of viral and cellular membranes (68). Infected cells expressing gp120-gp41 at their surface also interact with CD4-positive target cells in a relatively similar process of cell-cell fusion. These events may lead to the formation of giant cells (syncytia) (67) and ulti-mately to cell death. While the molecular events involved in membrane fusion remain obscure, the binding of gp120 to CD4 is believed to initiate a cascade of interactions and changes in conformation that result in the activation of the fusion poten-tial of the viral transmembrane glycoprotein, gp41 (17, 18, 35, 61). Evidence for the induction of conformational changes upon ligand binding has been demonstrated following soluble CD4-mediated exposure of several cryptic epitopes, in both gp120 and gp41 (34, 63, 65, 71). This process, described as receptor-mediated activation of fusion (1, 51), is triggered by the initial CD4-gp120 binding interactions, which probably loosen the contacts between gp120 and gp41 (22, 63) to allow exposure of the fusion peptide located in the amino terminus of gp41 (15, 21, 30).
CD4 is a transmembrane (TM) glycoprotein composed of four extracellular immunoglobulin-like domains (D1 to D4)
(12) anchored to the cell membrane by a 22-amino-acid TM domain and extended intracellularly by a short cytoplasmic domain. CD4 is thought to possess mobility in physiologically important regions of the molecule, including the D2-D3 junc-tion and the short stretch of five amino acids linking D4 to the TM domain (39). The CD4 residues involved in binding gp120 have been mapped to the first domain (D1), primarily in a
complementary cleft located in the C9C0ridge of the
CDR2-like region (11, 48, 73). Several lines of evidence suggest that regions outside the CDR2 loop are involved in postbinding events leading to the internalization of HIV. Involvement of the CDR3 loop has been proposed (4, 8, 72); however, several more recent studies (6, 52) contest such a role for CDR3 in the fusion process. Recently, it has been suggested that CDR3 may be involved at a postinternalization stage of HIV-1 replication (3).
In a previous paper, we have described a hybrid CD4-ex-pressing cell line, constructed by fusing the CD4 D1-D2 to the CD8 hinge-TM-cytoplasmic domains, that demonstrated a re-duced capacity to undergo HIV-mediated cell fusion and cell-free infection (58). Similar results have been obtained with a video imaging system, revealing a greatly delayed fusion
ca-pacity for cells expressing the same CD4zCD8 chimeric
mol-ecule and suggesting that the carboxy-terminal half of CD4 may play a role in the changes in conformation that lead to membrane fusion (23). However, truncated forms of the CD4 molecule have been shown to be unperturbed in syncytium formation, suggesting that the cytoplasmic tail is not an essen-tial element in the fusion process (58). Monoclonal antibodies (MAbs) directed against epitopes outside D1 have also been shown to block fusion without affecting the initial binding step (7, 10, 29, 54). While these include MAbs directed against D3
* Corresponding author. Mailing address: Infectiologie, Centre Hos-pitalier de U. Laval, 2705, Boul. Laurier, Ste-Foy, Que´bec, Canada G1V 4G2. Phone: (418) 654-2788. Fax: (418) 654-2786. Electronic mail address: [email protected].
† Present address: De´partement de Biochimie, RSVS, Pavillon C.E. Marchand, Universite´ Laval, Ste-Foy, Que´bec, Canada G1K 7P4.
8019
on November 9, 2019 by guest
http://jvi.asm.org/
(29), a more recent interpretation suggests that MAbs which bind to epitopes in D3 may be causing steric interference rather than masking residues involved in the fusion reaction (5).
Several recent studies have provided indications that the
fusion defect observed in the CD4zCD8 hybrid may map to a
region of CD4 located within or immediately external to the TM domain. Cells expressing a glycosylphosphatidylinositol-anchored CD4 have been shown to be defective in syncytium formation (45). The modification also resulted in an increased sensitivity to ligand-mediated downregulation of glycosylphos-phatidylinositol-linked CD4 (45), suggesting that the turnover of the cell surface receptor is an important determinant of syncytium formation. However, these findings conflict with those of an earlier study that found similar levels of fusion for cells expressing either wild-type CD4 (CD4wt) or glycosylphos-phatidylinositol-anchored CD4 (37). On the other hand, it has recently been suggested, from the available information on the structural attributes of CD4, that the interdomain regions, including the short linker region immediately adjacent to the TM domain, may be involved in postbinding events leading to membrane fusion (5, 39). The proposed rotational freedom thought to originate at the base of D4 (5, 39) may mediate the postbinding conformational changes that have been suggested in the various models of membrane fusion (17, 18, 64).
In this study, we have sequentially mutated the CD4 TM domain and the D4-TM linker region to investigate their con-tributions to HIV-mediated cell fusion and infectivity. Our findings indicate that the replacement of these two CD4 ele-ments with CD8 counterparts has no measurable influence on HIV-mediated syncytium formation. More importantly,
dele-tion of the D4-TM linker region severely perturbs the capacity of the cells to form syncytia and sustain infection with cell-free virus.
MATERIALS AND METHODS
Recombinant DNA constructs.Mutations in the CD4-coding sequence (Fig. 1A) were generated by overlapping PCR as described previously (14). The template plasmids originated from clone pT4B, containing the entire human CD4 cDNA, and from clone pT8F1, containing the entire human CD8 cDNA. These plasmids were obtained through the National Institutes of Health (NIH) AIDS Research and Reference Repository Program from Richard Axel. The strategy, illustrated schematically in Fig. 1B for the replacement of the CD4 TM domain, involved three rounds of PCR amplifications. The CD8 TM domain, flanked by short CD4 extensions (fragment AB), was amplified in the first reaction with chimeric primers A (59 -ACC-CCG-GTG-CAG-CCA-ATC-TAC-ATC-TGG-GCG-CC-39) and B (59 -CCG-GCA-CCT-GAC-ACA-GTG-GTT-GCA-GTA-AAG-GG-39). The 39halves of primers A and B corresponded to the extremities of the CD8 TM domain, whereas the 59halves corresponded to the TM flanking sequences of CD4. In the second PCR, fragment AB served as primer along with either primer C (59-TCT-AGA-ACT-AGT-GGA-TC-39) or D (59-CGA-GGT-CGA-CGG-TAT-CG-39), which map to sequences in the cloning vector pBluescript. Primer C combined with fragment AB generated a fragment corresponding to the 59half of the CD4-coding sequence and the TM sequence of CD8 (fragment CB), whereas primer D combined with fragment AB gener-ated a fragment corresponding to the 39half of the CD4 sequence, again with the TM sequence of CD8 at its extremity (fragment AD). Finally, the complete CD4zTMCD8 chimeric sequence (fragment CD) was generated in a third PCR by mixing fragments CB and AD, which annealed through the common TM sequence.
A slightly adapted version of the PCR strategy described above was used to generate mutants s367-371 and d367-371 (Fig. 1A). The first PCR was eliminated because the modifications were short enough to be completely incorporated into primers A and B. For the substitution of five CD8 amino acid residues, the chimeric primers A and B were, respectively, 59 -(1248)CGA-CTT-CGC-CTG-TGA-TAT-GGC-CCT-GAT-TGT-GC(1279)-39and 59 -(1265)ATA-TCA-CAG-GCG-AAG-TCG-GAC-CAT-GTG-GGC-AG(1234)-39, where the underlined portions represent the CD8 sequences and the numbers in parentheses represent FIG. 1. (A) Schematic representation of the mutations introduced in the CD4 molecule, with the numbers above the drawing denoting amino acid numbering and relative positioning of the epitopes recognized by various anti-CD4 MAbs. (B) Schematic representation of the PCR-based strategy used to generate the CD4zTMCD8 hybrid molecule (not drawn to scale). The relative positioning of each primer used (described in Materials and Methods) is shown by the lettered arrows. EXT, extracellular domains; CYT, cytoplasmic domain; a.a., amino acids.
on November 9, 2019 by guest
http://jvi.asm.org/
the nucleotide numbers from the CD4 sequence. The numbering of the CD4 nucleotide sequence conforms to that of the originally published sequence (44) (GenBank accession no. M35160). For the deletion reactions, primers A and B were, respectively, 59 -(1234)CTG-CCC-ACA-TGG-TCC-ATG-GCC-CTG-ATT-GTG(1278)-39and 59 -(1279)GCA-CAA-TCA-GGG-CCA-TGG-ACC-ATG-TGG-GCA-G-(1234)-39.
