Copyright © 2001, American Society for Microbiology. All Rights Reserved.
Origin Binding Protein-Containing Protein-DNA Complex Formation
at Herpes Simplex Virus Type 1 oriS: Role in
oriS-Dependent DNA Replication
JENNIFER A. ISLERANDPRISCILLA A. SCHAFFER*
Department of Microbiology and Cell and Molecular Biology Graduate Group, University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania 19104
Received 23 April 2001/Accepted 8 May 2001
Initiation of herpes simplex virus type 1 (HSV-1) DNA replication during productive infection of fibroblasts and epithelial cells requires attachment of the origin binding protein (OBP), one of seven essential virus-encoded DNA replication proteins, to specific sequences within the two viral origins, oriL and oriS. Whether initiation of DNA replication during reactivation of HSV-1 from neuronal latency also requires OBP is not known. A truncated protein, consisting of the C-terminal 487 amino acids of OBP, termed OBPC, is the product of the HSV UL8.5 gene and binds to origin sequences, although OBPC’s role in HSV DNA replication is not yet clear. To characterize protein-DNA complex formation at oriS in cells of neural and nonneural lineage, we used nuclear extracts of HSV-infected nerve growth factor-differentiated PC12 and Vero cells, respectively, as the source of protein in gel shift assays. In both cell types, three complexes (complexes A, B, and C) which contain either OBP or OBPC were shown to bind specifically to a probe which contains the highest-affinity OBP binding site in oriS, site 1. Complex A was shown to contain OBPC exclusively, whereas complexes B and C contained OBP and likely other cellular proteins. By fine-mapping the binding sites of these three complexes, we identified single nucleotides which, when mutated, eliminated formation of all three complexes, or com-plexes B and C, but not A. In transient DNA replication assays, both mutations significantly impaired oriS-dependent DNA replication, demonstrating that formation of OBP-containing complexes B and C is required for efficient initiation of oriS-dependent DNA replication, whereas formation of the OBPC-containing complex A is insufficient for efficient initiation.
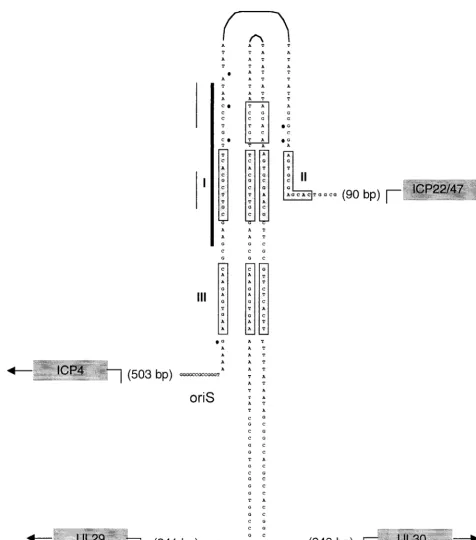
Herpes simplex virus type 1 (HSV-1) encodes seven proteins required for replication of its 152-kb double-stranded DNA genome (27). During productive infection, initiation of HSV DNA replication occurs at viral origins which include one copy of oriL, located in the unique long region of the genome, and two copies of oriS, located in the repeat sequences flanking the unique short region of the genome (22, 25). The core element of oriS consists of a 90-bp sequence that includes a 45-bp imperfect palindrome, whereas the core element of oriL con-sists of a 144-bp perfect palindrome (Fig. 1). Despite these differences, oriL and oriS share extensive nucleotide sequence homology, and both origins contain binding sites for the origin binding protein (OBP), encoded by the UL9 gene (8, 9, 19). OBP binds specifically to sequences within the origins, termed sites I, II, and III, which differ slightly in nucleotide sequence and thus in binding affinity for OBP (site I⬎site II⬎site III) (11). The imperfect oriS palindrome contains one copy each of sites I, II, and III, whereas the perfect oriL palindrome con-tains two copies each of sites I and III (Fig. 1).
The functional significance of the presence of three origins of DNA replication within the HSV-1 genome is not clear. Mutant viruses lacking either oriL or both copies of oriS have no obvious growth defects during productive infection of cells in culture, suggesting that the two types of origin are able to
substitute functionally for each other (13, 21). However, there is evidence that oriL and oriS differ with respect to the effi-ciency of origin function in neural (PC12) and nonneural (Vero) cells. Specifically, in undifferentiated PC12 cells and in Vero cells, oriL and oriS function with similar efficiency in in vitro assays. In nerve growth factor (NGF)-differentiated PC12 (Nd-PC12) cells, the efficiency of oriS function is significantly reduced whereas oriL function is the same as in undifferenti-ated PC12 or Vero cells. Furthermore, addition of the syn-thetic glucocorticoid dexamethasone (DEX) enhances oriL function and further represses oriS function. Although the mechanism responsible for the repression of oriS function in PC12 cells in response to NGF-induced differentiation is un-clear, the enhancement of oriL function in Nd-PC12 cells in response to DEX was shown to be mediated through a perfect glucocorticoid response element (GRE) present in oriL (10). oriS contains a degenerate GRE that may be responsible for the DEX-induced repression of oriS function in Nd-PC12 cells. Based on these findings, the differences in nucleotide sequence between oriL and oriS clearly have significant functional con-sequences. Moreover, cell-type-specific functional differences likely have significant implications for the biology of HSV (e.g., the ability to establish and reactivate from latent infection in neurons). One possible explanation for these functional differ-ences between oriL and oriS is that formation of OBP-con-taining protein-DNA complexes differs at oriL versus oriS and/or in cells of neural versus nonneural lineage.
In the current model of HSV DNA replication, OBP func-tions as a DNA replication initiator protein by binding to HSV
* Corresponding author. Mailing address: Harvard Medical School at the Beth Israel Deaconess Medical Center, 330 Brookline Ave., RN123, Boston, MA 02215. Phone: (617) 2958. Fax: (617) 667-8540. E-mail: [email protected].
