Copyright © 1997, American Society for Microbiology
Identification of Sequences Downstream of the Primer Binding Site
That Are Important for Efficient Replication of
Human Immunodeficiency Virus Type 1
XUGUANG LI,
1,2CHEN LIANG,
1YUDONG QUAN,
1RAVI CHANDOK,
2MICHAEL LAUGHREA,
1,2MICHAEL A. PARNIAK,
1,2LAWRENCE KLEIMAN,
1,2,3ANDMARK A. WAINBERG
1,2,3*
McGill University AIDS Centre, Jewish General Hospital,
1and Departments of Medicine
2and Microbiology,
3McGill University, Montreal, Quebec, Canada
Received 16 December 1996/Accepted 26 March 1997
Reverse transcription of retroviruses is initiated from an 18-nucleotide (nt) primer binding site (PBS),
located within the 5
*
region of viral genomic RNA, to which the host cell-derived tRNA primer is annealed and
also involves viral genomic sequences outside the PBS. We constructed proviral DNA clones of human
immunodeficiency virus (HIV) that had selective deletions of either a 7-nt segment found immediately
down-stream of the PBS or an extended nontranslated 54-nt stretch located immediately downdown-stream of the PBS and
containing the aforementioned 7-nt segment. Synthesis of minus-strand strong-stop DNA was assessed with
MT-4 cells infected with viruses derived from COS-7 cells that had been transfected with these various
constructs. We found that similar levels of minus-strand strong-stop DNA as well as DNA produced after
template switching were expressed in MT-4 cells infected with COS-7-derived wild-type viruses or with viruses
that had the 7-nt segment deleted. In contrast, significantly lower levels of viral DNA were detected in MT-4
cells after infection with viruses that had deletions of the 54-nt stretch. Furthermore, the molecular clone
containing the 7-nt deletion was able to replicate with wild-type kinetics, while that containing the 54-nt
deletion displayed a significantly diminished capacity in this regard. Further deletion analysis showed that a
16-nt segment at the 3
*
end of this 54-nt segment was largely responsible for these effects. We also conducted
studies to determine levels of viral mRNA in COS-7 cells that had been transfected with equivalent amounts
of DNA derived from either a wild-type HIV construct or our various deletion mutants. In the case of
transfections performed with the 7-nt deletion mutant and wild-type HIV DNA, high levels of viral mRNA
transcripts were detected, which was not the case for the 54 nt-deletion mutant. However, these various mRNAs
possessed similar stabilities, as shown through studies in which transcript formation was arrested by
treat-ment of cells with actinomycin D. Thus, the 54-nt segtreat-ment of 5
*
nontranslated RNA, located downstream of the
PBS, is involved in efficient expression of each of viral DNA, mRNA, and infectious virus.
Reverse transcription begins at the primer binding site
(PBS) of unspliced retroviral RNA, to which a tRNA primer is
bound (38). The PBS of human immunodeficiency virus type 1
(HIV-1) is located approximately 180 nucleotides (nt) from the
5
9
terminus of genomic RNA and is flanked at its 5
9
end by a
region referred to as R/U5 (49). This R/U5 region possesses a
number of functional activities, including a role in packaging of
viral RNA, a role in binding of the Tat transactivator protein,
and involvement in reverse transcription and integration of
proviral DNA (1, 7, 12, 13, 14, 21, 24, 25, 28, 29, 34, 43, 44, 52,
56, 57). A 133-nt noncoding and untranslated region is located
downstream of the PBS and upstream of the gag initiation
codon (49). The function of this sequence, especially its 5
9
portion, is not well understood, even though its 3
9
end is
thought to be involved in packaging, splicing, and dimerization
of genomic RNA and in translation of viral proteins (2, 9, 11,
15, 32, 35, 41, 42, 45, 51).
The PBS region of HIV-1 RNA and surrounding sequences
appear to be highly structured as determined by computer
modelling and chemical analysis (6, 8, 22). The unfolding of the
tRNA primer and of the RNA template is thought to be
me-diated by the viral nucleocapsid protein (NCp) (30, 31, 37).
Formation of the reverse transcription initiation complex
in-volves base pairing between the PBS and a complementary
18-nt region at the 3
9
end of tRNA, as well as additional
interactions between sequences that neighbor the PBS and the
remainder of the tRNA primer. In avian retroviruses, the
ef-ficiency of a tRNA
Trp-PBS complex in initiation of reverse
transcription was enhanced by inclusion of viral genomic
se-quences upstream of the PBS and the T
C
C loop of tRNA
Trp(1, 34). Furthermore, disruption of a stem-loop structure, i.e.,
the U-IR stem near the PBS, caused diminished reverse
tran-scription in both avian and murine retroviruses (12, 13, 44, 48).
We have studied the role in viral replication of noncoding
sequences that lie downstream of the PBS by introducing a
deletion of 7 nt immediately downstream of this region (33),
designated pHIV/del-7. Alternatively, we generated a 54-nt
deletion of the 5
9
portion of the noncoding region, designated
pHIV/del-LD, located immediately downstream of the PBS
and containing the aforementioned 7-nt sequence. Previous
studies by our group have shown that the first of these
dele-tions is not required for cell-free synthesis of minus-strand
strong-stop DNA in reactions performed with recombinant
reverse transcriptase (RT), NCp, tRNA
3Lysand viral RNA
tem-plate (37). We now show that deletion of the 7-nt segment
(pHIV/del-7) had relatively minor effects on in vivo reverse
transcription of the viral DNA product in MT-4 cells, in
con-trast to results obtained with deletion of the 54-nt segment, i.e.,
pHIV/del-LD. The latter sequence was also independently
in-* Corresponding author. Mailing address: McGill University AIDS
Centre, Lady Davis Institute-Jewish General Hospital, 3755
Cote-Ste-Catherine Rd., Montreal, Quebec H3T 1E2, Canada. Phone: (514) 340
8260. Fax: (514) 340 7537. E-mail: [email protected].
6003
on November 9, 2019 by guest
http://jvi.asm.org/
volved in efficient expression of viral mRNA. The pHIV/del-7
virus displayed replication kinetics similar to those of wild-type
viruses, while the pHIV/del-LD virus, as well as viruses
con-taining other deletions in this region, were significantly
im-paired in this regard.
(The research performed by Xuguang Li was in partial
ful-fillment of the requirements for a Ph.D. from the Faculty of
Graduate Studies and Research, McGill University, Montreal,
Canada.)
MATERIALS AND METHODS
Molecular clones with deletion mutations in sequences surrounding the PBS. The HxB2D recombinant clone of infectious DNA, obtained from the National Institutes of Health reagent repository, was used as a starting material for further genetic alteration. We modified a previously described PCR-based megaprimer mutagenesis procedure to generate deletions in the vicinity of the PBS (47). The primer selected for the 7-nt deletion (pHIV/del-7), immediately downstream of the PBS (nt 654 to 660), was 59-TGGCGCCCGAACAGGGACCTGAAAGGG AAACCAGAG-39. The primer for deletion of the 54-nt segment (pHIV/del-LD), also downstream of the PBS (nt 654 to 707), was 59-TGGCGCCCG AAC AGGGACCGCGCACGGCAAGAGGCG-39. These primers were used as forward primers in conjunction with a backward primer (termed Pst 1) (nt 1405 to 1422; 59-CCATTCTGCAGCTTCCTC-39) to specifically amplify sequences in regard to each of these deletions. The resulting amplified products were used as megaprimers with an additional primer, termed UPBS (upstream of PBS), lo-cated at the 59terminus of the R region (59-AGACCAGATCTGAGCCTGGG AG-39). Amplified fragments were then digested with BglII and PstI and were inserted into a pSVK3 vector (Pharmacia Biotech, Montreal, Quebec, Canada). The cloned fragments were sequenced to verify that correct modifications of viral gene sequences had been made and were inserted into the HXB2D clone of infectious DNA as described previously (36).