PCRs were carried out with 50 ng of starting plasmid or 5 to 10% of band-purified PCR products. The DNA was added to a reaction mixture containing 13 PCR buffer (10 mM Tris-HCl, 1.5 mM MgCl2, 50 mM KCl [pH 8.3]), 0.2 mM each deoxynucleoside triphosphate, 50 pmol of each oligonucleotide primer, and 2.5 U of Taq polymerase in a final volume of 100ml. Reactions were performed according to the following parameters: denaturation at 948C for 1 min, annealing at 458C for 2 min, and elongation at 728C for 1 min for 30 cycles followed by a 10-min incubation at 728C. The PCR products were digested with EcoRI and HindIII restriction endonucleases and ligated with T4 DNA ligase into the respective sites of pBluescript. After transformation in Escherichia coli JM109, several clones were sequenced by the dideoxy chain termination method with the T7 sequencing kit (Pharmacia, Uppsala, Sweden) in accordance with the instruc-tions of the manufacturer. The mutated CD4 inserts, along with the CD4wt insert isolated directly from clone pT4B, were each subcloned in the expression vector pTEJ-8, which carries the gene for resistance to neomycin (33).
Cell line transfections and surface expression.The CD4-negative T-cell line A2.01, obtained through the NIH AIDS Research and Reference Repository Program from Thomas Folks, was transfected by electroporation with the various CD4 constructs. Resistant cell clones were isolated by limiting dilution following selection in RPMI 1640 medium supplemented with 10% fetal calf serum, 2 mM glutamine, 100 U of penicillin per ml, 100mg of streptomycin per ml, and 1000
mg of geneticin (GIBCO Laboratories, Grand Island, N.Y.) per ml. Selected clones were screened by flow cytometry for cell surface expression of chimeric CD4 or CD4wt. Briefly, cells were incubated with the anti-CD4 monoclonal antibody Leu3A (Becton Dickinson, Mountain View, Calif.) and then stained with a fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse antibody (Becton Dickinson).
Epitope mapping.The selected clones were incubated with one or several of the following anti-CD4 MAbs, stained with a fluorescein-conjugated second antibody (Caltag Laboratories, San Francisco, Calif.), and analyzed by flow cytometry. The anti-D1D2 MAbs used were Leu3A (Becton Dickinson), B66.6 (provided by R.-P. Se´kaly) (20), and OKT4e and -f (Pat Rao, Ortho Pharma-ceuticals, Raritan, N.J.). The anti-D3D4 MAbs included Q428 (obtained through the NIH AIDS Research and Reference Repository Program from P. Kwong and Q. Sattentau) and M429 (provided by R.-P. Se´kaly) (20).
gp120 binding assay.Cells expressing modified CD4 or CD4wt were incubated with increasing concentrations of soluble recombinant HIV-1 IIIB gp120 (rgp120) purified from CHO cells (Genentech, San Francisco, Calif.) or with 10
mg of rgp120 per ml for different periods of time. After incubation at either 4 or 378C, the cells were washed twice, and cell-bound gp120 was revealed by flow cytometry with anti-gp120 MAb NEA-9305 (Dupont, Wilmington, Del.) followed by an R-phycoerythrin-conjugated second antibody (Caltag). Cell-bound fluo-rescence was detected with an EPICS XL cytometer (Coulter Corporation, Miami, Fla.). Alternatively, bound gp120 was revealed with a 1:50 dilution of a polyclonal antiserum from an individual with prescreened HIV-1 Env-reactive antibodies or the human MAb 48d, obtained through the NIH AIDS Research and Reference Repository Program from J. E. Robinson. FITC-conjugated goat anti-human immunoglobulin G (Jackson ImmunoResearch Laboratories, West Grove, Pa.) was used as a second antibody. For the kinetic evaluations of bound gp120, values were reported as MFI (at a given time point)/maximal MFI, where MFI is the mean fluorescence intensity after subtraction of binding in the ab-sence of the first ligand.
Virion binding assay.A high-titer stock of HIV-1 cell-free virus was prepared by transfecting 293T cells (56) with the infectious molecular clone pNL4-3, obtained through the NIH AIDS Research and Reference Repository Program from M. Martin. At 48 h after transfection, the culture supernatant was har-vested, filtered, and assayed for HIV-1 p24 antigen by enzyme-linked immu-nosorbent assay (ELISA). Cells expressing modified CD4 or CD4wt were incu-bated with the undiluted supernatant (typically at 1mg/ml in p24) for 2 h at 48C. After several washes to remove unbound virus, cell pellets were resuspended in lysis buffer for measurement of the p24 antigen level. The background level was calculated from binding of the viral particles to parental A2.01 cells.
Syncytium formation assay.Cells expressing modified CD4 or CD4wt were coincubated with HIV-1SF33 Env-expressing effector cells (A2.01-Env) (49). Briefly, the effector cells were produced by cotransfecting A2.01 cells with the expression vector pTEJ-8, encoding the cDNA of HIV-1SF33env, along with the
rev-encoding plasmid pCV1. The presence of Rev was included for efficient transport and expression of env mRNA (25). The fusion assay consisted of coincubating effector and target cells at a ratio of 1:1 for a total of 105cells per well of a microtiter plate. The cells were placed at 378C and monitored micro-scopically for the appearance of syncytia over a period of 20 to 24 h. Syncytia were defined by cell membrane ballooning and by the presence of giant cells with a diameter at least four times that of single cells (43). As suggested by Golding et al. (23), for unambiguous identification of syncytia, the counts were made after cell suspension mixing with an Eppendorf pipettor.
HIV-1 infectivity assay.A stock of cell-free HIV-1 strain SF33 was produced from chronically HIV-1SF33-infected Jurkat cells, kindly provided by Jay Levy (University of California, San Francisco). Cells expressing modified CD4 or CD4wt were incubated with HIV-1SF33at an input multiplicity of infection of 0.5 for 2 h. After being washed twice, 53104
cells per well were cultured in triplicate in 48-well plates, and the supernatant was collected periodically. Production of virus was evaluated by measuring the level of either p24 antigen or reverse transcriptase activity in the supernatant.
RESULTS
The CD8 TM-substituted CD4 molecule demonstrates a level of activity toward HIV-1 equivalent to that of CD4wt. (i) Cell surface expression.A potential role for the TM domain of CD4 in HIV-1-mediated cell fusion and infectivity was previ-ously suggested in papers reporting on the functional attributes of various CD4 chimeric molecules. However, of these studies, those investigating glycosylphosphatidylinositol-anchored CD4 presented contradictory findings (37, 45), whereas those
focus-ing on CD4zCD8 chimeras covered several potentially
impor-tant domains of the CD4 molecule, including the TM domain (23, 58). To clarify the role of the CD4 TM domain in HIV infectivity and syncytium formation, we produced a T-cell line expressing a CD8 TM-substituted CD4 molecule (Fig. 1A,
CD4zTMCD8). From the numerous clones expressing
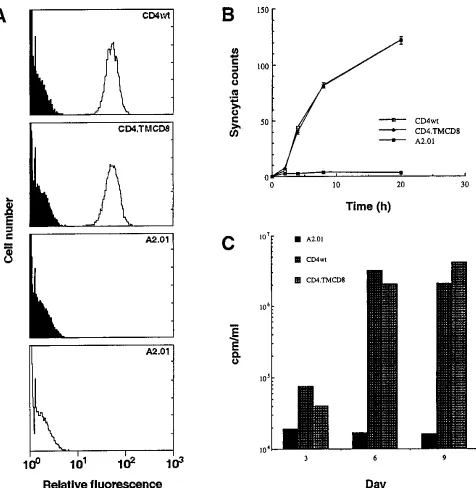
differ-ent levels of hybrid CD4 or CD4wt obtained, two were retained for further analysis on the basis of high and comparable levels of cell surface expression. The profiles depicted in Fig. 2A
indicate that both clones CD4wt and CD4zTMCD8 expressed
high and similar levels of the glycoprotein, while absence of expression was observed for the negative control parental cell line, A2.01. As in previous reports (2, 23, 58), these results indicate that a CD4 molecule possessing a CD8 transmem-brane domain can be as efficiently processed and presented at the cell surface as its CD4wt counterpart. The comparison of cells expressing similar amounts of modified CD4 and CD4wt molecules at the cell surface eliminated variations in activity attributable to differences in expression levels.