6808
on November 9, 2019 by guest
http://jvi.asm.org/
origins, initiating unwinding of origin DNA, and recruiting additional viral DNA replication proteins to the initiation site (reviewed in reference 4). Thus, the ability of OBP to bind to viral origins was shown to be essential for HSV DNA
[image:2.612.62.538.63.603.2]replica-tion, as mutations within the origins themselves or within OBP that abrogate origin binding inhibit origin-dependent DNA replication (12, 23, 24). DNA footprinting and electron micros-copy have demonstrated that binding of OBP to sites I and II
FIG. 1. Comparison of sizes and sequences in HSV-1 oriL and oriS. The DNA sequences of the core origin of oriL (a 144-bp perfect palin-drome) and oriS (a 45-bp imperfect palinpalin-drome) of HSV-1 KOS are shown. Black dots indicate the nucleotide differences between oriL and oriS. OBP binding sites are shaded in light gray and labeled I, II, and III. The GRE in oriL is boxed, and the origin factor 1 (OF-1) bipartite binding site in oriS is indicated by the thin black line. The sequence of the degenerate GRE in oriS is adjacent to the perfect GRE in oriL. The early genes that flank oriL (UL29 and UL30) and the immediate-early genes that flank oriS (ICP4 and ICP22/47) are shown in dark gray. The locations of the transcriptional start sites of these genes relative to the base of the origin palindrome are also shown. The 24-bp oriS site I probe used in gel shift assays throughout this study is represented by the thick vertical line.
on November 9, 2019 by guest
http://jvi.asm.org/
in oriS loops and distorts the A⫹T-rich apex of the origin, which is thought to allow for subsequent DNA unwinding via the ATP-dependent helicase activity of OBP (14, 15). Binding of OBP to viral origins is also thought to facilitate recruitment of other essential replication proteins (i.e., the product of the UL8, UL42, and UL29 genes) to the sites of initiation via direct protein-protein interactions (3, 16, 17).
A truncated form of OBP, termed OBPC, has also been shown to bind to oriL and oriS (2). OBPC, encoded by the UL8.5 gene, is the product of a unique delayed-early transcript that originates within the open reading frame (ORF) of the gene encoding OBP (UL9) (1). Because the genes encoding OBP and OBPC are translated in the same reading frame, the amino acid sequence of OBPC is identical to the C-terminal 487 amino acids of OBP; however, OBPC lacks the N-terminal domain of OBP, which includes five of the six conserved heli-case motifs, the ATP-binding and leucine zipper motifs, as well as domains that mediate interactions with a component of the helicase-primase complex (UL8) and the DNA polymerase processivity factor (UL42). Based on the observations that OBPC localizes to the nuclei of HSV-infected cells and binds to origin DNA, it has been postulated that OBPC may play a role in HSV DNA replication (2). Notably, overexpression of OBPC or C-terminal peptides of OBP has been shown to inhibit origin-dependent viral DNA replication and plaque for-mation by infectious HSV-1 DNA, presumably by occupying OBP binding sites and interfering with the initiation process (2, 20). Ultimately, determination of the precise role of OBPC in HSV DNA replication will require isolation of an OBPC mu-tant. Construction of this virus has not yet been achieved, however, because the genes encoding OBP and OBPC over-lap and their ORFs are translated in the same reading frame. Consequently, mutagenesis of the nucleotide sequence of the OBPC promoter and transcriptional and translational start sites (which would be needed to abrogate expression of OBPC) also alters the amino acid sequence of OBP. Since maintaining a functional form of OBP is critical to differentiating the roles of OBP and OBPC, construction of an OBP⫹OBPC⫺virus is a formidable challenge under these circumstances. Numerous efforts to introduce mutations into the wobble position of OBP codons (i.e., into the promoter and start sites of the OBPC gene) that do not alter the amino acid sequence of OBP but eliminate OBPC expression have met with only limited success. Given the difficulty in isolating a suitable OBPC mutant virus, we have been forced to evaluate OBPC function by using al-ternative approaches, including characterization of mutations in site I that eliminate binding of OBP but not OBPC to HSV origins.
To characterize the formation of protein complexes at oriS and to determine whether these complexes differ when pro-teins are derived from neural versus nonneural cells, we used nuclear extracts of HSV-infected Nd-PC12 and Vero cells as the source of protein and a DNA sequence containing oriS site I (Fig. 1) as the probe in gel shift assays. Using nuclear extracts of both cell types tested, three HSV-specific protein com-plexes, A, B, and C, were shown to bind specifically to the site I probe. The three complexes exhibited similar migration pat-terns when proteins were derived from Nd-PC12 or Vero cells. Mapping of the precise nucleotide binding sites of the three complexes revealed that complex A (containing only OBPC)
binds to a site lying entirely within the shared binding site of two OBP-containing complexes, B and C. We generated single-nucleotide substitution mutations which eliminate formation of all three complexes or of complexes B and C only and determined their effects on oriS-dependent DNA replication. Both mutations reduced oriS-dependent DNA replication sig-nificantly in in vitro assays.
MATERIALS AND METHODS
Cells and virusesPC12 cells (a gift from John Wagner, Cornell University Medical College, New York, N.Y.) were grown as described previously (10). PC12 cells were induced to differentiate by incubation in medium containing NGF (2.5S; Collaborative Biomedical Products, Bedford, Mass.) at a concentra-tion of 100 ng/ml for 6 days, with one medium change on day 3 postplating. In experiments in which PC12 cells were treated with DEX, 0.5M DEX was added to the medium at the time of infection as previously described (10). Vero cells (ATCC CCL-81) were propagated and maintained as described previously (7). The wild-type strain of HSV-1, KOS, was grown and assayed as previously described (7).
Plasmids and mutagenesis.A plasmid containing wild-type oriS (pOS822 [26]) was used in this study. Two single-nucleotide substitution mutations described below, termed mutOBP and mutR(C-G), were introduced into pOS822 by the Quick Change mutagenesis method (Stratagene, La Jolla, Calif.), with the addi-tion of 8l of 25% glycerol and 3l of dimethyl sulfoxide to each 50-l reaction. Reactions underwent 12 amplification cycles in a thermal cycler block (MHJ Research, Watertown, Mass.), using the following parameters: 95°C for 30 s, 55°C for 1 min, and 68°C for 12 min.
Gel mobility shift assays.Nuclear extracts were prepared from 3⫻106 HSV-infected (multiplicity of infection of 10 PFU/cell) Nd-PC12 and Vero cells at 12 h postinfection (hpi), and protein concentrations were measured as described previously (10).