To further define minimal sequences in the 54-nt deletion (pHIV/del-LD), three additional constructs that had deletions at nt 654 to 671, 672 to 691, and 692-707, respectively, were generated. These were constructed by using the primers 59-GAGAGAGCTCTGGGTCCCTGTTCGGCG-39, 59-CCGTGCGC GCTTCAGCAAGCCGAGTCTTTCCCTTTCGCTTTC-39, and 59-CCGTGCG CGCCTGCGTCGAGAGAGC-39 in conjunction with the primer UPBS (see above). Figure 1 shows a graphic description of the mutant viruses generated. Wild-type HXB2D viral DNA was designated pHIV/WT.
Replication potential of viral constructs.Molecular constructs containing the above-described mutations in leader regions surrounding the PBS were purified twice by CsCl2gradient ultracentrifugation. These plasmids were transfected into
COS-7 cells by using a standard calcium coprecipitation procedure (40). Virus-containing culture fluids were harvested approximately 72 h after transfection and were clarified by centrifugation for 30 min at 4°C at 3,000 rpm in a Beckman GS-6R centrifuge, prior to filtration with a 0.2mm-pore-size sterile membrane. Viral preparations were stored at270°C until use.
For purposes of infection, the viral stock was thawed and treated with 100 U of DNase I in the presence of 10 mM MgCl2at 37°C for 1 h to ensure that any
contaminating plasmids had been eliminated from the transfection inocula (36). Infection of MT-4 cells was performed by incubating cells at 37°C for 2 h with virus (50 ng of p24), following which the cells were washed three times with phosphate-buffered saline and incubated at 37°C with fresh medium. In some experiments, HIV-IIIB, kindly provided by R. C. Gallo, National Institutes of
Health, Bethesda, Md., was used as a positive control. Culture fluids were monitored for virus production by RT assay (10) and by p24 (capsid protein [CA]) antigen detection enzyme-linked immunosorption assay (Abbott Labora-tories, Abbott Park, Ill.).
Detection of viral DNA.At various times after infection (4 to 8 h), MT-4 cells were collected and washed extensively with serum-free medium. To ensure that no contaminating plasmids were present, fluids from each wash were routinely checked by PCR with HIV-specific primers (36). Total cellular DNA was then isolated from these cells (40) and analyzed by PCR with specific primer pairs to amplify minus-strand strong-stop DNA (20, 60). Cellular DNA isolated from cells inoculated with heat-inactivated wild-type viruses served as a negative control to ensure that potentially contaminating plasmids had been eliminated. For minus-strand strong-stop DNA, UPBS, located at the 59terminus of the R region (nt 468 to 489) (49), was employed as a forward primer, while the backward primer was AA559(nt 621 to 604), which was modified from a previ-ously published procedure (60). The expected product of this primer pair (i.e., UPBS-AA559) is 153 bp in length.
To amplify viral DNA generated after the first template switch, we employed U3 (nt 1 to 21) as a forward primer and AR (nt 532 to 511) as a backward primer. To amplify viral DNA made after the second template switch, we used UPBS as a forward primer and PST, in the gag gene (nt 1422 to 1398), as a backward primer. As a negative control, we also employed cells that had been pretreated with 2mM zidovudine (AZT) for 3 h prior to viral inoculation and maintained these cells in the presence of the drug for an additional 4 to 8 h prior to extraction of total DNA. PCR assays were performed with 50mg of sample DNA,
50 mM Tris-Cl (pH 8.0), 50 mM KCl, 2.5 mM MgCl2, 2.5 U of Taq polymerase,
0.2 mM deoxynucleoside triphosphates, 10 pmol of32P-end-labelled forward
primer, and 20 pmol of unlabelled backward primer. Reactions were standard-ized by simultaneous amplification ofb-globin sequences as an internal control (36, 60) and involved 30 cycles in which samples were subjected to 94°C (1 min), 60°C (1 min), and 72°C (1 min).
Analysis of viral RNA by Northern and slot blotting.Analysis of viral mRNA expression in COS-7 cells, transfected with various DNA constructs, was per-formed by slot and Northern blotting procedures as described previously (10). The efficiency of transfection was routinely monitored by detection of viral CA, using monoclonal anti-p24 antibodies in an immunofluorescence assay (10). For Northern blots, total cellular RNA extracted from COS-7 cells was purified with a commercial RNA extraction kit (Biotecs, Houston, Tex.). The extracted RNA was treated with 100 U of DNase I, followed by phenol-chloroform extraction and ethanol precipitation, to ensure removal of any contaminating plasmids and cellular DNA. The RNA pellets were resuspended in diethylpyrocarbonate-treated double-distilled water. RNA samples (up to 20mg) were fractionated on 1% agarose gels containing formaldehyde as a denaturant (10). RNA molecules were transferred to a Hybond-N nylon membrane (Amersham, Toronto, Can-ada) and hybridized with pBH10 viral DNA as a radiolabelled probe (nick translation system; Life Technologies, Toronto, Canada) as described previously (10).
To quantify viral RNA transcripts derived from COS-7 cells, total cellular RNA (harvested at various times after transfection) was immobilized on nylon membranes, using a slot blot apparatus, followed by UV irradiation (Amer-sham). Hybridization reactions were performed as described for Northern blots (10). The quantity of viral RNA was determined by counting the radioactivity on the relevant filter pads by liquid scintillation.
In some cases, viral RNA that had been packaged into virions (purified by sucrose gradient centrifugation) was also quantified by the slot blot protocol. To rule out the possibility that the samples tested also contained residual DNA, which might have been hybridized by the radiolabelled DNA probe, RNase digestion of RNA extracted from virions was performed with RNase A (Boehringer-Mannheim, Montreal, Canada) at a final concentration of 10mg/ml at 37°C for 30 min, following which phenol-chloroform extraction was per-formed.
[image:2.612.319.550.69.332.2]RNA stability assay.Thirty-six hours after transfection, actinomycin D was added to the culture medium to block the transcriptional activity of RNA poly-merase II (19). At different times, e.g., 0, 1, 3, and 6 h after addition of the drug, total cellular RNA was extracted by using an ultraspecTM-II RNA isolation system (Biotecs) and was treated with 100 U of RNase-free DNase I which was FIG. 1. Schematic depiction of deletion mutations surrounding the PBS of HIV-1 proviral DNA. pHIV/del-7 represents a 7-nt deletion immediately down-stream of the PBS; pHIV/del-LD represents a 54-nt deletion also immediately downstream of the PBS and containing the aforementioned 7-nt sequence. The initiation codon of the gag gene is indicated, along with relevant nucleotide positions.
on November 9, 2019 by guest
http://jvi.asm.org/
then removed by phenol-chloroform extraction. Two micrograms of RNA was used in reverse transcription reactions, with 59-TTTATTGAGGCTTAAGCAG TGGG-39(nt 56 to 78) as an antisense primer in a total volume of 20ml. One microliter of product was then amplified in a 15 cycle-PCR with 59-AGACCA GATCTGAGCCTGGGAG-39(nt 14 to 35) as a sense primer and the same antisense primer mentioned above to yield a 65-bp DNA fragment. Products were analyzed on 5% polyacrylamide gels and further quantified by molecular imaging analysis.