(ii) Syncytium formation. The fusion capacities of CD4wt and TM-substituted CD4 molecules were compared by
coin-cubating each target cell with the HIV-1SF33Env-expressing
effector cell line A2.01-Env, which was previously developed for rapid and reproducible assaying of cell-cell fusion (49). The coincubation was monitored microscopically for the appear-ance of syncytia over a period of 20 h. As illustrated in Fig. 2B,
the CD4zTMCD8 mutant was indistinguishable from the
CD4wt-expressing clone in terms of both the rate of syncytium formation and the number of syncytia counted. Upon coincu-bation of either target cell line with A2.01-Env effector cells, syncytia became visible within 1 h and reached a maximum level at 20 h. On the other hand, there was a total absence of syncytia when negative control A2.01 cells served as targets.
(iii) Cell-free infectivity.Considering that virus-cell interac-tions may differ from cell-cell interacinterac-tions, we were also in-terested in comparing cell-free HIV-1 interactions with cells
expressing either the chimeric CD4zTMCD8 or CD4wt
mol-ecules. To this end, a productive infection of each cell line was
established with HIV-1SF33. As can be observed in Fig. 2C,
CD4wt- and chimeric CD4-expressing cells displayed similar infection kinetics, whereas a productive infection was absent in A2.01 negative control cells. From these results indicating that the TM domain of CD4 is dispensable for viral replication, we can indirectly infer that the domain does not specifically con-tribute to the virus-cell interactions leading to viral entry.
Cells expressing CD4 with a deleted or CD8-substituted D4-TM linker region demonstrate reduced activities toward HIV-1. (i) Cell surface expression. In a subsequent line of
VOL. 70, 1996 CD4 IN HIV-MEDIATED FUSION AND INFECTION 8021
on November 9, 2019 by guest
http://jvi.asm.org/
investigation, we wanted to consider the proposal that the short CD4 segment linking D4 to the TM domain may be involved in the postbinding modulations leading to HIV-me-diated fusion (5, 39). To determine whether this interdomain was involved in the fusion process, we created a deletion mu-tant (Fig. 1A). To restore the original length of the molecule, and hence distinguish between the possibilities that fusion may
[image:4.612.72.548.66.554.2]be impeded with a shortened target molecule or with a mole-cule lacking residues directly involved in the fusion reaction, we also replaced the sequence with the heterologous counter-part from CD8. Modified CD4 cDNA was produced by over-lapping PCR (Fig. 1B), subcloned in the eukaryotic expression vector pTEJ-8, and transfected into the CD4-negative T-cell line A2.01. Independently derived clones were selected and
FIG. 2. Functional characterization of CD4zTMCD8 cDNA introduced in CD4-negative T cells. (A) Cell surface expression of CD4wt and chimeric CD4zTMCD8 in A2.01 cells evaluated with an EPICS 753 flow cytometer (Coulter). Cells were exposed to the MAb Leu3A followed by FITC-labeled goat anti-mouse antibodies (unfilled profile). FITC-labeled goat anti-mouse antibodies were used as a negative control in each reaction (filled profile). Profiles are shown for the CD4wt and chimeric CD4zTMCD8 cell clones as well as the parental cell line, A2.01. (B) Fusion kinetics of the various target cells coincubated with effector A2.01 cells expressing HIV-1 envelope glycoproteins. Syncytium formation was monitored periodically by microscopic observations. The values indicate the number of syncytia counted in four fields per well and represent the means6standard errors calculated from three separate wells. (C) Infection of the different cell lines with HIV-1 strain SF33. Cells were infected at day 0, and virus production was evaluated at 3-day intervals by measuring reverse transcriptase activity. Datum points were calculated from duplicate cultures and are representative of two independent experiments.
on November 9, 2019 by guest
http://jvi.asm.org/
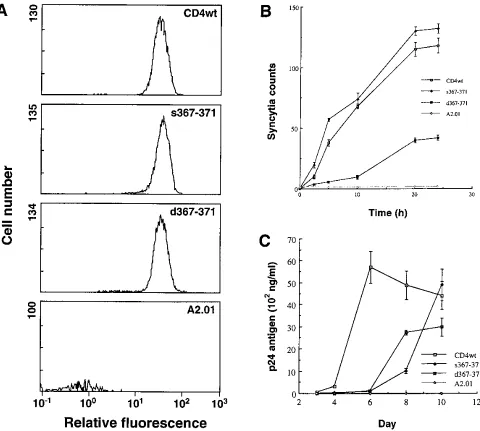
screened for cell surface expression of modified CD4. Illus-trated in Fig. 3A with anti-CD4 MAb Leu3A surface staining are profiles of the clones retained for further analysis on the basis of comparable levels of expression with CD4wt-express-ing cells.
(ii) Syncytium formation.To determine whether the dele-tion and/or substitudele-tion of five D4-TM interdomain residues of CD4 affected HIV-mediated syncytium formation, we coincu-bated the various target cells with the HIV-1 Env-expressing A2.01-Env effector cells. The kinetic profile in Fig. 3B illus-trates that, in comparison to that in cells expressing the CD4wt molecule, syncytium formation was significantly delayed in
[image:5.612.67.547.68.499.2]cells expressing the CD4 deletion mutation d367-371. Further-more, when the number of syncytia formed had leveled off after 24 h of coincubation, the counts for the d367-371 mutant represented about one-third of the counts obtained with the CD4wt cells. On the other hand, when the deletion mutant target cells were replaced by the substitution mutant s367-371 (Fig. 1A), both the rate and level of syncytium formation were restored to wild-type levels (Fig. 3B). To confirm these find-ings, we repeated the assay with two other series of cell clones, expressing similarly higher and lower levels of surface target ligand than the set represented in Fig. 3A. As expected, the profile differential observed in Fig. 3B was maintained, albeit
FIG. 3. Functional characterization of CD4 D4-TM interdomain-mutated cDNA introduced in CD4-negative T cells. (A) Cell surface expression of CD4wt and modified CD4 in A2.01 cells was evaluated with an EPICS XL flow cytometer (Coulter). Cells were exposed to the MAb Leu3A followed by FITC-labeled goat anti-mouse antibodies. Profiles, depicting expression after subtraction of the background level, are shown for CD4wt and D4-TM linker mutants as well as the parental cell line, A2.01. (B) Fusion kinetics of the various target cells coincubated with effector A2.01 cells expressing HIV-1 envelope glycoproteins. Syncytium formation was monitored periodically by microscopic observations. The values indicate the number of syncytia counted in four fields per well and represent the averages obtained from three separate wells. (C) Infection of the different cell lines with HIV-1 strain SF33. Cells were infected at a day 0, and the presence of virus in the culture supernatant was periodically evaluated by measuring the level of p24 antigen. Mean datum points with standard errors were calculated from triplicate cultures and are representative of three independent experiments.
VOL. 70, 1996 CD4 IN HIV-MEDIATED FUSION AND INFECTION 8023
on November 9, 2019 by guest
http://jvi.asm.org/
123 123
123
with set-dependent differences in the rates and levels of syn-cytium formation which reflected the spread in cell surface expression between the sets (data not shown).