The oligonucleotide probes used in gel shift assays were synthesized by the Nucleic Acid Facility at the University of Pennsylvania Cancer Center. All probes were double-stranded 24-mer oligonucleotides consisting of either wild-type or mutant oriS sequences containing the highest-affinity 10-bp OBP-binding site, site I. The sequences of these wild-type and mutant site I probes are shown in Fig. 4 and 5. Each oligonucleotide and its complement were gel purified, an-nealed to each other, and labeled using T4 polynucleotide kinase and [␥-32P] ATP (Dupont NEN, Boston, Mass.) as described elsewhere (9). DNA binding reactions were performed as follows Nuclear extract (5g) was incubated with 105cpm of32P-labeled oligonucleotide probe (1 ng) and 1.5g of poly (dA-dT) in DNA binding buffer (10% glycerol, 50 mM HEPES [pH 7.9], 100 mM NaCl, 0.5 mM dithiothreitol) in a final volume of 10l. Binding reaction mixtures were incubated for 30 min at room temperature. Protein-DNA complexes were re-solved by electrophoresis on a 6% nondenaturing polyacrylamide (19:1, acryl-amide/bisacrylamide ratio) gel (PAGE) at 4°C. Competition experiments were performed by adding a 100-fold excess of unlabeled DNA probe to the binding reaction. In some experiments, a double-stranded oligonucleotide containing the consensus binding site for nuclear factor 1 (NF-1) was used as a nonspecific competitor (6). Antibody supershift reactions were performed by adding 1l of antibody specific for OBP (R250; generously provided by Mark Challberg, Na-tional Institutes of Health, Bethesda, Md.), 1l of antibody specific for the glu-cocorticoid receptor (GR) (generously provided by Paul Farrell, Ludwig Insti-tute for Cancer Research, London, United Kingdom), or 1l of antibody specific for ICP8 (generously provided by Martin Zweig, National Institutes of Health, Frederick, Md.) to the binding reaction after 5 min, and incubation was allowed to continue for 25 min. Gels were dried and exposed in a Phosphorlmager cassette (Molecular Dynamics, Sunnyvale, Calif.). To evaluate the mobility of OBPC alone bound to the site I probe, approximately 500 ng of bacterially ex-pressed, six-histidine-tagged OBPC (HisOBPC [see below]) was incubated with 32P-labeled site I probe, using the binding reaction parameters described above. Bacterial production of HisOBPC.The UL8.5 gene was cloned into the bac-terial expression vector pTrcHisA (Invitrogen, Carlsbad, Calif.) such that a six-histidine tag was translated in frame with OBPC. Induction of HisOBPC expression was performed by addition of isopropyl--D-thiogalactopyranoside (1 mM) to XL1-Blue cells (Stratagene) as outlined in the Xpress system protein expression manual (Invitrogen). After 4 h, cells were lysed by addition of 1 mg of lysozyme per ml. Sarkosyl (1.5%) was added to cell lysates to improve protein solubility. HisOBPC was bound to a Ni2⫹column containing a 50% slurry of ProBond resin (Invitrogen), and fractions were eluted by addition of 20 mM phosphate buffer (pH⫽4) as outlined in the ProBond resin purification manual
on November 9, 2019 by guest
http://jvi.asm.org/
(Invitrogen). The purity of OBPC in each fraction was verified by sodium dodecyl sulfate-PAGE followed by Coomassie blue staining. The fraction in which HisOBPC was the only protein detectable by Coomassie blue staining was col-lected, and its protein concentration was measured by the method of Bradford (Bio-Rad, Hercules, Calif.), using a standard curve generated with known amounts of bovine serum albumin as the standard.
In vitro origin-dependent DNA replication assays.In vitro oriS-dependent DNA replication assays were performed as described previously (10). Briefly, 3.5⫻106PC12 cells were seeded in collagen-coated 100-mm-diameter plates. Twenty-four hours after plating, cells were transfected with 10g of wild-type or mutant pOS822 by the Lipofectin method (Gibco, Grand Island, N.Y.). After 5 h, cells were washed, and fresh medium containing NGF (100 ng/ml) was added. NGF differentiation proceeded for 6 days, with one medium change on day 3. After 6 days, cells were infected with KOS at a multiplicity of 10 PFU/cell. Cells were harvested at 18 hpi, and total cellular DNA was isolated. For Vero cells, 3.5⫻108cells were plated, transfected 24 h later with the indicated plasmid, and infected 24 h after transfection. Five micrograms of total Nd-PC12 or Vero cell DNA was digested withHindIII (to linearize the vector) and eitherMboI (to cleave unmethylated DNA) or DpnI (to cleave methylated DNA). Digested DNA was resolved by electrophoresis on a 0.8% agarose gel and transferred to a nylon membrane. After UV cross-linking, the membrane was prehybridized for 1 h at 55°C in ExpressHyb solution (Clontech, San Francisco, Calif.) and hybrid-ized for 3 h at 55°C with a32P-labeled probe (3⫻106cpm/ml) generated by nick translation of pGEM7Zf⫹(vector backbone of pOS822). The membrane was then washed according to the ExpressHyb protocol (Clontech) and exposed in a Phosphorlmager cassette.
RESULTS
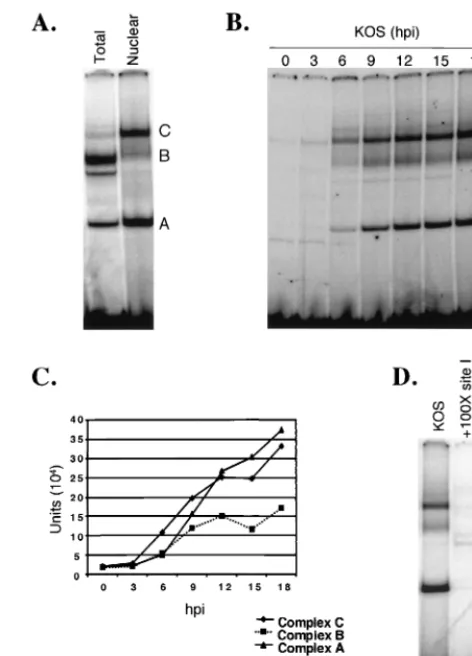
Three HSV-specific complexes bind to oriS site I when nu-clear extracts of Vero or Nd-PC12 cells are used as the source of protein.Previously published gel shift assays from this lab-oratory usingtotalVero cell extracts as the source of protein described binding of two HSV-specific complexes (complexes A and B) to oriS site I (5, 6, 9). In the present study, we used
nuclear extracts of Vero, Nd-PC12 cells, or Nd-PC12 cells
treated with DEX and observed binding of three HSV-specific complexes (complexes A, B, and C) to a double-stranded DNA probe containing oriS site I (Fig. 1). Specifically, to compare whole-cell versus nuclear extracts as the source of protein, we prepared extracts from KOS-infected Vero cells. Using the site I probe, the pattern produced using total Vero cell extract (Fig. 2A, lane 1) was nearly identical to that observed in previous studies using total Vero cell extract (5, 6, 9). Several differ-ences were observed, however, when total Vero cell extract was compared with nuclear extract as the source of protein (Fig. 2A). Complex C was much less prominent in total cell extract (lane 1) than in nuclear extract (lane 2), suggesting that nu-clear extracts are greatly enriched for the proteins that com-prise complex C. The intensity of complex A was also reduced in total cell relative to nuclear extracts, whereas the intensity of complex B was considerably greater in total cell than in nuclear extracts. Moreover, the mobility of complex B in the two types of extract differed. The enhanced intensity of complex B formed from total cell extracts suggests that these extracts are enriched for components of complex B relative to nuclear extracts. A fourth complex that migrated slightly below com-plex B was prominent in gel shifts using total cell extracts but barely visible in shifts using nuclear extracts. Taken together, these observations indicate that the efficiency of formation of specific complexes at oriS site I is dependent on the presence of individual proteins within the cytoplasm or nucleus and the concentrations of the proteins within these two compartments. To address the possibility that protein complexes that form at oriS site I differ in Vero versus PC12 cells or as a function
of NGF or DEX treatment of PC12 cells, we compared the profiles of site I-binding complexes that formed using nuclear extracts of Vero, untreated PC12, Nd-PC12, and DEX-treated Nd-PC12 cells infected with HSV (data not shown). All pro-files closely resembled that shown in Fig. 2A, lane 2. Thus, as measured by gel shift analysis, the differential effects of cell-type-specific factors on oriS function do not appear to be mediated by protein-DNA complex formation at oriS site I. The results of gel shift assays using Nd-PC12 cell nuclear extracts are shown throughout the remainder of this report.