Detection of viral proteins produced by transfected COS-7 cells.Expression of viral proteins in transfected COS-7 cells was determined by using a commercial kit for detection of p24 CA antigen and by RT assay as described previously (10). Both intracellular and extracellular CA levels were determined in order to shed light on the efficiency of viral assembly.
Viral proteins were also analyzed by Western blotting as described previously (10). For this purpose, protein samples (standardized on the basis of viral p24) were fractionated on sodium dodecyl sulfate–12% polyacrylamide gels and trans-ferred to nitrocellulose filters (10). The filters were then blocked with 5% skim milk–0.05% Tween 20–phosphate-buffered saline at 37°C for 2 h, followed by exposure to sera obtained from HIV-1-seropositive individuals (10). After ex-tensive washing with 0.05% Tween 20–phosphate-buffered saline,125I-labelled
goat anti-human immunoglobulin G (ICN, Mississauga, Canada) was added and left for 1 h at 37°C. The filters were then washed three times, dried, and exposed to Kodak X-Omat film at270°C.
RESULTS
Replication of virus deletion mutants.
The mutations
intro-duced into proviral DNA constructs (Fig. 1) include a deletion
of the conserved 7-nt stretch located immediately downstream
of the PBS (pHIV/del-7) and an extensive 54-nt deletion
down-stream of the PBS containing the aforementioned 7-nt
seg-ment (pHIV/del-LD) (Fig. 1). In addition, the 54-nt deletion
region was subdivided by smaller deletions termed
pHIV/del-LD1, pHIV/del-LD2, and pHIV/del-LD3 (Fig. 1).
To investigate the replication potentials of these constructs,
viruses (containing 50 ng of p24) derived from COS-7 cells that
had been appropriately transfected were used to infect MT-4
cells. Figure 2 shows that wild-type virus (pHIV/WT) and one
of the deletion mutants (pHIV/del-7) replicated efficiently, as
determined by levels of RT activity in culture fluids after 3 and
7 days. In contrast, the pHIV/del-LD mutant was significantly
impaired in the ability to produce viral progeny (Fig. 2).
Fur-ther analysis revealed that the pHIV/del-LD3 mutant was most
severely diminished in its ability to replicate.
We also studied the ability of viruses derived from
transfec-tions of COS-7 cells to infect MT-4 cells, using a p24 antigen
capture assay. In this instance, we also examined viruses that
had been subjected to more extensive deletion mutagenesis
than that for pHIV/del-LD. Two different concentrations of
viral inoculum were used in each case. The results in Table 1
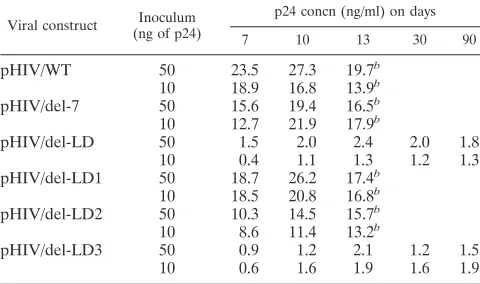
show that the pHIV/del-7 construct yielded levels of p24
sim-ilar to those from wild-type virus over 13 days, while both
pHIV/del-LD and the pHIV/del-LD3 construct, with a
dele-tion of 16 nt at the 3
9
end of the 54-nt LD deletion, were
severely impaired and produced only low levels of p24 for at
least 90 days in culture. In contrast, little or no effect was
observed when pHIV/del-LD1, with a deletion of 18 nt at the
start of this 54-nt segment, was employed. Moderate inhibition
of p24 synthesis was noted when pHIV/del-LD2 was studied;
this construct lacks a stretch of 20 nt at the center of this 54-nt
region. These findings are consistent with the data in Fig. 2 and
with the work presented below on synthesis of viral DNA in
infected cells.
Production of minus-strand strong-stop DNA in infected
cells.
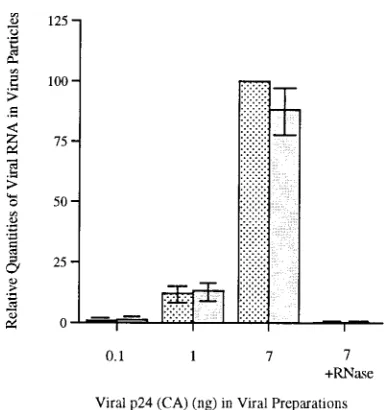
We found that similar levels of viral RNA were packaged
into viruses derived from COS-7 cells that had been
trans-fected 72 h earlier with our various constructs (Fig. 3) (results,
based on RNA/p24 ratios, are shown for pHIV/WT and pHIV/
del-LD). When these RNA preparations were digested with
RNase as a negative control, little or no hybridizable material
remained, indicating that contaminating viral DNA was not
present in these preparations.
[image:3.612.74.281.67.256.2]Since previous work had shown that certain sequences
sur-rounding the PBS were involved in reverse transcription in
cell-free systems (24, 25, 33), we asked whether the
modifica-tions introduced into our constructs would result in impaired
generation of viral DNA. Toward this end, total cellular DNA
was isolated at 4 and 8 h after infection of MT-4 cells with
viruses derived from COS-7 cells and was analyzed by PCR
with primer pairs that specifically amplify minus-strand
strong-stop DNA as well as viral DNA that is generated after each of
the first and second template switch events. We found that
similar levels of minus-strand strong-stop DNA were present in
MT-4 cells infected by pHIV/del-7 (Fig. 4A, lanes 6 and 12)
and by wild-type virus (Fig. 4A, lanes 4 and 10) after 4 to 8 h.
In contrast, MT-4 cells infected with the pHIV/del-LD mutant
contained significantly decreased levels of minus-strand
strong-stop DNA (Fig. 4A, lanes 8 and 14) (i.e., about 10 times
FIG. 2. Viral replication capacities of various constructs. Cell-free viruses harvested from COS-7 cells transfected with various molecular constructs (72 h posttransfection) were used to infect MT-4 cells. Culture fluids were collected and monitored for RT activity. The decreased viral production in MT-4 cultures after 1 week in the case of cells infected by pHIV/WT (h) and pHIV/del-7 ({) was due to viral cytopathology; fresh cells were not added to these cultures.E, cells infected by pHIV/del-LD;Ç, mock-infected cells.