(iii) Cell-free HIV-1 infectivity.To determine whether the differences in syncytium formation extended to virus-cell inter-actions, we investigated HIV-1 replication in cells expressing modified CD4 and CD4wt molecules. To this end, cells were
infected with cell-free HIV-1SF33, and the culture supernatant
was periodically evaluated for the presence of virus. As indi-cated by the infectivity curves in Fig. 3C, significant delays in virus replication were observed for both modified CD4-ex-pressing cells in comparison with CD4wt-exCD4-ex-pressing cells. While the latter cells produced high levels of virus within 5 days postinfection, the mutants required up to 10 days before releasing significant levels of virus into the culture supernatant. These data suggest that, in contrast to syncytium formation, both types of mutations introduced in the D4-TM linker have deleterious effects on cell-free infectivity.
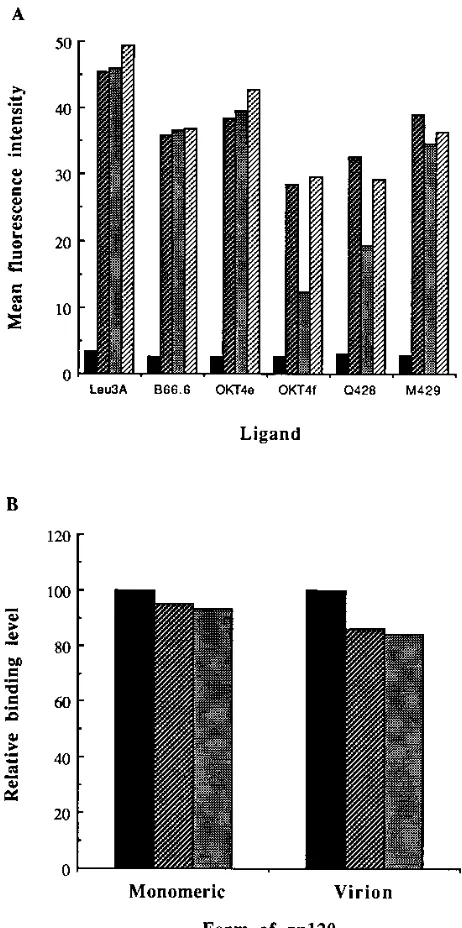
Certain epitopes in the D4-TM-deleted CD4 molecules are significantly perturbed. To determine whether alterations in the structural integrity of the CD4 mutants were responsible for the functional defects observed, we probed the cells with a panel of anti-CD4 MAbs, which, as indicated in Fig. 1A, in-cluded MAbs or each of the extracellular domains of CD4. As illustrated by the histogram in Fig. 4A, the mutations in the D4-TM interdomain did not significantly alter affinities for MAbs directed against either D1 or D4. Recognition by the reference anti-D1 MAb Leu3A was comparable for all three clones assayed. Similarly, reaction with anti-D4 MAb M429 was only slightly reduced for the modified-CD4-expressing cells. Furthermore, both the mutants and CD4wt cells reacted equivalently with B66.6, an MAb that maps to several discon-tinuous residues of D1 (20), as well as with OKT4e, an MAb that recognizes residues brought into proximity by the immu-noglobulin-like fold in D1 (31). Taken together, these results indicate that the deletion or replacement of five CD4 D4-TM interdomain residues probably had minimal effects on the con-formations of the proximal domain, D4, and the most distant extracellular domain, D1.
In contrast to MAbs directed against D1 and D4, those recognizing residues at the D1-D2 interface (OKT4f) (31) and within the D3 domain (Q428) (29) were found to be consis-tently less reactive with d367-371 (Fig. 4A). Recognition of the deletion mutant by OKT4f was perturbed by over 50% com-pared with that of CD4wt, whereas recognition by Q428 was reduced by approximately 40%. On the other hand, the sub-stitution mutant s367-371 reacted with OKT4f at a level similar to that of CD4wt yet seemed slightly perturbed in its capacity to react with Q428. These results indicate that the deletion of five CD4 D4-TM interdomain residues significantly perturbed the structural integrity of the D1-D2 interface and, to a lesser extent, D3.
[image:6.612.319.550.68.532.2]Finally, the region involved in binding gp120 was also checked for possible perturbations by incubating the cells with a saturating concentration of soluble gp120. As indicated in Fig. 4B, a very slight difference in binding capacity was ob-served for CD4wt and modified CD4 molecules. No further difference in binding capacity was observed when cells were incubated with increasing concentrations of rgp120 (data not shown). Since binding to monomeric gp120 may not ade-quately reflect interactions with the naturally occurring oligo-meric form, we also verified that virion binding capacities were not significantly altered by the mutations in CD4. To this end, cells were incubated with culture medium containing cell-free virus, and bound particles were quantified in a p24 antigen-based ELISA. In using this approach, we were certain that the
measurements reflected virion-bound gp120 and not the bind-ing of soluble gp120 also present in the culture supernatant. In comparison to CD4wt (Fig. 4B), both modified CD4-express-ing cells demonstrated a slight drop in virion bindCD4-express-ing capacity, which probably reflected an oligomer-mediated amplification of the differences observed for monomeric binding. However,
FIG. 4. (A) Differences in cell surface reactivity to various ligands detected by flow cytometry (EPICS XL). Cells were incubated at 48C with various CD4 MAbs. Bound MAb was detected with FITC-conjugated secondary anti-bodies. Values shown are the relative mean fluorescence intensities after sub-traction of background (second antibody alone). , A2.01; , CD4wt; , d367-371; , s367-371. (B) Binding of monomeric gp120 and virus particles to CD4wt and D4-TM linker mutants. Cells were incubated at 48C with either monomeric ligand or cell-free virus. Cell-bound rgp120 was revealed in flow cytometry after staining with an anti-HIV-1 antiserum followed by an FITC-conjugated secondary antibody. Cell-bound virions were revealed by p24 antigen assay. Values shown were normalized to binding for CD4wt cells after subtrac-tion of background binding. , CD4wt; , d367-371; , s367-371. The results shown, from one experiment, are representative of three repetitions.
on November 9, 2019 by guest
http://jvi.asm.org/
these minor reductions in binding levels cannot account for the pronounced defects observed in cell fusion and cell-free infec-tivity (Fig. 3). Furthermore, while the binding assays were done with a source of HIV-1 (IIIB/LAV) which differed from that used in the cell fusion and infectivity assays (SF33), mutant d367-371 has been previously shown to be similarly defective towards both strains (50), suggesting that the defect is gener-alized and not attributable to reduced binding to gp120.
Binding of soluble gp120 to cell surface CD4 mutated in the D4-TM linker region leads to decreased exposure of V3 loop.
To exclude the possibility that the delay in syncytium formation or cell-free infectivity could be explained by a delayed capacity to bind gp120, we measured the kinetics of soluble gp120 binding for both modified CD4- and CD4wt-expressing cells. To this end, target cells were incubated for different periods of
time at 378C with a saturating concentration of rgp120. Values
at each time point were expressed as a fraction of the maximal binding attained. In agreement with an earlier report (23), the CD4wt-expressing cells exhibited rapid gp120 binding followed by progressive tapering to the 2-h maximal binding point (Fig. 5A). Similarly, the d367-371 and s367-371 expressers also dis-played rapid binding of rgp120. In contrast to CD4wt mole-cules, however, peak binding with both mutants was reached within 20 min of incubation, after which binding levels declined sharply. Since the absolute binding values for the mutants at
378C and 60 min were significantly lower than those for the
CD4wt cells, compared with equivalent binding at 48C and 60
min (Fig. 4B), we initially suspected that the decrease in sur-face gp120 was due to rapid ligand internalization. In line with
this hypothesis, the kinetics of binding of gp120 at 48C for both
mutants were restored to a typical saturation profile (data not shown). However, when we measured modulation of surface CD4 or intracellular gp120, no correlation with the decreasing extracellular gp120 could be observed (data not shown).