To determine the kinetics of protein-DNA complex forma-tion at oriS site I during HSV infecforma-tion, we performed a time course experiment in which Nd-PC12 cells were infected with KOS, nuclear extracts were prepared at 3-h intervals postin-fection, and complex formation with the site I probe was
eval-FIG. 2. Identification and specificity of oriS binding complexes. (A) Vero cells were infected with KOS at a multiplicity of 10 PFU/cell. At 12 hpi, total cell or nuclear extracts were prepared and used in gel shift assays. (B) NGF-differentiated PC12 cells were infected with KOS at a multiplicity of 10 PFU/cell and harvested at the times indicated. Nu-clear extracts were prepared, and protein-DNA complexes were ana-lyzed as described in Materials and Methods. The letters to the right correspond to specific complexes (A, B, and C). (C) Quantitation and comparison of the intensities of bands corresponding to complexes A, B, and C observed during the time course shown in panel B, expressed as Phosphorlmager units. (D) HSV-infected Nd-PC12 cell nuclear extracts were incubated with the site I probe either alone (KOS) or in the presence of a 100-fold molar excess of unlabeled specific compet-itor DNA (⫹100X site I) or unlabeled nonspecific competitor DNA (⫹100X NF-1).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.310.546.69.397.2]uated by gel shift analysis. At 0 and 3 hpi, formation of two complexes with the probe was detected. As viral DNA repli-cation begins at approximately 3 hpi, and the 0- and 3-hpi profiles were the same, it is likely that these complexes contain primarily cellular proteins. Efficient formation of three pro-tein-DNA complexes, designated complexes A, B, and C, was first observed at 6 hpi. The intensity of these complexes in-creased throughout the course of infection (through 18 hpi), indicating that their formation was dependent on viral infec-tion. Although the intensity of complex C was always greater than that of complex B, the increase in the intensities of the two complexes over time (i.e., the kinetics of their formation) paralleled each other at all times postinfection (Fig. 2C). Spe-cifically, for both complexes the most rapid increase in band intensity occurred between 3 and 9 hpi. In contrast, the most rapid increase in the intensity of complex A was evident slightly later, between 6 and 12 hpi. Notably, material that did not enter the gel is visible in this and other gels; it is not known if this material contains OBP or OBPC.
To determine whether binding of complexes A, B, and C to oriS site I was specific, complex formation was evaluated in the presence of unlabeled specific and nonspecific competitor DNA (Fig. 2D). Whereas addition of a 100-fold molar excess of unlabeled oriS site I DNA abrogated formation of com-plexes A, B, and C (lane 2), addition of a 100-fold molar excess of nonspecific DNA (NF-1 binding site) had no effect on com-plex formation (lane 3), indicating that comcom-plexes A, B, and C bind specifically to the site I probe.
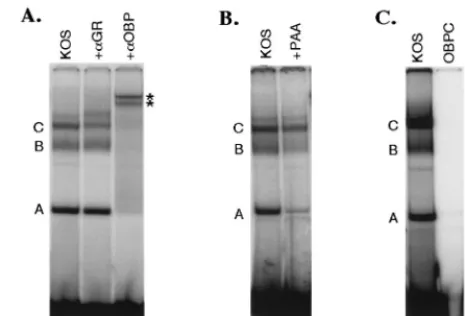
Do complexes A, B, and C contain OBP and/or OBPC?To determine whether complexes A, B, and C contain OBP and/or OBPC, we performed antibody supershift experiments in which antibodies specific for OBP and OBPC were added to gel shift binding reactions (Fig. 3A). Addition of antibody specific for OBP and OBPC shifted the mobility of all three complexes (lane 3, two supershifted bands are marked with asterisks; a third is frequently visible below the second super-shifted band), indicating that complexes A, B, and C contain OBP and/or OBPC. Although addition of the OBP/OBPC-specific antibody also appears to shift the faint band which migrates slightly more slowly than complex C, this observation was not reproducible. Addition of antibody specific for ICP8 (provided by M. Zweig) had no effect on complex formation (data not shown).
Based on previous studies by Hardwicke and Schaffer (10) demonstrating a functional role for GR binding to the consen-sus GRE in oriL, we investigated the possibility that GR may participate in complex formation at the degenerate GRE se-quence present in oriS. For this purpose, antibody to GR was added to oriS site I binding reactions. Addition of antibody to GR had no discernible effect on complexes A and B but re-sulted reproducibly in the presence of an additional band above complex C and a slight decrease in the intensity of complex C, suggesting that GR may be a component of com-plex C (Fig. 3A, lane 2).