TABLE 1. Levels of p24 antigen expression in infected MT-4 cells
aViral construct (ng of p24)Inoculum p24 concn (ng/ml) on days
7 10 13 30 90
pHIV/WT
50
23.5
27.3
19.7
b10
18.9
16.8
13.9
bpHIV/del-7
50
15.6
19.4
16.5
b10
12.7
21.9
17.9
bpHIV/del-LD
50
1.5
2.0
2.4
2.0
1.8
10
0.4
1.1
1.3
1.2
1.3
pHIV/del-LD1
50
18.7
26.2
17.4
b10
18.5
20.8
16.8
bpHIV/del-LD2
50
10.3
14.5
15.7
b10
8.6
11.4
13.2
bpHIV/del-LD3
50
0.9
1.2
2.1
1.2
1.5
10
0.6
1.6
1.9
1.6
1.9
aMT-4 cells were infected with various viral constructs, and p24 levels in culture fluids were measured.
bAfter day 13, cytotoxicity resulted in the death of cultures that produced relatively high levels of p24. In contrast, cultures infected by the pHIV/del-LD and pHIV/del-LD3 viruses continued to generate low levels of p24 activity over extensive periods.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.316.556.535.677.2]less than with wild-type virus as quantified by densitometry).
As a control, we performed mock infections with culture fluids
of COS-7 cells that had been transfected with DNA from cells
inoculated with heat-inactivated viruses and were unable to
de-tect a DNA signal (Fig. 3A, lanes 1, 2, and 3 for wild-type, pHIV/
del-7, and pHIV/del-LD, respectively). Consistent results were
obtained with total cellular DNA by using primer pairs that
am-plify DNA that is present after the first template switch (Fig.
4B) as well as full-length reverse-transcribed DNA (Fig. 4C) (60).
As an additional important control, we treated cells with 2
m
M AZT in order to prevent synthesis of the viral DNA
prod-uct generated after the first template switch. Indeed, we found,
as expected, that such treatment did not affect levels of
minus-strand strong-stop DNA in the case of either wild-type virus
(Fig. 4A, lanes 5 and 11) or pHIV/del-7 (Fig. 4A, lanes 7 and
13), nor, in fact, did the presence of AZT affect the already
diminished levels of minus-strand strong-stop DNA found in
cells infected by pHIV/del-LD (Fig. 4A, lanes 9 and 15).
In contrast, treatment with 2
m
M AZT significantly impaired
synthesis of DNA products generated after both the first and
template switch events for each of the viruses tested (Fig. 4B
and C) (compare lanes 4 and 5, lanes 6 and 7, lanes 8 and 9,
lanes 10 and 11, lanes 12 and 13, and lanes 14 and 15). In the
case of the full-length product (Fig. 4C), it should be noted
that pHIV/del-LD, as expected, yielded a DNA product (lanes
8 and 14) smaller than that obtained with wild-type virus. In
this case, treatment with AZT prevented the appearance of
any detectable DNA product (Fig. 4C, lanes 9 and 15).
Thus, the 54-nt untranslated sequence, located immediately
downstream of the PBS, is necessary for both efficient reverse
transcription and efficient infectivity. As expected from the
results shown in Fig. 4, far less proviral DNA became
inte-grated into MT-4 cells after infection with pHIV/del-LD than
after infection with pHIV/WT or pHIV/del-7, but the pHIV/
del-LD DNA persisted for up to 3 months (data not shown).
No evidence of revertant virus was observed as determined by
sequencing during extensive cultivation, although p24 antigen
could be detected at low levels for as long as 3 months.
To further define the minimal necessary sequences within
this 54-nt region, the pHIV/del-LD1, pHIV/del-LD2, and
pHIV/del-LD3 viruses were used to infect MT-4 cells, and
levels of reverse-transcribed DNA were determined (Fig. 5).
Molecular imaging analysis showed that, in comparison with
wild-type virus (Fig. 5, lanes 4 and 8), pHIV/del-LD3 was
severely (i.e.,
.
90%) impaired in synthesis of minus-strand
strong-stop DNA (Fig. 5, lanes 3 and 7). Only a modest
dim-inution (
'
50%) in generation of such material occurred when
pHIV/del-LD2 was studied (Fig. 5, lanes 2 and 6), while no
effect whatever was seen in the case of pHIV/del-LD1 (Fig. 5,
lanes 1 and 5). These findings are consistent with the results on
viral replication described above.
[image:4.612.81.273.64.269.2]Role of the untranslated region downstream of the PBS in
viral gene expression.
The data described above indicate that
the 54-nt region is involved in generation of viral DNA,
con-sistent with observations for cell-free systems (37). However,
the observed reductions (
'
10-fold) might not have led directly
to the near lethality of pHIV/del-LD, since postintegrational
effects, e.g., generation of viral mRNA and proteins, might also
have played a role. We therefore assessed what role the
un-translated region downstream of the PBS might play in
expres-sion of viral mRNA. Figure 6 depicts the results of Northern
blot analysis of viral RNA extracted from COS-7 cells
trans-fected with either mutant or wild-type constructs. Levels of
viral RNA transcripts in cells transfected with pHIV/del-LD
were much lower than those in cells transfected with pHIV/
FIG. 3. Relative quantities of viral RNA packaged into viral structures. COS-7 cells were transfected with either pHIV/del-LD ( ) or pHIV/WT (u). After 60 h, viruses in culture fluids were purified by sucrose gradient ultracentrifugation. RNA was extracted from equal amounts (on the basis of p24 content) of viruses and quantified by slot blot and liquid scintillation analysis (see Materials and Methods). Experiments were performed with three replicate sam-ples; error bars represent standard deviations. In some cases, viral RNA was digested with RNase, and all hybridizable material was eliminated. Results are standardized to 100 for pHIV/WT (7 ng).
FIG. 4. Detection of viral DNA. Viruses harvested from culture fluids of COS-7 cells that had been transfected with various molecular constructs were standardized on the basis of p24 content and used to infect MT-4 cells. Total cellular DNA (approximately 50mg) was isolated from infected cells at 4 and 8 h after infection and subjected to PCR analysis with primers that specifically amplify minus-strand strong-stop DNA (59). Primers amplifyingb-globin were used as an internal control to monitor the input of sample DNA (59). Mock infections involved culture fluids derived from COS-7 cells that had been trans-fected with DNA from cells inoculated with heat-inactivated viruses. Lanes 1 to 3, cells exposed to heat-inactivated viruses HIV/WT, 7, and HIV/del-LD, respectively; lanes 4, 6, and 8, cells infected with HIV/WT, HIV/del-7, and HIV/del-LD, respectively; lanes 5, 7, and 9, cells infected with HIV/WT, HIV/ del-7, and HIV/del-LD in the presence of 2mM AZT, respectively. Lanes 1 to 9, cells were maintained for 4 h after exposure to virus prior to extraction of DNA; lanes 10 to 15, same order of experiments as in lanes 4 to 9 except that DNA was extracted after 8 h, lanes 16 to 19, several dilutions of HxB2D plasmid as a positive control (i.e., 10-fold dilutions of plasmids in terms of copy numbers [53 102, 53103, 53104, and 53105, respectively]). (A) Detection of minus-strand
strong-stop DNA; (B) Detection of viral DNA generated after the first template switch. (C) Detection of viral DNA generated after the second template switch. (D) PCR amplification ofb-globin DNA as an internal control.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.317.555.382.552.2]WT, although the major three bands representing unspliced,
singly spliced, and multiply spliced RNA were present in each
case (Fig. 6A). Similar viral RNA transcript patterns were
observed in COS-7 cells transfected with pHIV/del-7 and
wild-type virus (data not shown).