Knowing that binding of CD4 to gp120 has a modulatory effect on the exposure of the V3 loop (63), we considered the possibility that the level of gp120 detected was dropping simply because the epitope probed to reveal bound ligand became less accessible. To verify this possibility, we replaced the anti-V3 loop MAb initially used to detect surface-bound gp120 with a polyclonal antiserum to HIV-1 derived from an HIV-infected individual. As shown by the kinetic profiles in Fig. 5B, for this less selective method of detecting bound gp120, a typical time-dependent saturation curve was observed for both mutants and CD4wt cells. This result indicates that the decline in the amount of gp120 detected with the anti-V3 loop MAb was probably due to a decrease in the exposure of this particular epitope and not to a decrease in the amount of ligand bound to the cells. To determine whether this epitope loss occurred in other regions of gp120, we extended our kinetic assays to include detection with MAb 48d, which recognizes conforma-tional epitopes close to the CD4 binding region that are known to be up modulated by the binding of CD4 (71). The detection of bound gp120 with 48d failed to reveal variations in epitope exposure between the CD4wt and modified CD4 molecules (data not shown), suggesting that the aberrant effects of the mutations on postbinding modulations are not generalized.
DISCUSSION
While the human CD4 molecule is believed to play an im-portant role in HIV-1 Env-mediated fusion and syncytium formation (7, 10, 23, 29, 54, 58), its precise contribution to postbinding events remains nebulous. In this study we at-tempted to clarify the role of the CD4 molecule in the contexts of both cell-to-cell interactions leading to syncytium formation
and virus-to-cell interactions leading to entry and productive replication. In light of some contradictory findings (37, 45) and recent proposals (5), we focused our attention on the CD4 TM domain and its short adjoining extracellular region. In agree-ment with several reports (16, 32, 37), we found that the CD4 TM domain could be replaced with the equivalent domain of a heterologous molecule without inhibiting the processes of HIV-mediated cell fusion and cell-free infectivity. More im-portantly, our findings indicate that mutations in the short region linking D4 to the TM domain have a profound impact on the capacity of the CD4 molecule to sustain the events of
FIG. 5. Effects of mutations in the CD4 D4-TM interface on gp120 binding kinetics. Cells were incubated with a saturating concentration of gp120 at 378C for incremental periods of time. Cell-bound gp120 was detected by flow cytom-etry after staining with the anti-V3 loop MAb NEA-9305 followed by an Rphy-coerythrin-conjugated secondary antibody (A) or after staining with an anti-HIV-1 antiserum followed by an FITC-conjugated secondary antibody (B). Experiments were repeated more than three times, and the results shown are from one representative experiment. max, maximum.
VOL. 70, 1996 CD4 IN HIV-MEDIATED FUSION AND INFECTION 8025
on November 9, 2019 by guest
http://jvi.asm.org/
membrane fusion in the contexts of both cell-cell and virus-cell interactions. Therefore, we suggest that this region may be involved in mediating the conformational changes that are thought to activate the fusion potential in gp41 (61).
The concept that highly dynamic interactions between HIV-1 Env and CD4 occur has been well documented and lends credence to the model of receptor-mediated activation of fusion (1, 51), whereby the binding step triggers a complex series of changes in conformation that culminates in the un-masking of the fusion peptide located in the amino terminus of gp41. This model of receptor-based modulations is supported by the enhanced detection, upon treatment with soluble or cell-associated CD4, of epitopes normally buried in both gp41 and gp120 (34, 47, 63, 65, 71). Modulation of epitopes in the CD4 molecule upon ligand binding, while not as well docu-mented as for Env, has also been reported (10, 24, 61, 69, 74). Furthermore, structure-based data generated from the super-position of several independently crystallographed fragments of CD4 (5, 39, 59, 75) have revealed patterns of deviation indicative of concentrated areas of mobility in the molecule. Some of the flexibility observed in CD4 has been ascribed to the junctions of the extracellular domains (5, 26, 38, 39, 60), particularly the D2-D3 junction, which demonstrates extensive susceptibility to enzymatic proteolysis (29). The base of D4, although not resolved in the rat D3-D4 model because of proximity to the TM domain, is also thought to contribute to the mobility of the extracellular domains of CD4 (5). The premise that these flexible junctions may contribute to biolog-ical functions of the CD4 molecule (5, 38) is now substantiated by our findings that a modified D4-TM linker leads to reduced activity toward HIV-1. Finally, a recent study on the thermo-dynamic properties of soluble CD4 has revealed extensive in-terdomain interactions, especially between D1, D2, and D3, interactions which are thought to stabilize D4 (70). If such extensive interdomain contacts do exist, this further implies that many of the biological properties ascribed to CD4 are highly sensitive to conformational perturbations.
The D4-TM interdomain deletion mutant found to be defi-cient in syncytium formation and cell-free infection exhibited certain structural aberrations. By probing all four extracellular domains of the mutated CD4 and CD4wt molecules with var-ious anti-CD4 MAbs, we established that certain epitopes were perturbed by the deletion, namely, in the D1-D2 interface and to a lesser extent in D3. When the deletion was replaced by a heterologous substitution, all epitopes demonstrated CD4wt-like levels of reactivity towards the panel of MAbs, with the exception of a very slight perturbation in D4. The replacement also restored fusion capacity to CD4wt levels, strongly suggest-ing that there is a link between the various perturbations ob-served in the structure of the deletion mutant and its deficiency in cell-cell fusion. The CD4 domains perturbed by the deletion may be involved in specific postbinding interactions or may mediate conformational changes that allow exposure of the fusion peptide in gp41. As a consequence, structural perturba-tions such as those observed in the deletion mutant would lead to a delayed and decreased activation of the gp41 fusion po-tential. In agreement with this proposal are indications that certain MAbs shown to inhibit fusion without affecting fusion operate by limiting postbinding conformational changes (7, 54). Furthermore, since there is no apparent homology be-tween the five CD4 residues deleted in mutant d367-371 (TPVQP) and the five CD8 substitution residues in mutant s367-371 (DFACD) that restored fusion, we can speculate that the decrease in fusion capacity was probably not the result of deleting residues involved in specific interactions of the fusion process. However, the possibility that shortening the molecule
by five residues also contributed to the perturbations observed cannot be excluded. Thus, in addition to the structural alter-ations identified and in agreement with structure-based pro-posals (5, 38), it may be that part of the deficiency in fusion potential was due to a restriction in the mobility of a molecule that had been amputated at its base.
Epitopes in gp120-gp41 modulated upon CD4 binding in-clude the V3 loop, as demonstrated by enhanced anti-V3 loop MAb staining (63) and increased susceptibility to exogenous enzymes (13). It has been suggested that cleavage of the V3 loop may be the trigger of the fusion process (53, 64); however, that such a phenomenon actually occurs remains to be clearly established (42). In probing cell-bound gp120 with an anti-V3 MAb to verify that the delay in syncytium formation observed with mutant d367-371 was not attributable to a lower rate of gp120 binding, we discovered that both CD4 deletion and substitution mutants demonstrated a gradual decrease in the amount of surface-bound gp120 detected. However, when the cells were probed with a polyclonal antiserum to HIV instead of the anti-V3 loop MAb, all curves followed the typical satu-ration pattern previously described by others (23), indicating that the level of gp120 binding to mutant CD4 cell surfaces was not actually declining and that the global conformation of the ligand was being maintained over time. The gradual decline in reactivity towards the anti-V3 loop MAb may thus be ex-plained by a regression in the accessibility of the V3 epitope following binding of gp120 to either d367-371 or s367-371. This
downward modulation, not observed at 48C, was unexpected
considering that we used monomeric gp120, a form not nor-mally susceptible to significant conformational changes (47, 65). Although not verified in this context, oligomeric gp120, the form found at the virion or cell surface and much more conducive to conformational changes, would be expected to be even more extensively affected by the mutations in the CD4 molecule.