To distinguish between complexes that contain OBP and those that contain OBPC, we took advantage of the fact that the synthesis of the delayed-early protein, OBPC, but not the early protein, OBP, is largely but not entirely dependent on HSV DNA synthesis. Thus, OBPC transcript and protein levels are greatly reduced in the presence of phosphonoacetic acid
(PAA) (2). PAA used at 400g/ml inhibits HSV DNA syn-thesis by inhibiting the activity of the virus-encoded DNA polymerase. PAA was therefore used to reduce the synthesis of OBPC in an effort to distinguish between complexes contain-ing OBP and OBPC. For this purpose, Nd-PC12 cells were infected with KOS in the absence (Fig. 3B, lane 1) or presence (lane 2) of 400g of PAA per ml, and nuclear extracts were used in gel shift assays. As expected, addition of PAA de-creased the intensities of all three complexes, as this drug inhibits viral DNA replication, resulting in fewer genomes from which viral proteins can be expressed. However, of the three complexes A, B, and C, addition of PAA decreased the intensity of complex A most markedly (12% of the no-PAA control), suggesting that the synthesis of the primary compo-nent of complex A is most dependent on HSV DNA synthesis and thus contains OBPC. Addition of PAA also reduced the intensity of complex B (34% of the no-PAA control), suggest-ing that in addition to OBP, this complex may also contain OBPC or another PAA-sensitive protein. Addition of PAA had the least effect on formation of complex C (56% of the no-PAA control), suggesting that this complex does not con-tain OBPC.
[image:5.612.316.549.74.232.2]To determine whether PAA-sensitive complex A contained exclusively OBPC, we compared its mobility with that of a band produced by bacterially expressed OBPC bound to the site I probe (Fig. 3C). HisOBPC was synthesized in bacteria and purified by metal chelate affinity chromatography. Incuba-tion of HisOBPC with the site I probe resulted in the forma-tion of a complex (lanes 2 and 3) that migrated with mobility identical to that of complex A from KOS-infected cells (lane 1;
FIG. 3. Characterization of HSV-specific complexes that form at oriS. (A) HSV-infected PC12 cell nuclear extracts were incubated with the site I probe. After 5 min of incubation, no antibody (lane 1), antibody specific for GR (lane 2), or antibody specific for OBP (lane 3) was added to the binding reaction. Incubation was continued for an additional 25 min, and protein-DNA complexes were resolved by non-denaturing PAGE. Complexes are labeled A, B, and C, and super-shifted complexes are indicated with asterisks. (B) Nd-PC12 cells were infected with KOS either in the absence (lane 1) or presence (lane 2) of PAA (400 g/ml), cells were harvested at 12 hpi, and nuclear extracts were prepared; 5g of nuclear extract was incubated with the site I probe, and complexes were resolved and visualized as described for panel A. (C) HSV-infected PC12 cell nuclear extract (lane 1) or bacterially expressed His OBPC (lane 2 and 3) was incubated with the oriS site I probe. Complexes were resolved and visualized as described for panel A. Lane 3 is a darker exposure of lane 2.
on November 9, 2019 by guest
http://jvi.asm.org/
lane 3 is a longer exposure of lane 2). Together, these results suggest that complex A contains OBPC and no other viral or cellular proteins and that complexes B and C contain OBP. Notably, the presence of OBPC in complex B remains a pos-sibility.
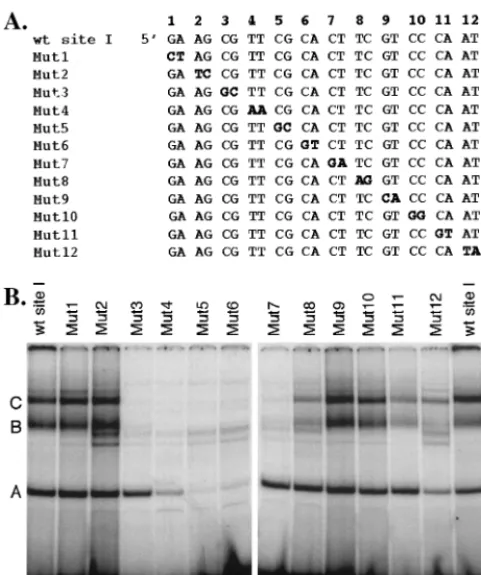
Nucleotide mapping of the binding sites of complexes A, B, and C.To identify the precise nucleotides required for forma-tion of complexes A, B, and C with the site I probe, we ana-lyzed the ability of these complexes to form with probes that contained sequential two-base-pair substitution mutations (Fig. 4A, mutations are indicated in bold). In contrast to com-plexes produced using the wild-type probe, comcom-plexes B and C were barely detectable upon incubation of nuclear extracts with probes Mut3 through Mut7, indicating that the mutations contained within these probes abrogate formation of these complexes. The intensity of complexes B and C was reduced to a lesser extent upon incubation of extracts with probes Mut8, -11, and -12 relative to the wild-type probe, implicating the wild-type nucleotides mutated in these probes as essential for efficient formation of complexes B and C with site I DNA. Formation of complex A was reduced significantly upon incu-bation of extracts with probes Mut4, -5, and -6 and slightly
upon incubation with Mut12. The enhanced intensity of two bands that migrate below complex B in tests using probes Mut3 through -8 and Mut12 was notable. The composition of these bands (i.e., whether they contain viral or cellular proteins) is unknown. These results indicate that 10 nucleotides (repre-sented by probes Mut3 through Mut7) are required for forma-tion of complexes B and C, whereas only 6 nucleotides (rep-resented by probes Mut4 through Mut6) are required for efficient formation of complex A. Interestingly, the dinucleo-tide represented by Mut12 appeared to be required for effi-cient formation of all three complexes. Whether this result is real or a consequence of the location of the mutated nucleo-tides at the 3⬘ end of the probe remains to be determined. Additional tests in which unlabeled mutant probes were used to compete for formation of complexes A, B, and C to the wild-type site I probe confirmed these results (data not shown). Based on these findings and as shown schematically in Fig. 5B, the binding site for complex A is contained entirely within the shared binding site for complexes B and C.
[image:6.612.55.296.70.358.2]A point mutation eliminates formation of complexes B and C without affecting formation of complex A at oriS site I.To further test the observation that the binding site for complex A is contained entirely within the shared binding site of com-plexes B and C, we generated a probe containing a single-nucleotide substitution mutation outside the binding site for complex A but within the binding site for complexes B and C, this mutant probe was designated mutOBP (Fig. 5B). Similarly, we generated a probe that contained a single-nucleotide sub-stitution mutation within the binding site for all three com-plexes; this probe was designated mutR(C-G). mutR(C-G) differs from mutR(C-A) described previously by Dabrowski et al. (5) in that in this study, C was changed to G rather than A
FIG. 4. Mapping of the nucleotide binding sites of complexes A, B, and C. (A) Nucleotide sequence of the wild-type (wt) and mutant site I probes. Two-base-pair substitution mutations (indicated in bold) were introduced throughout the probe, yielding a total of 12 mutant probes (labeled Mut1 through Mut12). (B) HSV-infected PC12 cell nuclear extract was incubated with equal counts of labeled wild-type or mutant site I probe; protein-DNA complexes were resolved by non-denaturing PAGE, gels were dried, and complexes were visualized with a Phosphorlmager. The profile of complexes that form on the wild-type probe is shown in the far left and right lanes. Complexes A, B, and C are indicated in these lanes.