These results were further confirmed by quantitative slot
blot analysis. Figure 6B shows that dramatically reduced levels
of RNA transcript were present in cells transfected with pHIV/
del-LD compared with those transfected with wild-type virus
(pHIV/WT) or pHIV/del-7. The differences were most
pro-nounced at early time points after transfection (16 h).
To exclude the possibility that the reduced levels of viral
mRNA in pHIV/del-LD-transfected cells were due to
instabil-ity, we determined the half-lives of the viral mRNA molecules
produced following transfection of COS-7 cells by wild-type
and mutated constructs. Toward this end, cells were treated
with actinomycin D at 36 h after transfection, as described in
Materials and Methods, and total RNA was extracted at 0, 1,
3, and 6 h thereafter and reverse transcribed to yield DNA.
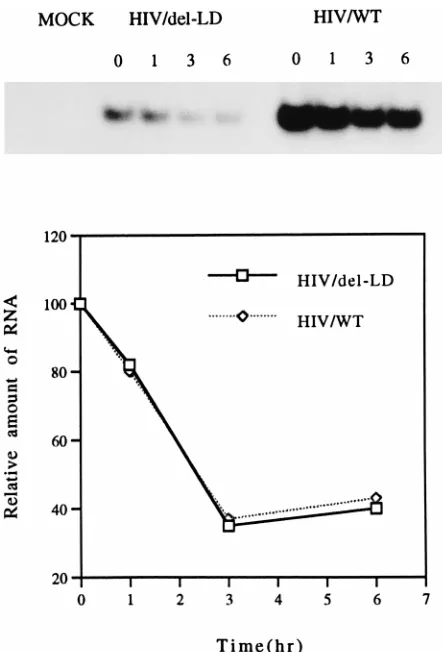
The results of specific PCR amplifications revealed an
ex-pected disappearance of relevant amplified genetic material
over time (Fig. 7, top). Consistent with the results shown in
Fig. 6, cells transfected with the HIV/del-LD construct
pro-duced much lower overall levels of mRNA than did those
transfected by wild-type material. However, the rates of
disap-pearance of viral RNA in both cases were nearly identical as
shown by molecular imaging analysis (Fig. 7, bottom). Indeed,
no differences in regard to stability were observed among
mRNA molecules derived from the wild-type, pHIV/del-LD,
pHIV/del-LD1, pHIV/del-LD2, or pHIV/del-LD3 construct
(data not shown).
Effects on viral protein synthesis.
We next wished to
inves-tigate protein expression and viral assembly in COS-7 cells that
had been transfected with wild-type DNA and the pHIV/
del-LD construct. Toward this end, p24 detection and Western
blot analyses were performed with culture fluids and cell
ly-sates. As expected on the basis of the RNA transcript results
described above, COS-7 cells transfected with pHIV/del-LD
produced lower levels of both intracellular and extracellular
p24 after 16 h than cells transfected with pHIV/WT (Table 2).
Interestingly, transfection with pHIV/del-LD did not result in
an excess accumulation of intracellular p24 relative to that with
other transfections, suggesting that viral protein assembly had
proceeded normally.
It was also important to determine whether the diminished
synthesis of viral proteins associated with pHIV/del-LD would
affect the profiles of the viral proteins produced by transfected
COS-7 cells, in a system in which the same total amount of
[image:5.612.61.297.69.184.2]protein was analyzed in each case by gel electrophoresis.
Fig-ure 8 shows a Western blot analysis of proteins produced by
COS-7 cells that had been transfected by pHIV/del-LD (lane
1) or pHIV/WT (lane 2) or mock transfected (lane 4). Lane 3
of Fig. 8 represents MT-4 cells infected by wild-type HIV. The
contents of lanes 1 to 3 were equalized on the basis of the
amount of p24 as determined by enzyme-linked
immunosor-bent assay. We found that viral protein profiles were essentially
nondistinguishable among COS-7 cells transfected by the
mu-tant (Fig. 8, lane 1) or the wild-type construct (lane 2) or MT-4
cells infected by wild-type virus (lane 3), nor were differences
observed in regard to transfections by the pHIV/del-LD1,
pHIV/del-LD2, or pHIV/del-LD3 construct (data not shown).
Thus, deletion of the 54-nt stretch downstream of the PBS did
not affect patterns of viral protein synthesis but rather led to a
FIG. 5. Detection of minus-strand strong-stop DNA [(2)ssDNA]. Lanes: 1 and 5, infection by pHIV/del-LD1; 2 and 6, infection by pHIV/del-LD2; 3 and 7, infection by pHIV/del-LD3; 4 and 8, infection by pHIV/WT; 9, inoculation of cells with heat-inactivated wild-type virus. Positive controls of serially diluted HXB2D plasmids are as shown in Fig. 4.
FIG. 6. (A) Northern blots for detection of viral RNA. Total cellular RNA was purified from COS-7 cells 16 h after transfection with either pHIV/del-LD or pHIV/WT. Lane 1, RNA (20mg) from cells transfected with pHIV/del-LD; lane 2, RNA (20mg) from cells transfected with pHIV/WT; lane 3, RNA (10mg) from cells transfected with pHIV/del-LD; lane 4, RNA (10mg) from cells transfected with pHIV/WT; lane 5, RNA (20mg) from mock-transfected COS cells. Molec-ular size markers are indicated. (B) Quantitative determination of viral RNA transcripts by slot blot analysis. Total cellular RNA was harvested from COS-7 cells and purified at 16, 24, 48, and 72 h, respectively, after transfection with various molecular constructs. Relative intensities were calculated by comparison with levels of radioactivity obtained with wild-type transfections after 72 h, which were defined as 100 (i.e., 2,478 cpm). Standard deviations (for four separate experiments) are indicated by error bars.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.317.552.236.618.2]marked decrease in levels of all viral proteins produced. This is
because the untranslated sequences downstream of the PBS
can influence the production of infectious progeny virus by
affecting both reverse transcription and the expression of viral
mRNA.
DISCUSSION
Reverse transcription is initiated at the PBS to which the
tRNA primer is bound. The HIV-1 PBS is located about 180 nt
from the 5
9
terminus of unspliced RNA (23). The PBS is
flanked by the R/U5 region at its 5
9
border and by a 133-nt
untranslated sequence at its 3
9
end (23). Increasing evidence
suggests that the untranslated sequences that flank the PBS are
involved in several steps of viral replication, including reverse
transcription, integration, expression of the proviral genome,
and packaging. Two lines of data prompted us to initiate an
identification of regions downstream of the PBS that are
es-sential for viral replication. First, efficient retroviral reverse
transcription requires interaction between primer tRNA and
the RNA at multiple sites (1, 4, 5, 12, 13, 24–26, 51). Second,
HIV-1 has evolved to choose tRNA
3Lysas a primer for optimal
growth (16, 27, 36, 58).
As an additional control, we also employed AZT, as
de-scribed in Materials and Methods, to block synthesis of viral
DNA products. Consistent with previous observations, we
found that AZT had no effect on the presence of minus-strand
strong-stop DNA, which is carried into cells by the virions in
which it is made (39, 54). However, the use of AZT efficiently
interfered with production of DNA products that are
gener-ated after each of the first and second template switch events,
consistent with previous observations (3).
[image:6.612.333.535.66.369.2]Nonessentiality of a small, restricted region downstream of
the PBS.