Considering that the substitution completely restored fusion capacity, we have discarded the possibility that the altered modulatory effect on the V3 loop, observed with both mutants, has a significant effect on cell-cell fusion. On the other hand, a decrease in the exposure of the V3 loop may have a significant impact on postbinding events of virus-cell contacts, since both mutants displayed significantly delayed infectivity curves com-pared with CD4wt cells. While the virus-cell and cell-cell in-teractions that characterize membrane fusion are believed to be similar, there have been several reports suggesting that differences between these two processes exist (9, 27, 36, 53, 55, 57, 66). We have described a modification which appears to be more inhibitory toward virus-cell interactions than toward cell-cell interactions leading to syncytium formation. Consistent with this notion, anti-V3 loop MAbs and the V3 loop-selective inhibitor heparin have been shown to neutralize cell-free in-fectivity without inhibiting syncytium formation (27, 53). Fur-thermore, Hasunuma et al. (28) have described MAbs against D3-D4 of CD4 that show inhibitory activity toward cell-free infection but not syncytium formation.
In summary, our findings indicate that the TM domain of CD4 may be replaced with that of a heterologous protein without affecting HIV-1-mediated processes of syncytium for-mation and cell-free infectivity. As suggested by others (45), stabilization of cell surface expression for interaction with HIV-1 envelope glycoproteins may be the key attribute pro-vided by the TM domain in the fusion process. Furthermore, we have provided evidence that the short region linking D4 to the TM domain is an important determinant of postbinding events leading to viral internalization and syncytium formation. Deletion of the linker region creates perturbations in the
on November 9, 2019 by guest
http://jvi.asm.org/
tral domains of the extracellular moiety of CD4 and possibly affects the mobility of the entire molecule. These alterations appear to be responsible for the diminished fusion capacity observed in cells expressing the CD4 deletion variant. Finally, both the deletion and substitution of the linker region lead to decreased accessibility of the V3 loop in gp120 following re-ceptor binding. This perturbation, which does not appear to extend its effect to other epitopes of gp120 known to be mod-ulated by CD4, appears to be at least partly responsible for the delay in cell-free infectivity observed in cells expressing either CD4 variant. How modifications at the base of the CD4 extra-cellular moiety translate into an underexposed V3 loop and reduced infectivity is not clear. Nonetheless, our findings sug-gest, as others have (63, 65, 71), that exposure of the V3 loop is an essential element of the postbinding events leading to virus internalization. Whether the recently identified cofactor participates at this level warrants further investigation (19). However, we would need to reconcile the cofactor requirement with our finding that the epitope loss affects only virus-cell interactions and to extend our investigation of other cell types and HIV-1 isolates to address differences in cofactor usage (19).
These findings contribute to the concept that receptor bind-ing must be followed by an intricate cascade of changes in conformation before the fusion potential in gp41 can be acti-vated (17, 18, 64). It remains to be determined whether the modifications made in the D4-TM linker region restrict inter-face flexibility or affect postbinding modulations. The potential interactions between CD4, Env, and an accessory molecule warrant further investigation in the contexts of both sequential events and formation of oligomeric fusion complexes (19, 40, 46). Nonetheless, this work clearly establishes that the role of the CD4 molecule in HIV-1 entry and syncytium formation goes beyond that of serving as a receptor for gp120.
ACKNOWLEDGMENTS
We extend our thanks to Maurice Dufour for performing the cyto-fluorometric analyses.
This work was funded in part by the National Health Research and Development Program (NHRDP) of Canada and the Fonds de la Recherche en Sante´ du Que´bec (FRSQ) to L. Poulin. L. Poulin is a recipient of an AIDS Research Scholar Award from the NHRDP. S. Moir is a Ph.D. student supported by the NHRDP.
REFERENCES
1. Allan, J. S. 1991. Receptor-mediated activation of immunodeficiency viruses in viral fusion. Science 252:1322–1323.
2. Bedinger, P., A. Moriarty, R. C. von Borstel II, N. J. Donovan, K. S. Steimer,
and D. Littman.1988. Internalization of the human immunodeficiency virus does not require the cytoplasmic domain of CD4. Nature (London) 334:162– 165.
3. Benkirane, M., M. Hirn, D. Carrie`re, and C. Devaux. 1995. Functional epitope analysis of the human CD4 molecule: antibodies that inhibit human immunodeficiency virus type 1 gene expression bind to the immunoglobulin CDR3-like region of CD4. J. Virol. 69:6898–6902.
4. Berger, E. A., J. D. Lifson, and L. E. Eiden. 1991. Stimulation of glycoprotein gp120 dissociation from the envelope glycoprotein complex of human im-munodeficiency virus type 1 by soluble CD4 and CD4 peptide derivatives: implications for the role of the complementarity-determining region 3-like region in membrane fusion. Proc. Natl. Acad. Sci. USA 88:8082–8086. 5. Brady, R. L., E. J. Dodson, G. G. Dodson, G. Lange, S. J. Davis, A. F.
Williams, and A. N. Barclay.1993. Crystal structure of domains 3 and 4 of rat CD4: relation to the NH2-terminal domains. Science 260:979–983. 6. Broder, C. C., and E. A. Berger. 1993. CD4 molecules with a diversity of
mutations encompassing the CDR3 region efficiently support human immu-nodeficiency virus type 1 envelope glycoprotein-mediated cell fusion. J. Vi-rol. 67:913–926.
7. Burkly, L. C., D. Olson, R. Shapiro, G. Winkler, J. J. Rosa, D. W. Thomas,
C. Williams, and P. Chisholm.1992. Inhibition of HIV infection by a novel CD4 domain 2-specific monoclonal antibody. Dissecting the basis for its inhibitory effect on HIV-induced cell fusion. J. Immunol. 149:1779–1787.
8. Camerini, D., and B. Seed. 1990. A CD4 domain important for HIV-medi-ated syncytium formation lies outside the virus binding site. Cell 60:747–754. 9. Cao, J., L. Bergeron, E. Helseth, M. Thali, H. Repke, and J. Sodroski. 1993. Effects of amino acid changes in the extracellular domain of the human immunodeficiency virus type 1 gp41 envelope glycoprotein. J. Virol. 67:2747– 2755.
10. Celada, F., C. Cambiaggi, J. Maccari, S. Burastero, T. Gregory, E. Patzer, J.
Porter, C. McDanal, and T. Matthews.1990. Antibody raised against soluble CD4-rgp120 complex recognizes the CD4 moiety and blocks membrane fusion without inhibiting CD4-gp120 binding. J. Exp. Med. 172:1143–1150. 11. Choe, H. R., and J. Sodroski. 1992. Contribution of charged amino acids in the CDR2 region of CD4 to HIV-1 gp120 binding. J. Acquired Immune Defic. Syndr. 5:204–210.
12. Clark, S. J., W. A. Jefferies, A. N. Barclay, J. Gagnon, and A. F. Williams. 1987. Peptide and nucleotide sequences of rat CD4 (W3/25) antigen: evi-dence for derivation from a structure with four immunoglobulin-related domains. Proc. Natl. Acad. Sci. USA 84:1649–1653.
13. Clements, G. J., M. J. Price-Jones, P. E. Stephens, C. Sutton, T. F. Schulz,
P. R. Clapham, J. A. McKeating, M. O. McClure, S. Thomson, M. Marsh, J. Kay, R. A. Weiss, and J. P. Moore.1991. The V3 loops of the HIV-1 and HIV-2 surface glycoproteins contain proteolytic cleavage sites: a possible function in viral fusion? AIDS Res. Hum. Retroviruses 7:3–16.
14. Darveau, A., A. Pelletier, and J. Perreault. 1995. PCR-mediated synthesis of chimeric molecules. Methods Neurosci. 26:77–85.