FIG. 5. Point mutations in the site I probe eliminate formation of specific protein-DNA complexes. (A) HSV-infected Nd-PC12 cell nu-clear extract was incubated with wild-type (wt) or mutant [mutOBP or mutR(C-G); sequences shown in panel B] site I probes. Binding reac-tions were subjected to nondenaturing PAGE, gels were dried, and complexes were visualized with a Phosphorlmager. Complexes are marked A, B, and C. (B) DNA sequences of the wild-type, mutOBP, and mutR(C-G) probes. Single-base-pair substitution mutations are indicated in bold. The binding sites for complex A (smaller box) and complexes B and C (larger box) are indicated (based on Fig. 4B). Above each probe sequence are indicated the complexes that form with each probe (an “X” through the complex designates the failure of the complex to form with that probe).
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.313.548.447.605.2](Fig. 5B). Nuclear extracts from KOS-infected Nd-PC12 cells were incubated with the wild-type oriS site I, mutOBP, or mutR(C-G) probe, and protein-DNA complex formation was evaluated by gel shift analysis (Fig. 5A). As expected, com-plexes A, B, and C formed efficiently with the wild-type probe; however, complex A, but not complex B or C, formed with mutOBP, and none of the three complexes formed with mutR (C-G). These findings confirm those presented in Fig. 4 and demonstrate that a single-nucleotide substitution mutation is sufficient to eliminate binding of all three complexes. The nucleotide sequences of the wild-type and mutant probes and of the complexes that bind to each probe are summarized schematically in Fig. 5B.
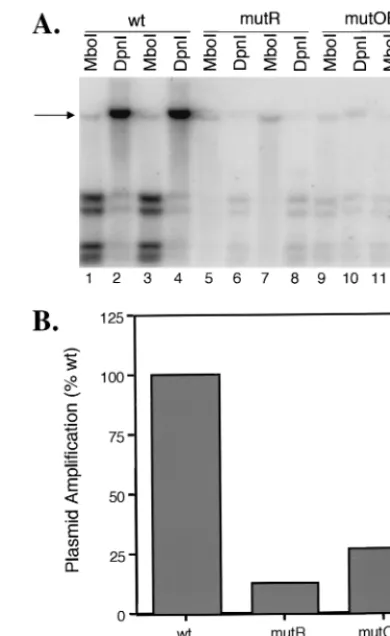
Effect of mutOBP and mutR (C-G) on oriS-dependent HSV DNA replication.To examine the effect of formation of com-plexes A, B, and C at site I on origin-dependent HSV DNA replication, the mutOBP and mutR(C-G) mutations were in-troduced into the wild-type oriS-containing plasmid, pOS822, and the effects of these mutations were evaluated in in vitro origin-dependent DNA replication assays (Fig. 6A). Undiffer-entiated PC12 cells were transfected with pOS822 (lanes 1 to 4) or pOS822 containing the mutR(C-G) (lanes 5 to 8) or mutOBP (lanes 6 to 10) mutation. PC12 cells transfected with the pOS822 backbone (pGEM) which does not contain oriS were used as a negative control for plasmid amplification (data not shown). Following transfection, PC12 cells were differen-tiated with NGF for 6 days and then infected with KOS. At 18 hpi, total cell DNA was extracted, subjected to restriction enzyme digestion withMboI or DpnI to distinguish between input or newly replicated plasmid DNA, respectively, and an-alyzed by Southern blot. The probe used in these tests hybrid-izes to a DNA sequence contained in the vector component of all pOS822-derived plasmids. The blot in Fig. 6A shows the results of each plasmid tested in duplicate, and the data are presented quantitatively in Fig. 6B. The intensity of the band corresponding to newly replicated pOS822 (DpnI resistant) (Fig. 6A, lanes 2 and 4, arrow) was⬃11-fold greater than that of input pOS822 (MboI resistant (lanes 1 and 3), indicating that the wild-type oriS-containing plasmid was amplified effi-ciently (Fig. 6B). In contrast, amplification of plasmids con-taining the mutR(C-G) (lanes 6 and 8) or mutOBP (lanes 10 and 12) mutation was considerably less than that of the wild-type plasmid (lanes 1 and 3) (Fig. 6). Similar results to mutR (C-G) were obtained for mutR(C-A) by Dabrowski et al. (5). The levels of newly replicated mutant plasmids were only 1.4-and 2.0-fold, respectively, above the input plasmid levels. There-fore, the single-nucleotide substitution mutations in mutR(C-G), mutR(C-A), and mutOBP reduced the efficiency of plas-mid replication to 10% G); Fig. 6B], 4% [mutR(C-A)] (5), and 25% (mutOBP; Fig. 6B) of the wild-type level, indicating that mutations in the shared region of the binding sites of complexes A, B, and C [mutR(C-A), mutR(C-A)] or B and C (mutOBP) greatly reduce the ability of oriS to support efficient origin-dependent DNA replication.
DISCUSSION
OBP and OBPC-containing complexes that form at oriS site I.The results presented here confirm and extend previous studies in which total Vero cell extracts were used to
demon-strate formation of two protein complexes, complexes A and B, at oriS site I by gel shift analysis (5, 6, 9). Comparison of the site I-binding complexes from total cell and nuclear extracts revealed differences in the intensities of complexes A and B as well as the presence of an additional complex, complex C, whose formation appears to be dependent on nuclear factors (Fig. 2A). The differences in complex formation noted when total cell versus nuclear extracts were used may reflect differ-ences in the presence or concentration of nucleus- or cyto-plasm-specific proteins present in these complexes.