A short, 6-nt sequence, located immediately
down-stream of the PBS, was previously shown to be important in
specifying utilization of the tRNA primer (33). We have now
shown that this sequence is apparently unnecessary either for
synthesis of minus-strand strong-stop DNA in infected cells
(pHIV/del-7) after transfection (Fig. 2) or for viral replication,
[image:6.612.64.285.67.393.2]FIG. 7. RNA stability assay. Actinomycin D was added to culture medium at 36 h after transfection of COS-7 cells, and total cellular RNA was extracted at 0, 1, 3, and 6 h thereafter. Levels of viral RNA were determined by RT-PCR (top) and analyzed by molecular imaging (bottom). Mock, RT-PCR performed with wild-type HIV RNA in the absence of RT; HIV/del-LD and HIV/WT, infections performed with HIV/del-LD and wild-type constructs, respectively.
FIG. 8. Viral protein analysis by Western blotting. Proteins isolated from COS-7 cells were analyzed by Western blotting as described in Materials and Methods. Lanes: 1, proteins from COS cells transfected with pHIV/del-LD; 2, proteins from COS cells transfected with pHIV/WT; 3, positive control, using proteins derived from MT-4 cells infected by HIV-IIIB; 4, proteins from
mock-transfected COS-7 cells.
TABLE 2. Intracellular and extracellular p24 levels in COS-7 cells
transfected with pHIV/del-LD or pHIV/WT
h after transfection
p24 level (ng/ml)a
Intracellular Extracellular
pHIV/del-LD
transfection transfectionpHIV/WT pHIV/del-LDtransfection transfectionpHIV/WT
16 5.2260.48 170615.2 4.4960.32 187616.8 24 17.961.55 241622.2 18.7616.2 258625.3 48 64.866.02 233624.5 97.0610.2 304629.8 72 62.965.89 245623.3 145613.5 300631.2
aAt various times after transfection, both intracellular and extracellular viral p24 levels were determined. Data are means6standard deviations from four separate experiments.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.57.299.607.701.2]consistent with in vitro studies that employed the nucleocapsid
protein (37). Others have reported that nucleotide
substitu-tions downstream of the PBS may be observed in HIV-1
re-vertants that have deletions within the PBS; these rere-vertants
appeared to have wild-type replication capacity (50). Thus, this
small, 6- or 7-nt sequence may not be required in either
main-tenance of viral secondary structure or interaction with
tRNA
3Lys. Likewise, an 18-nt segment encompassing the
above-mentioned 7 nt downstream of the PBS is not essential for
synthesis of viral DNA and viral replication, despite being
DNase sensitive (17). It should be noted that our experiments
were performed with a transient-transfection system, while
others employed “integrated templates” (17). While studies on
integrated templates would have provided additional
informa-tion, the results in Fig. 2 show that virus infectivity was not
severely affected by deletion of a 7-nt stretch downstream of
the PBS.
Identification of sequences important for viral replication
downstream of the PBS.
An extended 54-nt deletion, which
does not compromise packaging or dimerization signals or the
splice donor (2, 11, 15, 35, 41, 52), was found to significantly
diminish virus replication (pHIV/del-LD) (Fig. 2). The
prox-imity of this 54-nt region to the PBS might play a role in
limiting viral replication, due to a defect in reverse
transcrip-tion (37). Measurement of reverse-transcribed DNA, shortly
after viral entry, revealed only low levels of product (Fig. 4).
This was most pronounced when an 18-nt segment at the 3
9
end of the 54-nt region was removed (Fig. 5). This deficit in
reverse transcription could be due to poor NCp-mediated
in-teractions between the tRNA primer and the RNA template
(37).
In the case of minus-strand strong-stop DNA, it can be
argued that the above-described experiments do not
distin-guish between synthesis of material that is present in virions
versus that produced in cells after infection (39, 54). For this
reason, we employed AZT to influence the synthesis of viral
DNA products that are generated after the first template
switch. As previously demonstrated, we found that the use of
AZT in our protocols did not result in chain termination until
after the first template switch event (3, 60). The potential role
of contaminating DNA plasmids was also ruled out through
mock infection protocols performed with DNA from cells
in-oculated with heat-inactivated viruses (see Materials and
Methods).
We also found that deletion of the 54-nt region resulted in a
significant decrease in accumulation of viral RNA transcripts
in transfected cells (Fig. 6). The relationship between defects
in reverse transcription and mRNA production is unknown.
The 54-nt segment could conceivably have affected both of
these activities. Of course, the observed differences could be
explained if pHIV/del-LD mRNA was less stable than
wild-type viral RNA. However, we did not observe differences in
stability between the various wild-type and mutated RNA
tran-scripts studied. It is also unlikely that this region would
influ-ence efficiency of integration, since the latter process is
pri-marily affected by the termini of reverse-transcribed viral DNA
(46, 56). Indeed, MT-4 cells infected with the pHIV-1/del-LD
virus contained integrated proviral DNA. Our results show
that the 54-nt stretch downstream of the PBS is important for
virus replication and that a 16-nt sequence located at the 3
9
end of this segment is principally involved. Further studies on
nucleosome structure and/or identification of potential
tran-scription factor binding sites, through the use of reporter
genes, will yield additional functional information, as will
fur-ther deletion analysis of the 16-nt segment itself (17, 18, 53,
55). Most binding elements for inducible or constitutive
cellu-lar transcripts are located upstream of the PBS (for a review,
see reference 13). However, untranslated sequences
down-stream of the PBS can also affect viral transcription activity, as
documented here.
It is also interesting that the 54-nt deletion had little effect
on packaging of viral RNA, despite overlap at its 3
9
end with a
substitution mutation that extends further downstream and
that can affect RNA encapsidation (32). Differences between
these constructs could have resulted in RNA structures that
may have been differentially recognized by viral proteins
in-volved in selection and encapsidation. The use of different viral
strains and cell lines might also have contributed to differences
between the studies.
In summary, we have shown that a 54-nt segment in the
noncoding region of the HIV-1 genome, downstream of the
PBS, is important for viral replication in two principal ways.
First, this region clearly is necessary for efficient reverse
tran-scription of the viral DNA product, including that which is
generated both prior to and after each of the two template
switch events. In addition, this region is important for the
efficient generation of viral mRNA and consequently for the
synthesis of viral protein and infectivity. Each of these effects,
i.e., on reverse transcription and on synthesis of viral
tran-scripts, seems to be independent of the other. This was shown
through (i) studies in which transfection of cells with deleted
DNA constructs failed to generate significant levels of viral
mRNA and (ii) independent experiments in which infection by
viruses containing relevant deletions in viral RNA yielded
ex-tremely low levels of viral DNA products generated both
be-fore and after template switching.
ACKNOWLEDGMENTS
Xuguang Li and Chen Liang contributed equally to this project.
This research was supported by grants from the Medical Research
Council of Canada and by Health Canada. Xuguang Li was supported
by a postdoctoral fellowship from the National Health Research
De-velopment Programme of Health Canada. Mark A. Wainberg is a
National AIDS Scientist of Health Canada.
