Vol.61, No. 11
Herpes Simplex Virus Type
1oriL
Is
Not
Required for Virus
Replication
or
for the Establishment
and Reactivation of
Latent
Infection in Mice
MARYELLEN POLVINO-BODNAR, PAULO K. ORBERG,AND PRISCILLA A. SCHAFFER*
Laboratory of Tumor Virus Genetics, Dana-Farber CancerInstitute, andDepartment of Microbiology andMolecular
Genetics, Harvard Medical School, Boston, Massachusetts 02115
Received 11May 1987/Accepted 31 July 1987
During thecourseofexperiments designed toisolate deletionmutantsofherpes simplexvirustype1 inthe geneencoding themajor DNA-binding protein, ICP8,amutant,d61, thatgrewefficiently in ICP8-expressing Vero cells butnotin normal Verocellswasisolated(P. K. Orberg andP. A.Schaffer, J.Virol.61:1136-1146,
1987). d61wasderived by cotransfection ofICP8-expressing Vero cells with infectious wild-type viral DNA and a plasmid, pDX, that contains an engineered 780-base-pair (bp) deletion in the ICP8 gene, as well as a
spontaneous -55-bp deletioninOriL. Gel electrophoresis and Southern blot analysisindicated thatd61 DNA
carried both deletionspresentinpDX. The ability of d61toreplicate despitethe deletion inOriL suggested that
afunctionalOriLisnotessential forvirusreplicationinvitro. Becaused61 harbored twomutations,asecond
mutant,ts+7, withadeletioninoriL-associated sequencesandanintact ICP8genewasconstructed. Bothd61 andts+7 replicated efficiently in their respective permissive host cells, althoughtheiryieldswereslightly lower
than those of control viruses with intact oriLsequences. An in vitro test oforigin function of isolated OriL sequences from wild-type virus and ts+7 showed that wild-typeOHiL, butnotts+7 OHIL, wasfunctional upon infection with helpervirus. Inaneffort todetermine therequirement for OriLinlatency, ts+7wascompared withwild-type virus for its abilitytoestablish, maintain, and be reactivatedfrom latent infection inamurine
eye model. The mutant was reactivated as efficiently as was wild-type virus from trigeminal ganglia after
cocultivation withpermissive cells, and each of thesevenreactivatedisolateswasshowntocarrythe-150-bp deletion characteristic ofts+7. These observations demonstratethatOHLis notrequiredfor virusreplicationin
vitroorfor theestablishment and reactivation of latent infectioninvivo.
The genomeof herpes simplex virus type 1 (HSV-1) is a
linear, double-stranded DNA molecule of approximately 160,000 base pairs (bp). It consists ofalong unique region
(UL) flanked by invertedrepeatsequencesaband b'a' andan
Scomponentconsisting ofashortunique region (Us) flanked
by the invertedrepeatsacand c'a'(Fig. 1, line 1). During the processof viral DNA synthesis, molecules aregenerated in which the long and short components of the genome are inverted relative to one another such that approximately equimolaramountsof the fourpossible isomers ofviral DNA
areproduced (1, 36). The viralgenomeisthoughttoreplicate byarolling-circle mechanism which yields large head-to-tail
concatemeric intermediates thataresubsequently cleavedto generate unit-length molecules (1, 3). Early electron micro-scopic studies provided evidence for the existence oftwo
origins of DNA synthesis in the HSV-1genome, one nearthe
centerofUL and another nearone end of the molecule (9, 14). Studies of thegenomesof defective interfering particles
generated during serialpassage of virus at high multiplicity of infection and tests of origin function with cloned viral DNAfragments have shown that HSV-1DNA contains two
copies of one origin, termed
oris,
located in the c and c'invertedrepeats, and one copyofa second, more complex
origin, termed oriL, located near thecenter of UL (2, 7, 8, 16-18, 25, 35, 37, 43).
Fororigin function, the diploid
oris
requiresno morethan 90 bp, which includes an almost perfect palindromicse-quenceof 45 bp (40).oriLshares considerablehomology with
oris
and contains a perfect 144-bp palindrome (12, 28, 45).* Corresponding author.