Alterna-FIG. 6. Effects of mutations mutOBP and mutR(C-G) on oriS-dependent HSV DNA replication. (A) PC12 cells were transfected with pOS822-derived plasmids containing wild-type (wt; lanes 1 to 4), mutR(C-G) (lanes 5 to 9), and mutOBP (lanes 9 to 12) or mu-tant [mutR(C-G) and mutOBP] oriS sequence. The pOS822 vector (pGEM) plasmid was used as a negative control (data not shown). Following transfection, PC12 cells were differentiated with NGF for 6 days and infected with KOS at a multiplicity of 10 PFU/cell. At 18 hpi, total cellular DNA was extracted, subjected to digestion with
MboI (lanes 1, 3, 5, 7, 9, and 11) orDpnI (lanes 2, 4, 6, 8, 10, and 12) to differentiate newly replicated DNA, and analyzed by South-ern blot hybridization using a32P-labeled, nick-translated probe which recognizes pGEM vector sequence present in the wild-type and mutant oriS-containing plasmids. Each sample was tested in duplicate. oriS-containing plasmid DNA, which is resistant to enzyme digestion, is indicated by an arrow. (B) The bands shown in panel A were quantitated by Phosphorlmager analysis, and fold replication was calculated by dividing the sum of theMboI-resistant andDpnI-resistant bands by theMboI-resistant band for each sample. Mutant plasmid amplification is expressed as a percentage of wild-type plasmid ampli-fication (set to 100%). The graph shown represents the average of the duplicate samples shown in panel A.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.333.528.71.389.2]tively, the conformation, oligomerization, or posttranslational modification of proteins contained within these complexes may differ in the cytoplasm versus the nucleus. That the interaction of OBP-containing complexes with oriS is dependent on fac-tors present in the nucleus and/or cytoplasm likely has impor-tant implications for the functional role of OBP during HSV infection. Notably, however, no differences in complex forma-tion were noted when proteins used in gel shift assays were derived from nuclei of Vero or Nd-PC12 cells or Nd-PC12 cells treated with DEX. Thus, the basis for the differential effects of NGF and DEX on oriS function noted previously (10) remains unclear.
Of the three HSV-specific complexes that form at site I, gel shift analysis using nuclear extracts of PAA-treated cells or bacterially expressed OBPC suggests that the sole protein com-ponent of complex A is OBPC. In contrast, several lines of evidence suggest that complexes B and C contain OBP: these complexes (i) supershift upon addition of antibody to OBP, (ii) are first detectable and increase in intensity at times consistent with E protein expression, and (iii) share a nucleotide binding site previously shown to be the binding site for OBP (6, 11).
The electrophoretic mobilities of complexes B and C differ, however, suggesting that they are composed of different oligo-meric forms of OBP or that they contain additional viral or cellular proteins. Incubation of the site I probe with nuclear extracts of Vero cells infected with an OBP-expressing recom-binant adenovirus resulted in the formation of two complexes that migrate with mobilities identical to those of complexes B and C (unpublished observations), indicating that otherviral
proteins are not required for formation of these complexes, and additional protein components of complexes B and C are therefore likely to be cellular in origin. This hypothesis is further supported by the finding that GR, a cellular protein known to affect the efficiency of HSV origin function (10), may be a component of complex C. Given that several members of the HSV DNA replication complex (i.e., UL8, UL42, and ICP8) have been shown to interact with OBP in assays using purified proteins (3, 16, 17), it is somewhat surprising that these proteins have not yet been detected in OBP-containing complexes bound to oriS by gel shift assay. Specifically, addi-tion of antibody to ICP8 to binding reacaddi-tions had no detectable affect on site I complex formation (data not shown), yet ICP8 has been shown to bind specifically to OBP (3), and the inter-action site in OBP has been mapped. One possible explanation for our inability to detect ICP8 within these complexes is that the amounts of DNA replication proteins in HSV-infected cell nuclear extracts are limited. It is also possible that conditions compatible with formation of the replication complex at the origin are not duplicated in our in vitro assay. Identification of other protein components within the complexes that form at oriS will contribute to our understanding of initiation of HSV DNA replication at HSV-1 origins.
Roles of OBP and OBPC in HSV DNA replication.Extensive mutagenesis of the oriS site I probe revealed that complexes B and C share a 10-bp binding site which was previously de-scribed as OBP binding site I (11). Despite the fact that OBP and OBPC share the same C-terminal DNA binding domain, the DNA binding site of complex A, which appears to consist solely of OBPC, was found to comprise only 6 bp which lie totally within the 10-bp binding site of complexes B and C.
That the OBP-containing complexes require a larger binding site than OBPC may reflect the larger size of OBP relative to OBPC, dimer formation of OBP but not OBPC, or the pres-ence of additional cellular proteins in complexes B and C but not A which are necessary for efficient contact with origin DNA. Notably, the sizes of complex A and B binding sites determined in this study are contrary to previous findings in which the nucleotide binding site of complex A was several nucleotides larger than that of complex B (6). These differ-ences are likely due to differdiffer-ences in the composition of the cell extract used (total cell extract [6] versus nuclear extract [this study]) as well as differences in the mutant probes used for mapping studies (nucleotide deletions [6] versus dinucleotide substitutions [this study]).
Given that the binding site of complex A (OBPC) lies en-tirely within the binding site of OBP-containing complexes B and C, a single base pair was identified which, when mutated (mutOBP), eliminated binding of complexes B and C without affecting binding of complex A to site I. The mutOBP mutation reduced the replication efficiency of an oriS-containing plas-mid to 25% of that of the wild-type plasplas-mid, indicating that binding of OBPC alone is insufficient to support origin-depen-dent DNA replication. This finding is not unexpected, given that OBPC lacks multiple domains present in OBP that are thought to be necessary for HSV DNA replication. The loca-tion of the complex A binding site relative to that of complexes B and C does not allow us to make the reciprocal mutation in site I (i.e., eliminate binding of OBPC without affecting bind-ing of OBP-containbind-ing complexes), and thus we cannot rule out the possibility that binding of OBPC is important for HSV DNA replication. Based on previous studies that describe a strong dominant-negative effect of OBPC on the replication of HSV (2), it has been postulated that OBPC may act as a repressor of origin-dependent DNA replication by competing with OBP for origin binding. As noted above, elucidation of the precise function of OBPC will require isolation and char-acterization of OBP⫹OBPC⫺plasmids and viruses.
ACKNOWLEDGMENTS
This study was funded by NIH grant R01-A128537 from the Na-tional Institute of Allergy and Infectious Diseases. J.A.I. was sup-ported by NIH training grant T32-AI007325.
We gratefully acknowledge John Wagner for providing PC12 cells, Mark Challberg for providing antisera to OBP, Paul Farrel for pro-viding antisera to GR, the University of Pennsylvania Nucleic Acid Facility for assistance in sequencing problematic regions of oriS, and members of the Schaffer laboratory for helpful discussions and ideas.