REFERENCES
1. Aiyar, A., D. Cobrinik, Z. Ge, H. J. Kung, and J. Leis. 1992. Interaction between U5 viral RNA and the TCC loop of the tRNATrpprimer is required
for efficient initiation of reverse transcription. J. Virol. 66:2464–2472. 2. Aldovini, A., and R. Young. 1990. Mutations of RNA and protein sequences
involved in human immunodeficiency virus type 1 packaging result in pro-duction of noninfectious virus. J. Virol. 64:1920–1926.
3. Arts, E. J., and M. A. Wainberg. 1994. Preferential incorporation of nucle-oside analogs after template switching during human immunodeficiency vi-rus reverse transcription. Antimicrob. Agents Chemother. 38:1008–1016. 4. Arts, E. J., X. Li, Z. Gu, L. Kleiman, M. A. Parniak, and M. A. Wainberg.
1994. Comparison of deoxy-oligonucleotide and tRNALys.3as primers in an
endogenous HIV-1 in vitro reverse transcription/template switching reac-tion. J. Biol. Chem. 269:14672–14680.
5. Arts, E. J., S. R. Strtor, X. Li, J. W. Rausch, K. J. Howard, B. Ehresmann, T. W. North, B. M. Wohrl, R. Goody, M. A. Wainberg, and S. F. J. LeGrice. 1996. Initiation of (2) strand DNA synthesis from tRNALys.3on lentiviral
RNAs: implications of specific HIV-1 RNA-tRNALys.3interactions
inhibit-ing primer utilization by retroviral reverse transcriptions. Proc. Natl. Acad. Sci. USA 93:10063–10068.
6. Baudin, F., R. Marquet, C. Isel, J.-L. Darlix, B. Ehresmann, and C. Ehres-mann.1993. Functional sites in the 59region of HIV-1 RNA form defined structural domains. J. Mol. Biol. 229:382–397.
7. Berkhout, B., R. H. Silverman, and K. T. Jeang. 1989. Tat transactivates the human immunodeficiency virus through a nascent RNA target. Cell 59:273– 282.
8. Berkhout, B., and L. Schoneveld. 1993. Secondary structure of the HIV-2 leader RNA comprising the tRNA-primer binding site. Nucleic Acids Res. 21:1171–1178.
9. Berlioz, C., and J.-L. Darlix. 1995. An internal ribosome entry mechanism promotes translation of murine leukemia virus gag polyprotein procursors. J. Virol. 69:2214–2222.
10. Boulerice, F., S. Bour, R. Geleziunas, A. Lvovich, and M. A. Wainberg. 1990.
on November 9, 2019 by guest
http://jvi.asm.org/
High frequency of isolation of defective human immunodeficiency virus type 1 and heterogeneity of viral gene expression in clones of infected U937 cells. J. Virol. 64:1745–1755.
11. Clavel, F., and J. M. Orenstein. 1990. A mutation of human immunodefi-ciency viruses with reduced RNA packaging and abnormal particle morphol-ogy. J. Virol. 64:5230–5234.
12. Cobrinik, D., A. Aiyar, Z. Ge, M. Katzman, H. Huang, and J. Leis. 1991. Overlapping U5 sequence elements are required for efficient integration and initiation of reverse transcription. J. Virol. 65:3864–3872.
13. Cobrinik, D., L. Soskey, and J. Leis. 1988. A retroviral RNA secondary structure required for efficient initiation of reverse transcription. J. Virol. 62:3622–3630.
14. Cullen, B. R. 1991. Regulation of HIV-1 gene expression. FASEB J. 5:2361–2368. 15. Darlix, J.-L., C. Gubas, M.-T. Nugeyre, F. Clavel, and F. Barre-Sinoussi. 1990. Cis elements and trans-acting factors involved in the RNA dimeriza-tion of HIV-1. J. Mol. Biol. 216:689–699.
16. Das, A. T., B. Klaver, and B. Berkhout. 1995. Reduced replication of HIV-1 mutants that use reverse transcription primers other than the natural tRNA3Lys. J. Virol. 69:3090–3097.
17. El Kharronbi, A., and M. A. Martin. 1996. cis-Acting sequences located downstream of the human immunodeficiency virus type 1 promotor affect its chromatin structure and transcriptional activity. Mol. Cell. Biol. 16:2958– 2966.
18. El Kharronbi, A., and E. Verdin. 1994. Protein-DNA interactions with DNase-I hypersensitive sites located downstream of the HIV-1 promotor. J. Biol. Chem. 269:19916–19924.
19. Felber, B. K., M. Hadzopoulou-Cladaras, C. Cladaras, T. Copeland, and G. N. Pavlakis.1989. Rev protein of human immunodeficiency virus type 1 affects the stability and transport of the viral mRNA. Proc. Natl. Acad. Sci. USA 86:1495–1499.
20. Gilboa, E., S. W. Mitra, S. P. Goff, and D. Baltimore. 1979. A detailed model of reverse transcription and tests of crucial aspects. Cell 18:93–100. 21. Harrich, D., C. Ulich, and R. B. Gaynor. 1996. A critical role for the TAR
element in promoting efficient human immunodeficiency virus type 1 reverse transcription. J. Virol. 70:4017–4027.
22. Harrison, G. P., and A. M. L. Lever. 1992. The human immunodeficiency virus type 1 packaging signal and major splice donor region have a conserved stable secondary structure. J. Virol. 66:4144–4153.
23. Haseltine, W. A. 1991. Molecular biology of HIV-1. FASEB J. 5:2349–2360. 24. Isel, C., C. Ehresmann, G. Keith, B. Ehresmann, and R. Marquet. 1995. Initiation of reverse transcription of HIV-1: secondary structure of the HIV-1 RNA/tRNALys.3(template/primer) complex. J. Mol. Biol. 247:236–
250.
25. Isel, C., J.-M. Lanchy, S. F. J. LeGrice, C. Ehresmann, B. Ehresmann, and R. Marquet.1996. Specific initiation and switch to elongation of HIV-1 reverse transcription requires post-transcriptional modifications of primer tRNALys.3. EMBO J. 15:917–924.
26. Isel, C., R. Marquet, G. Keith, C. Ehresmann, and B. Ehresmann. 1993. Modified nucleotides of tRNALys.3modulate primer/template loop-loop
in-teractions in the initiation complex of HIV-1 reverse transcription. J. Biol. Chem. 269:1388–1993.
27. Jiang, M., J. Mak, A. Ladha, E. Cohen, M. Klein, B. Rovinski, and L. Kleiman.1993. Identification of tRNAs incorporated into wild-type and mutant human immunodeficiency virus type 1. J. Virol. 67:3246–3253. 28. Kang, S. M., J. K. Wakefield, and C. D. Morrow. 1996. Mutations in both the
U5 region and the primer-binding site influence the selection of the tRNA used for the initiation of HIV-1 reverse transcription. Virology. 222:401–414. 29. Kang, S. M., Z. Zhang, and C. D. Morrow. 1997. Identification of a sequence within the U5 required for human immunodeficiency virus type 1 to stably maintain a primer binding site complementary to tRNAMet. J. Virol. 71:207–
217.
30. Karpel, R. L., L. Henderson, and L. E. Oroszlan. 1987. Interactions of retroviral structural proteins with single stranded nucleic acids. J. Biol. Chem. 262:4961–4967.
31. Khan, R., and D. Giedroc. 1992. Recombinant human immunodeficiency virus type 1 nucleocapsid protein unwinds tRNA. J. Biol. Chem. 267:6689– 6695.