15. Dedera, D., and L. Ratner. 1991. Demonstration of two distinct cytopathic effects with syncytium formation-defective human immunodeficiency virus type 1 mutants. J. Virol. 65:6129–6136.
16. Diamond, D. C., R. Finberg, S. Chaudhuri, B. P. Sleckman, and S. J.
Bu-rakoff.1990. Human immunodeficiency virus infection is efficiently mediated by a glycolipid-anchored form of CD4. Proc. Natl. Acad. Sci. USA 87:5001– 5005.
17. Eiden, L. E., and J. D. Lifson. 1992. HIV interactions with CD4: a continuum of conformations and consequences. Immunol. Today 13:201–206. 18. Ellens, H., and C. Larsen. 1993. CD4-induced changes in gp120/gp41
con-formation and its potential relationship to fusion, p. 291–312. In J. Bentz (ed.), Viral fusion mechanisms. CRC Press, Boca Raton, Fla.
19. Feng, Y., C. C. Broder, P. E. Kennedy, and E. A. Berger. 1996. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 272:872–877.
20. Fleury, S., D. Lamarre, S. Meloche, S. E. Ryu, C. Cantin, W. A. Hendrickson,
and R.-P. Sekaly.1991. Mutational analysis of the interaction between CD4 and class II MHC: class II antigens contact CD4 on a surface opposite the gp120-binding site. Cell 66:1037–1049.
21. Freed, E. O., D. J. Myers, and R. Risser. 1990. Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc. Natl. Acad. Sci. USA 87:4650–4654.
22. Fu, Y. K., T. K. Hart, Z. L. Jonak, and P. J. Bugelski. 1993. Physicochemical dissociation of CD4-mediated syncytium formation and shedding of human immunodeficiency virus type 1 gp120. J. Virol. 67:3818–3825.
23. Golding, H., R. Blumenthal, J. Manischewitz, D. R. Littman, and D. S.
Dimitrov.1993. Cell fusion mediated by interaction of a hybrid CD4. CD8 molecule with the human immunodeficiency virus type 1 envelope glycopro-tein does occur after a long lag time. J. Virol. 67:6469–6475.
24. Golding, H., D. S. Dimitrov, J. Manischewitz, C. C. Broder, J. Robinson, S.
Fabian, D. R. Littman, and C. K. Lapham.1995. Phorbol ester-induced down modulation of tailless CD4 receptors requires prior binding of gp120 and suggests a role for accessory molecules. J. Virol. 69:6140–6148. 25. Hammarskjo¨ld, M.-L., J. Heimer, B. Hammarskjo¨ld, I. Sangwan, L. Albert,
and D. Rekosh.1989. Regulation of human immunodeficiency virus env expression by the rev gene product. J. Virol. 63:1959–1966.
26. Harrison, S. C., J. Wang, Y. Yan, T. Garrett, J. Liu, U. Moebius, and E.
Reinherz.1992. Structure and interactions of CD4. Cold Spring Harbor Symp. Quant. Biol. 26:449–453.
27. Harrop, H. A., D. R. Coombe, and C. C. Rider. 1994. Heparin specifically inhibits binding of V3 loop antibodies to HIV-1 gp120, an effect potentiated by CD4 binding. AIDS 8:183–192.
28. Hasunuma, T., H. Tsubota, M. Watanabe, Z. W. Chen, C. I. Lord, L. C.
Burkly, J. F. Daley, and N. L. Letvin.1992. Regions of the CD4 molecule not involved in virus binding or syncytia formation are required for HIV-1 infection of lymphocytes. J. Immunol. 148:1841–1846.
29. Healey, D., L. Dianda, J. P. Moore, J. S. McDougal, M. J. Moore, P. Estess,
D. Buck, P. D. Kwong, P. C. Beverley, and Q. J. Sattentau.1990. Novel anti-CD4 monoclonal antibodies separate human immunodeficiency virus infection and fusion of CD41cells from virus binding. J. Exp. Med. 172: 1233–1242.
30. Helseth, E., U. Olshevsky, D. Gabuzda, B. Ardman, W. Haseltine, and J.
Sodroski.1990. Changes in the transmembrane region of the human immu-nodeficiency virus type 1 gp41 envelope glycoprotein affect membrane fu-sion. J. Virol. 64:6314–6318.
31. Jameson, B. A., P. E. Rao, L. I. Kong, B. H. Hahn, G. M. Shaw, L. E. Hood,
and S. B. H. Kent.1988. Location and chemical synthesis of a binding site for HIV-1 on the CD4 protein. Science 240:1335–1339.
VOL. 70, 1996 CD4 IN HIV-MEDIATED FUSION AND INFECTION 8027
on November 9, 2019 by guest
http://jvi.asm.org/
32. Jasin, M., K. A. Page, and D. R. Littman. 1991. Glycosylphosphatidylinosi-tol-anchored CD4/Thy-1 chimeric molecules serve as human immunodefi-ciency virus receptors in human, but not mouse, cells and are modulated by gangliosides. J. Virol. 65:440–444.
33. Johansen, T. E., M. S. Scholler, S. Tolstoy, and T. W. Schwartz. 1990. Biosynthesis of peptide precursors and protease inhibitors using new consti-tutive and inducible eukaryotic expression vectors. FEBS Lett. 267:289–294. 34. Kang, C. Y., K. Hariharan, M. R. Posner, and P. Nara. 1993. Identification of a new neutralizing epitope conformationally affected by the attachment of CD4 to gp120. J. Immunol. 151:449–457.
35. Klasse, P. J., and J. P. Moore. 1992. Kinetics of the HIV-CD4 interactions and virus-cell fusion. AIDS 6:325–327.
36. Konopka, K., E. Pretzer, F. Celada, and N. Duzgunes. 1995. A monoclonal antibody to the gp120-CD4 complex has differential effect on HIV-induced syncytium formation and viral infectivity. J. Gen. Virol. 76:669–679. 37. Kost, T. A., J. A. Kessler, I. R. Patel, J. G. Gray, L. K. Overton, and S. G.
Carter.1991. Human immunodeficiency virus infection and syncytium for-mation in HeLa cells expressing glycophospholipid-anchored CD4. J. Virol.
65:3276–3283.
38. Kwong, P. D., S. E. Ryu, W. A. Hendrickson, R. Axel, R. M. Sweet, G.
Folena-Wasserman, P. Hensley, and R. W. Sweet.1990. Molecular charac-teristics of recombinant human CD4 as deduced from polymorphic crystals. Proc. Natl. Acad. Sci. USA 87:6423–6427.
39. Lange, G., S. J. Lewis, G. N. Murshudov, G. G. Dodson, P. C. Moody, J. P.
Turkenburg, A. N. Barclay, and R. L. Brady.1994. Crystal structure of an extracellular fragment of the rat CD4 receptor containing domains 3 and 4. Structure 2:469–481.
40. Langedijk, J. P., W. C. Puijk, W. van Hoorn, and R. H. Meloen. 1993. Location of CD4 dimerization site explains critical role of CDR3-like region in HIV-1 infection and T-cell activation and implies a model for complex of coreceptor-MHC. J. Biol. Chem. 268:16875–16878.
41. Lasky, L. A., G. Nakamura, D. H. Smith, C. Fennier, C. Shimasaki, E.
Patzer, P. Berman, T. Gregory, and D. J. Capon.1987. Delineation of a region of the human immunodeficiency virus type 1 gp120 glycoprotein critical for interaction with the CD4 receptor. Cell 50:975–985.
42. Lazaro, I., D. Naniche, N. Signoret, A. M. Bernard, D. Marguet, D.
Klatz-mann, T. Dragic, M. Alizon, and Q. Sattentau.1994. Factors involved in entry of the human immunodeficiency virus type 1 into permissive cells: lack of evidence of a role for CD26. J. Virol. 68:6535–6546.
43. Lifson, J. D., G. R. Reyes, M. S. McGrath, S. B. Stein, and E. G. Engleman. 1986. AIDS retrovirus induced cytopathology: giant cell formation and in-volvement of CD4 antigen. Science 232:1123–1127.