While
oris
sequencesarestableuponcloning in bacteria, the larger oriL palindrome suffers deletions with high frequency when propagated inbacterial vectors(12, 45). Spontaneous or engineered deletions withinoris
and oriL palindromes result in loss of thecapacity forautonomous replication (38, 45).The existence of two classes of defective interfering particles (class I, whose genomes contain
oris
sequences, and class II, whose genomes contain oriL sequences)dem-onstratesthatbothoriginsareindeedcapableoffunctioning during productive infection in vitro (7, 8, 19). Little is known, however, about the requirement for each class of origin in the process of viral DNA synthesis (i.e., are oriL and bothcopies of
oris
essential for viral DNAsynthesis?)orwhether one class of origin is preferentially used during
latentinfection. The formerquestionwaspartially answered
inarecentreportthata mutantof HSV-1 lackingone copy of
oris
is viable(23). In thispaper,wedemonstrate thattheabsence ofafunctional oriL has little effectonviral
replica-tion in vitro.Furthermore,weshow that oriL isnotrequired for the establishmentorreactivation of latent infection ina
murineeyemodel.
MATERIALS AND METHODS
Cells and viruses. African green monkey kidney (CV-1,
Vero, and U-47) and human embryonic lung (HEL) cells were propagated and maintained as previously described
(44). ICP8-expressing U-47 cells usedaspermissive hosts for the derivation and propagation of ICP8 deletion mutants
werederived by cotransfection ofVerocells with the
ICP8-3528 JOURNALOFVIROLOGY, Nov. 1987,p. 3528-3535
0022-538X/87/113528-08$02.00/0
CopyrightC 1987, American Society forMicrobiology
on November 10, 2019 by guest
http://jvi.asm.org/
a b UL b'a'o' US co
. , . I
0.34 0.35 0.36 0.37
3.3kb gB
5.6kb ? 2
--- ---- -- --
-1 I 1
0.38 0.39 0.40 0.41 0.42 0.43 0.44 4.2 kb IC P8 4.3kb D
0
lOkb ICP8 -.. 4.2kb DNAMI.
9kb ?
tsA24-I
4
BSB T 5
'W
1 'I
II IXP K P TP S 6 pKEF-P4
i---p 7 pSGIS-SaIIA
8 pDX
B K
OxBomHI E E=EcoRI
---4 Kr-KpnI
s P-Pst I
--q
s SZSGII
TzBst E 11 Xz Xho I
i A
s x x
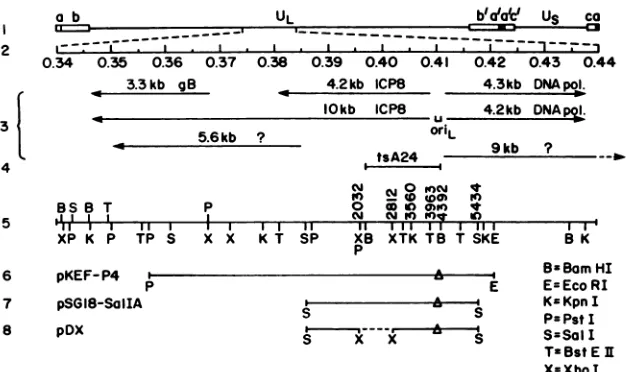
FIG. 1. Genomic location oforiLand
plasmids
used in thisstudy.
Theprototype arrangementof the HSV-1genomeis shown inline1.Themapunits of the
expanded,
centralportion
ofthelong
unique region
(UL)areshown inline2.Thetranscripts
knowntomaptothisregion
of thegenome(all
of theearly
or0
kinetic class[15])andthelocation of theoriL (12, 45)areshowninline3. Thelength
of thetranscript
isindicated in kilobases
(kb).
The locationof the mutation in the ICP8ts mutantused in thisstudy,
tsA24, appearsin line4(44). Relevantrestrictionsitesand the
corresponding
nucleotide numbers(28)aredepicted
in thenextthreelines;thesymbol
Arepresentsdeletion inoriL sequencesof -55bp
which ispresentin theparental plasmid, pKEF-P4,
from whichpSG18-Sa/I
AandpDX
werederived.containing plasmid pKEF-P4 (Fig.
1,
line6)
andpSV2neo
(27,
34).
TheKOS strain of HSV-1wasusedasthe
wild-type
virusfrom which
temperature-sensitive
QsA24
[44])
anddeletionmutantswerederived. Viruseswere
propagated
andassayed
as
previously
described(31).
Plasmids and
cloning.