32. Kim, H. J., K. Lee, and J. J. O’Rear. 1994. A short sequence upstream of the 59major splicing donor is important for encapsidation of human immuno-deficiency virus type 1 genomic RNA. Virology 198:336–340.
33. Kohlsteadt, L., and T. Steitz. 1992. Reverse transcriptase of HIV-1 can use either human tRNALys.3or E. coli tRNAGluas primer in an in vitro
utiliza-tion assay. Proc. Natl. Acad. Sci. USA 89:4652–4656.
34. Leis, J., A. Ashok, and D. Cobrinik. 1993. Regulation of initiation of reverse transcription of retroviruses, p. 33–47. In A. M. Skalka and S. P. Goff (ed.), Reverse transcriptase. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
35. Lever, A. M. L., H. Gottlinger, W. Haseltine, and J. Sodroski. 1989. Identi-fication of a sequence required for efficient packaging of human immuno-deficiency virus type 1 RNA into virions. J. Virol. 63:4085–4087. 36. Li, X., J. Mak, E. J. Arts, Z. Gu, L. Kleiman, M. A. Wainberg, and M. A.
Parniak.1994. Effects of alterations of primer binding site sequences on
human immunodeficiency virus type 1 replication. J. Virol. 68:6198–6206. 37. Li, X., Y. Quan, E. J. Arts, Z. Li, B. D. Preston, H. de Rocquigny, B. P.
Roques, J.-L. Darlix, L. Kleiman, M. A. Parniak, and M. A. Wainberg.1996. Human immunodeficiency virus type 1 nucleocapsid protein (NCp7) directs specific initiation of minus-strand DNA synthesis primed by human tRNA3Lys
in vitro. J. Virol. 70:4996–5004.
38. Litvak, S., L. Sarih-Cottin, M. Fournier, M. Andreola, and L. Tarrago-Litvak.1994. Priming of HIV-1 replication by tRNALys.3: role of reverse
transcriptase. Trends Biochem. Sci. 19:114–118.
39. Lori, F., F. Veronese, A. L. De Vico, P. Lusso, M. S. Reitz, and R. C. Gallo. 1992. Viral DNA carried by human immunodeficiency virus type 1 virions. J. Virol. 66:5067–5074.
40. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
41. Marquet, R., F. Baudin, C. Gabus, J.-L. Darlix, M. Mongel, C. Ehresmann, and B. Ehresmann.1991. Dimerization of HIV-1 RNA: stimulation by cat-ions and possible mechanism. Nucleic Acids Res. 19:2349–2357.
42. Miele, G., A. Mouland, G. P. Harrison, E. Cohen, and A. M. L. Lever. 1996. The human immunodeficiency virus type 1 59packaging signal structure affects translation but does not function as an internal ribosome entry site structure. J. Virol. 70:944–951.
43. Muesing, M. A., D. H. Smith, and D. J. Capon. 1987. Regulation of mRNA accumulation by a human immunodeficiency virus trans-activator protein. Cell 48:691–701.
44. Murphy, J. E., and S. P. Goff. 1989. Construction and analysis of deletion mutations in the U5 region of Moloney murine leukemia virus: effects on RNA packaging and reverse transcription. J. Virol. 63:319–327.
45. Paillart, J.-C., R. Marquet, E. Scripkin, B. Ehresmann, and C. Ehresmann. 1994. Mutational analysis of the bipartite dimer linkage structure of HIV-1 genomic RNA. J. Biol. Chem. 269:27486–27493.
46. Panganiban, A., and H. Temin. 1983. The terminal nucleotides of retrovirus DNA are required for integration but not for virus production. Nature (London) 306:155–160.
47. Picard, V., E. Ersdal-Badju, A. Lu, and S. C. Bock. 1994. A rapid and efficient one-tube PCR-based mutagenesis technique using Pfu DNA poly-merase. Nucleic Acids Res. 22:2587–2591.
48. Prats, A. C., L. Sarih, C. Gabus, S. Litvak, G. Keith, and J. L. Darlix. 1988. Small finger protein of avian and murine retroviruses has nucleic acid an-nealing activity and positions the replication primer tRNA onto genomic RNA. EMBO J. 7:1777–1783.
49. Ratner, L., W. Haseltine, R. Patarca, K. J. Livak, B. Starcich, S. F. Josephs, E. R. Doran, J. A. Rafalski, E. A. Whitehorn, K. Baumeister, L. Ivanoff, S. R. Petteway, Jr., M. L. Pearson, J. A. Lautenberger, T. S. Papas, J. Ghrayeb, N. T. Chang, R. C. Gallo, and F. Wong-Staal.1985. Complete nucleotide sequence of the AIDS virus, HTLV-III. Nature (London) 313:277–284. 50. Rhim, H., J. Park, and C. D. Morrow. 1991. Deletions in the tRNALys
primer-binding site of human immunodeficiency virus type 1 identify essen-tial regions for reverse transcription. J. Virol. 65:4555–4564.
51. Skripkin, E., C. Isel, R. Marquet, B. Ehresmann, and C. Ehresmann. 1996. Psoralen crosslinking between human immunodeficiency virus type 1 RNA and primer tRNALys.3. Nucleic Acids Res. 24:509–514.
52. Skripkin, E., J.-C. Pallart, R. Marquet, B. Ehresmann, and C. Ehresmann. 1994. Identification of the primary site of HIV-1 RNA dimerization in vitro. Proc. Natl. Acad. Sci. USA 91:4945–4949.
53. Soudeyns, H., R. Geleziunas, G. Shyamala, J. Hiscott, and M. A. Wainberg. 1993. Identification of a novel glucocorticoid response element within the genome of the human immunodeficiency virus type 1. Virology 194:758–768. 54. Trono, D. 1992. Partial reverse transcripts in virions from human
immuno-deficiency and murine leukemia viruses. J. Virol. 66:4893–4900.
55. Van Lint, C., J. Ghysdael, P. Paras, Jr., A., Burney, and E. Verdin. 1994. A transcriptional regulatory element is associated with a nuclease-hypersensi-tive site in the pol gene of human immunodeficiency virus type 1. J. Virol. 68:2632–2648.
56. Vicenzi, E., D. S. Dimitrov, A. Engelman, T.-S. Migone, D. F. J. Purcell, J. Leonard, G. Englund, and M. A. Martin.1994. An integration-defective U5 deletion mutant of human immunodeficiency virus type 1 reverts by eliminating additional long terminal repeat sequences. J. Virol. 68:7879– 7890.
57. Wakefield, J. K., S.-M. Kang, and C. D. Morrow. 1996. Construction of a type 1 human immunodeficiency virus that maintains a primer binding site complementary to tRNAHis. J. Virol. 70:966–975.
58. Wakefield, J. K., A. Wolf, and C. Morrow. 1995. Human immunodeficiency virus type 1 can use different tRNAs as primers for reverse transcription but selectively maintains a primer binding site complementary to tRNA3Lys. J.
Vi-rol. 69:6021–6029.
59. Whitcomb, J. M., B. A. Ortiz-Conde, and S. H. Hughes. 1995. Replication of avian leukosis viruses with mutations at the primer binding site: use of alternate tRNAs as primer. J. Virol. 69:6228–6238.
60. Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Y. Chen.1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell. 61:213–222.