REFERENCES
1.Baradaran, K., C. E. Dabrowski, and P. A. Schaffer.1994. Transcriptional analysis of the region of herpes simplex virus type 1 genome containing the UL8, UL9, and UL10 genes and identification of a novel delayed-early gene product, OBPC. J. Virol.68:4251–4261.
2.Baradaran, K., M. A. Hardwicke, C. E. Dabrowski, and P. A. Schaffer.1996. Properties of the novel herpes simplex virus type 1 origin binding protein, OBPC. J. Virol.70:5673–5679.
3.Boehmer, P. E., M. C. Craigie, N. D. Stow, and I. R. Lehman.1994. Asso-ciation of origin binding protein and single strand DNA-binding protein, ICP8, during herpes simplex virus type 1 DNA replication in vivo. J. Biol. Chem.269:29329–29334.
4.Boehmer, P. E., and I. R. Lehman.1997. Herpes simplex virus DNA repli-cation. Annu. Rev. Biochem.66:347–384.
5.Dabrowski, C. E., P. J. Carmillo, and P. A. Schaffer.1994. Cellular protein interactions with herpes simplex virus type 1 oriS. Mol. Cell. Biol.14:2545– 2555.
on November 9, 2019 by guest
http://jvi.asm.org/
6.Dabrowski, C. E., and P. A. Schaffer.1991. Herpes simplex virus type 1 origin-specific binding protein: oriS-binding properties and effects of cellular proteins. J. Virol.65:3140–3150.
7.DeLuca, N. A., and P. A. Schaffer.1985. Activation of immediate-early, early, and late promoters by temperature-sensitive and wild-type forms of herpes simplex virus type 1 protein ICP4. Mol. Cell. Biol.5:1997–2008.
8.Elias, P., and I. R. Lehman.1988. Interaction of the origin binding protein with an origin of replication of herpes simplex virus 1. Proc. Natl. Acad. Sci. USA85:2959–2963.
9.Hardwicke, M. A., and P. A. Schaffer.1995. Cloning and characterization of herpes simplex virus type 1 oriL: comparison of replication and protein-DNA complex formation by oriL and oriS. J. Virol.69:1377–1388. 10. Hardwicke, M. A., and P. A. Schaffer.1997. Differential effects of nerve
growth factor and dexamethasone on herpes simplex virus type 1 oriL- and oriS-dependent DNA replication in PC12 cells. J. Virol.71:3580–3587. 11. Hazuda, D. J., H. C. Perry, A. M. Naylor, and W. L. McClements.1991.
Characterization of the herpes simplex virus origin binding protein with oriS. J. Biol. Chem.266:24624–24626.
12. Hernandez, T. R., R. E. Dutch, I. R. Lehman, C. Gustafsson, and P. Elias. 1991. Mutations in a herpes simplex virus type 1 origin that inhibit interac-tion with origin binding protein also inhibit DNA replicainterac-tion. J. Virol.65: 1649–1652.
13. Igarashi, K., R. Fawi, R. J. Roller, and B. Roizman.1993. Construction and properties of a recombinant herpes simplex virus 1 lacking both S-compo-nent origins of DNA synthesis. J. Virol.67:2123–2132.
14. Koff, A., J. F. Schwedes, and P. Tegtmeyer.1991. Herpes simplex virus origin binding protein (UL9) loops and distorts the viral replication origin. J. Virol. 65:3284–3292.
15. Makhov, A. M., P. E. Boehmer, I. R. Lehman, and J. D. Griffith.1996. The herpes simplex virus type 1 origin-binding protein carries out origin specific DNA unwinding and forms stem-loop structures. EMBO J.15:1742–1750. 16. McLean, G. W., A. P. Abbotts, M. E. Parry, H. S. Marsden, and N. D. Stow.
1994. The herpes simplex virus type 1 origin-binding protein interacts spe-cifically with the viral UL8 protein. J. Gen. Virol.75:3699–2706.
17. Monahan, S. J., L. A. Grinstead, W. Olivieri, and D. S. Parris.1998. Inter-action between the herpes simplex virus type 1 origin-binding and DNA polymerase accessory proteins. Virology241:122–130.
18. Nguyen-Huynh, A. T., and P. A. Schaffer.1998. Cellular transcription factors enhance herpes simplex virus type 1 oriS-dependent DNA replication. J. Vi-rol.72:3635–3645.
19. Olivo, P. D., N. J. Nelson, and M. D. Challberg.1988. Herpes simplex virus DNA replication: the UL9 gene encodes and origin-binding protein. Proc. Natl. Acad. Sci. USA85:5414–5418.
20. Perry, H. C., D. J. Hazuda, and W. L. McClements.1993. The DNA binding domain of herpes simplex virus type 1 origin binding protein is a transdomi-nant inhibitor of virus replication. Virology193:73–79.
21. Polvino-Bodnar, M., P. K. Orberg, and P. A. Schaffer.1987. Herpes simplex virus type 1 oriL is not required for virus replication or for the establishment and reactivation of latent infection in mice. J. Virol.61:3528–3535. 22. Stow, N. D.1982. Localization of an origin of DNA replication within the
TRs/IRs repeated region of the herpes simplex virus type 1 genome. EMBO J.1:863–867.
23. Stow, N. D., G. Brown, A. M. Cross, and A. P. Abbotts.1998. Identification of residues within the herpes simplex virus type 1 origin binding protein that contribute to sequence-specific DNA binding. Virology240:183–192. 24. Weir, H. M., and N. D. Stow.1990. Two binding sites for the herpes simplex
virus type 1 UL9 protein are required for efficient activity of the oriS repli-cation origin. J. Gen. Virol.71:1379–1385.
25. Weller, S. K., S. Spadaro, J. E. Schaffer, A. W. Murray, A. M. Maxam, and P. A. Schaffer.1985. Cloning, sequencing, and functional analysis of oriL, a herpes simplex virus type 1 origin of DNA synthesis. Mol. Cell. Biol.5:930– 942.
26. Wong, S. W., and P. A. Schaffer.1991. Elements in the transcriptional regulatory region flanking herpes simplex virus type 1 oriS stimulate origin function. J. Virol.65:2601–2611.
27. Wu, C. A., N. J. Nelson, D. J. McGeoch, and M. D. Challberg.1988. Iden-tification of herpes simplex virus type 1 genes required for origin-dependent DNA synthesis. J. Virol.62:435–443.