Themaplocations oforiL-associated
viralDNAinsertsin
plasmids
usedin thisstudy
areshowninFig.
1.pKEF-P4
(4)
andptkCAToris
(not shown)
werekindly
provided
by
N. DeLuca. The latterplasmid
is aderivative of
ptkCAT
(6)
which contains a220-bp
SmaIfragment
thatcontainsoris.
pSG18-Sall
A(21;
Fig.
1,
line7)
contains the intactgenefor ICP8anda
spontaneous
-55-bp
deletion in
oriL-
pDX (Fig.
1,
line8)
was derived frompSG18-SalI
Aby
digestion
withXhol andreligation.
pUC18
(46)
wasobtained fromPharmacia,
Inc.(Piscataway,
N.J.).
Restriction endonucleases and T4 DNA
ligase
wereob-tained from New
England
BioLabs,
Inc.(Beverly,
Mass.)
and usedas
suggested
by
the manufacturer.Isolationof viral DNA.Infected-cellDNAwas
prepared
asdescribed
previously
(5).
Afterproteinase
Kdigestion,
viralDNAwas
separated
from cellularDNAby centrifugation
inCsCl
gradients
(11).
KOS DNA was isolated from infectedHEL
cells; tsA24,
tsC,,
ts+3,
andts'7
DNAs wereisolatedfrom infected Vero
cells;
and d6l DNA was isolated frominfected U-47cells.
Isolation of viralDNA
fragments.
HSV DNAfragments
for use asprobes
in Southern blothybridizations
wereisolated afterrestriction endonucleasedigestion
ofplasmid
DNAandelectrophoresis through
a 0.4% agarosegel.
Afterstaining
withethidium
bromide,
bands of interestwere excised andsubmitted to three
cycles
offreezing
andthawing
(32).
Agarose
wasseparated
fromthe DNA solutionby
centrifu-gation.
ViralDNAfragments
used intestsoforigin
functionwere isolated after
digestion, electrophoresis
through
a4%polyacrylamide
gel,
and electroelution into adialysis bag
(24).
Inallcases,DNAfragments
wereethanolprecipitated
after
purification
by
passagethrough
anElutip-d
column(Schleicher
&Schuell, Inc.,
Keene,
N.H.).
Blot
hybridization.
Specific
DNA sequences indigests
ofviral or cellular DNA were detected
by
the method ofSouthern
(33).
Probes were labeled with [12P]dCTP
and[32
P]dGTP
(Amersham
Corp., Arlington Heights,
111.)
by
nick translation
(24).
Marker transfer and marker rescue. Vero and U-47 cells
were cotransfected with infectious viral
(KOS
ortsA24)
DNA and
plasmid (pDX
orpKEF-P4)
DNAs aspreviously
described
(29).
Cloning
andsequencing0fiL
fromts'7.
Viral DNA fromts'7
wasdigested
tocompletion
withKpnI
and cloned intoKpnl-cleaved pUC18.
Clonescontaining
theKpnl
Vfrag-ment were identified
by
filterhybridization
(13)
with theKpnI
Vfragment
ofpKEF-P4
as theprobe.
TheRsal-BamHl
fragment
ofKpnl
V,
which contains thelarge
palindrome
oforiL,
wassubcloned intoBamHI-andHincll-cleaved
M13mp18.
The Rsal-BamHlfragment
cloned intoM13mp18
wasthesamesizeonacrylamide
gels (i.e.,
±5bp)
astheunclonedRsaI-BamHI
fragment
deriveddirectly
fromviral
DNA,
indicating
thatfurther deletionhadnotoccurredas aconsequence of
cloning
(data
notshown).
Sequence analysis
wascarried outusing
thechaintermi-nation method of
Sanger
et al.(30).