44. Maddon, P. J., D. R. Littman, M. Godfrey, D. E. Maddon, L. Chess, and R.
Axel.1985. The isolation and nucleotide sequence of a cDNA encoding the T cell surface protein T4: a new member of the immunoglobulin gene family. Cell 42:93–104.
45. Marshall, W. L., E. S. Mittler, P. Avery, J. P. Lawrence, and R. W. Finberg. 1994. Glycosylphosphatidylinositol-anchored CD4 supports human immuno-deficiency virus type 1 replication, but not cytopathic effect, in T-cell trans-fectants. J. Virol. 68:4039–4042.
46. Matthews, T. J., C. Wild, C. H. Chen, D. P. Bolognesi, and M. L. Greenberg. 1994. Structural rearrangements in the transmembrane glycoprotein after receptor binding. Immunol. Rev. 140:93–104.
47. McKeating, J. A., J. Cordell, C. J. Dean, and P. Balfe. 1992. Synergistic interaction between ligands binding to the CD4 binding site and V3 domain of human immunodeficiency virus type I gp120. Virology 191:732–742. 48. Moebius, U., L. K. Clayton, S. Abraham, S. C. Harrison, and E. L. Reinherz.
1992. The human immunodeficiency virus gp120 binding site on CD4: de-lineation by quantitative equilibrium and kinetic binding studies of mutants in conjunction with a high-resolution CD4 atomic structure. J. Exp. Med.
176:507–517.
49. Moir, S., and L. Poulin. 1996. Expression of HIV-1 env gene in a human T-cell line for a rapid and quantifiable fusion assay. AIDS Res. Hum. Ret-roviruses 12:811–820.
50. Moir, S., and L. Poulin. Unpublished observations.
51. Moore, J. P. 1991. Model for receptor mediated activation of immunodefi-ciency viruses in viral fusion. Science 252:1322–1323.
52. Moore, J. P. 1993. A monoclonal antibody to the CDR-3 region of CD4 inhibits soluble CD4 binding to virions of human immunodeficiency virus type 1. J. Virol. 67:3656–3659.
53. Moore, J. P., and P. L. Nara. 1991. The role of the V3 loop of gp120 in HIV infection. AIDS 5:S21–S33.
54. Moore, J. P., Q. J. Sattentau, P. J. Klasse, and L. C. Burkly. 1992. A
monoclonal antibody to CD4 domain 2 blocks soluble CD4-induced confor-mational changes in the envelope glycoproteins of human immunodeficiency virus type 1 (HIV-1) and HIV-1 infection of CD41cells. J. Virol. 66:4784– 4793.
55. Pantaleo, G., G. Poli, L. Butini, C. Fox, A. I. Dayton, and A. S. Fauci. 1991. Dissociation between syncytia formation and HIV spreading. Suppression of syncytia formation does not necessarily reflect inhibition of HIV infection. Eur. J. Immunol. 21:1771–1774.
56. Pear, W. S., G. P. Nolan, M. L. Scott, and D. Baltimore. 1993. Production of high-titer helper-free retroviruses by transient transfection. Proc. Natl. Acad. Sci. USA 90:8392–8396.
57. Pleskoff, O., M. Seman, and M. Alizon. 1995. Amphotericin B derivative blocks human immunodeficiency virus type 1 entry after CD4 binding: effect on virus-cell fusion but not on cell-cell fusion. J. Virol. 69:570–574. 58. Poulin, L., L. A. Evans, S. B. Tang, A. Barboza, H. Legg, D. R. Littman, and
J. A. Levy.1991. Several CD4 domains can play a role in human immuno-deficiency virus infection in cells. J. Virol. 65:4893–4901.
59. Ryu, S. E., P. D. Kwong, A. Truneh, T. G. Porter, J. Arthos, M. Rosenberg,
X. P. Dai, N. H. Xuong, R. Axel, R. W. Sweet, and W. A. Hendrickson.1990. Crystal structure of an HIV-binding recombinant fragment of human CD4. Nature (London) 348:419–426.
60. Ryu, S. E., A. Truneh, R. W. Sweet, and W. A. Hendrickson. 1994. Structures of an HIV and MHC binding fragment from human CD4 as refined in two crystal lattices. Structure 2:59–74.
61. Sattentau, Q. J. 1992. CD4 activation of HIV fusion. Int. J. Cell Cloning
10:323–332.
62. Sattentau, Q. J., A. G. Dalgleish, R. A. Weiss, and P. C. L. Beverly. 1986. Epitopes of the CD4 antigen and HIV infection. Science 234:1120–1123. 63. Sattentau, Q. J., and J. P. Moore. 1991. Conformational changes induced in
the human immunodeficiency virus envelope glycoprotein by soluble CD4 binding. J. Exp. Med. 174:407–415.
64. Sattentau, Q. J., and J. P. Moore. 1993. The role of CD4 in HIV binding and entry. Philos. Trans. R. Soc. Lond. B 342:59–66.
65. Sattentau, Q. J., J. P. Moore, F. Vignaux, F. Traincard, and P. Poignard. 1993. Conformational changes induced in the envelope glycoproteins of the human and simian immunodeficiency viruses by soluble receptor binding. J. Virol. 67:7383–7393.
66. Simmons, G., A. McKnight, Y. Takeuchi, H. Hoshino, and P. R. Clapham. 1995. Cell-to-cell fusion, but not virus entry in macrophages by T-cell line tropic HIV-1 strains: a V3 loop-determined restriction. Virology 209:696– 700.
67. Sodroski, J., W. C. Goh, C. Rosen, K. Campbell, and W. A. Haseltine. 1986. Role of the HTLV-III/LAV envelope in syncytium formation and cytopath-icity. Nature (London) 322:470–474.
68. Stein, B. S., S. D. Gowda, J. D. Lifson, R. C. Penhallow, K. G. Bensch, and
E. G. Engleman.1987. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 49:659–668. 69. Szabo, G. J., P. S. Pine, J. L. Weaver, P. E. Rao, and A. Aszalos. 1992. CD4
changes conformation upon ligand binding. J. Immunol. 149:3596–3604. 70. Tendian, S. W., D. G. Myszka, R. W. Sweet, I. M. Chaiken, and C. G.
Brouillette.1995. Interdomain communication of T-cell CD4 studied by absorbance and fluorescence difference spectroscopy measurements of urea-induced unfolding. Biochemistry 34:6464–6474.
71. Thali, M., J. P. Moore, C. Furman, M. Charles, D. D. Ho, J. Robinson, and
J. Sodroski.1993. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J. Virol. 67:3978–3988.
72. Truneh, A., D. Buck, D. R. Cassatt, R. Juszczak, S. Kassis, S. E. Ryu, D.
Healey, R. Sweet, and Q. Sattentau.1991. A region in domain 1 of CD4 distinct from the primary gp120 binding site is involved in HIV infection and virus-mediated fusion. J. Biol. Chem. 266:5942–5948.
73. Tsui, P., R. W. Sweet, G. Sathe, and M. Rosenberg. 1992. An efficient phage plaque screen for the random mutational analysis of the interaction of HIV-1 gp120 with human CD4. J. Biol. Chem. 267:9361–9367.
74. Walker, L., D. Wilks, J. O’Brien, J. Habeshaw, and A. Dalgleish. 1992. Localized conformational changes in the N-terminal domain of CD4 iden-tified in competitive binding assay of monoclonal antibodies and HIV-1 envelope glycoprotein. AIDS Res. Hum. Retroviruses 8:1083–1090. 75. Wang, J., Y. Yan, T. P. J. Garrett, J. Liu, D. W. Rodgers, R. L. Garlick, G. E.
Tarr, Y. Husain, E. L. Reinherz, and S. C. Harrison.1990. Atomic structure of a fragment of human CD4 containing two immunoglobulin-like domains. Nature (London) 348:411–418.