The Klenowfragment
reactions were conducted at 50'C to avoid artifacts
gener-ated
by secondary
structures.Theproducts
were run on8%polyacrylamide
gels containing
8 Mureaand 40% formam-ide.Invitrotestsof
origin
function. Theprocedure
usedtotestwhether
plasmid
DNAsequencescovalently
linkedto viralorigins
ofreplication
wereamplified
aftertransfection andinfectionwith
helper
viruswassimilar tothatdescribedby
other
investigators
(22, 39,
40,
45),
with onesignificant
modification. Since
oriL
suffers deletions whenpropagated
in bacteria
(12, 45),
gel-purified
viral DNAfragments
wereligated
tothetestplasmid,
and theligation
mixturewasuseddirectly
in the transfection ofCV-1 cells.The
gel-purified KpnI
Vfragment (map
units 0.406 to0.419)
of KOS DNA and ofts'7
DNA(80
ng)
wasligated
to420 ng of
Kpnl-cleaved pUC18,
and 1/20 of theligation
mixture was
electrophoresed
in a 0.6%gel
toverify
thatligation
had indeed occurred. One-half of theremaining
C*j C-i 0 to-i it
to 0 Wm M
p 0 E In (Alf qt
CM C*j it) 102 in
i
I I I 11'6
1 11 I I I 111-X X K T SP x XTK TB T SKEp
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.147.460.61.247.2]3530 POLVINO-BODNAR ET AL.
ligation
mixture or 110 ng of plasmid DNA (pUC18,ptkCAToris,
orpDX)wascoprecipitatedwith salmon spermDNA,
and thecoprecipitate
was added to each of tworeplicate
100-mmpetri dishescontaining2.5 x 106 CV-1cellsper dish. Four hours after
transfection,
cells were shockedwith15% glycerol for1min. At 10 hposttransfection, cells in
one of each
pair
of replicate dishes were infected with HSV-1(KOS),
at amultiplicity of10PFU percell. At 16 hpostinfection,
all cells wereharvested andwashed, and total cellular DNAwasextractedby usingsodiumdodecyl sulfate andproteinase
Kdigestion,
phenol and chloroformextrac-tion,
and ethanolprecipitation.
A 10-,ug portion of eachsample
was thendigested
with KpnI to excise unit-lengthpUC18
fromreplication
concatemersand withDpnI (whichdigests input, methylated plasmid
DNA but notnewly
rep-licated, unmethylated
DNA[20])
todistinguish newly
repli-cated DNA. Control
samples containing
135 to 270 ng ofplasmid
DNA were alsodigested
with KpnI alone or withKpnI
plus
DpnI.Allsamples
werethenelectrophoresedina0.8% agarose
gel
and transferred to a nitrocellulose filter,whichwas
probed
with32P-labeled pUC18, and anautoradi-ogramwas obtained.
Latency testing.
Groups
of 12 7-week-old CD-1 mice(Charles
RiverBreeding Laboratories,
Inc.,Kingston,
N.Y.)wereinoculated with2 x 106PFUin20
p.l
of eitherwild-typevirus or
ts+7
after corneal scarification. On day3postinoc-ulation,
eyeswabsweretaken from four miceandtrigeminalganglia
were removed from two mice for each virus (miceusedforeye swabs mayor may nothavebeen the same as
those sacrificed for
ganglion
assays). Eye swabs wereas-sayed
directly
for infectious virus in Verocells, andgangliawere
frozen,
thawed,sonicated,
clarifiedby low-speedcen-trifugation,
andassayed
in Vero cells. On day 30postinoculation, surviving
miceweresacrificed,
andgangliawere removed
immediately
(within 1 to 2minofdeath),cutinto
eight equal-sized pieces,
and cocultivated with Vero cells. Onday
5 ofcocultivation,
Vero cells andganglion
pieces
werescraped
intomedium, frozen,
thawed, andsonicated. The
suspension
was clarifiedby
low-speedcen-trifugation,
andthe supernatantfluidwas assayed forinfec-tious virusin Vero cell
monolayers.
RESULTS
Derivation of mutants ofHSV-1carrying deletions in OriL-associated sequences. (i) d61, a double mutant in ICP8 and
oriL.
Wehaveconstructedtwodeletionmutantsof HSV-1 in the geneencoding
themajor
DNA-binding protein, ICP8,andverifiedthatoneofthese, d61,hadincorporatedtheoriL
deletion,
aswellasthe ICP8deletionpresent in theplasmidused in itsconstruction (27).
Briefly,
a780-bp deletionwasintroduced into
ICP8-coding
sequences ofplasmidpSG18-Sall A to
yield pDX (Fig.
1, line 8). In addition to theengineered
deletion,pSG18-SaiI
A and consequently pDX also contain a spontaneous deletion in oriL-associated se-quences. Itwas assumedthat if oriL were essential for virusreplication, only
theICP8 deletion in pDX would beincor-porated
into the viral genome when ICP8-expressing cellswere cotransfected with pDX and infectious, wild-type
DNA. Mutant d61 was isolated from theresulting progeny
on thebasisof itscapacitytogrow inICP8-expressing Vero
cells
(U-47 [27])
but notinnormal Verocells. Examination ofthe DNAofd61revealed thatithadincorporated the 780-bp
engineered
deletionin theICP8gene (27). Unexpectedly, theKpnI
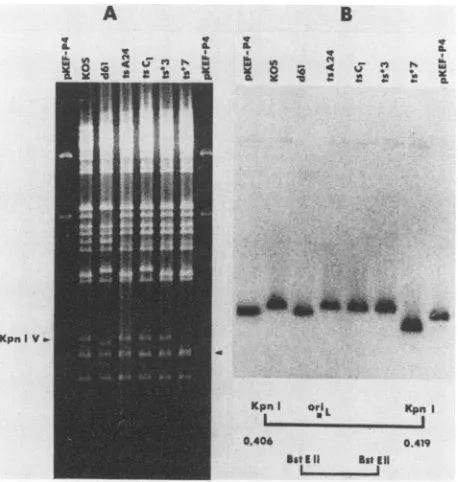
Vfragment
of d61 (Fig. 2, lane 2) exhibited greatermobility
than itsKOS counterpart did(lane 1). When KOS,~~~~IIIL.
0 o o w
X Z;
V
I-e04
832bp >
780bp 4 i
KpnI KpnI/ BamHi Xho I
KpnI or
_L
0.406 A
armHi
Sal I
[image:3.612.370.515.63.413.2]0.418
FIG. 2. Southern blot analysis of oriL-associated deletions in viral andplasmidDNAs.Viralorplasmid DNAs were digested with KpnIonly (leftmosttwolanes), KpnI and BamHI (next fourlanes),
orXhoI (rightmost lane), electrophoresed in an agarose gel, and transferredto anitrocellulose filter. Thefilter was then probed with theKpnI-SaIlfragment shownatthe bottomof the figure and inFig. 1, line5.On the leftmargin of thefigureareindicated thepositions of the oriL-containing KpnI V fragment and those of the KpnI-BamHI (0.406to0.411)fragments of KOS DNA (832 bp[28]) and of
d61, pKEF-P4, and pDX (780 bp). The size of the latter fragment
wasestimated bycomigration with the sequenced 780-bp (28)XhoI
fragmentof pKEF-P4, whichwasvisible in the rightmost lane of the ethidiumbromide-stainedgel but isnotdetected using thisprobe.
d61, pKEF-P4,andpDX DNAs were cleaved with KpnI and
BamHI and probed with the KpnI-SaII fragment shown at
the bottom of Fig. 2, the KpnI-BamHI fragments of d61,
pKEF-P4,andpDX exhibited greater mobility (780 bp)than
thecorresponding KOS fragment did (832 bp). These
obser-vations indicate that d61 had also incorporated the
sponta-neous oriL-associated deletion present in pDX and the
parental plasmid pSG18-SalI A. The efficient replicationof
d61 in ICP8-expressing cells suggested the possibility that
oriLmightnotberequiredfor viral replication in vitro. The
existence of multiple mutations in d61, however,
necessi-tated theisolation ofasecond mutant with a mutation inoriL only.
(ii) ts+7, a deletion mutant in OHL. A second mutant was
therefore constructedbymarker rescue of the ts mutation in theICP8mutant,tsA24,withplasmid pKEF-P4 (Fig. 1, line
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
B
Kp I Kp I
I a a
:.*X& ^
Kpn V
0.406
1ta11 lStall
J- )
FIG. 3. (A)KpnI restrictionpatternsofplasmid and viral DNAs. Theposition of the undeleted KpnIVfragment of KOS. tsA24. tsCl,
and ts+3 is shown onthe left. The arrow on the right side of the
figure indicates the position of the KpnI Vfragmentofts'7.ts.7 and ts+3werederivedfrom39°C plaques generated in the markerrescue
of tsA24withpKEF-P4. tsCt wasisolatedfrom 34°C plaques of the samemarkerrescue.(B) Southern blot analysis of the gel shown in
panelAprobed with the BstE IIfragment (shown in the diagramat
thebottom of thefigure) purified from pKEF-P4 and labeled with32P
bynicktranslation. Thediagram also illustrates the location of MriL within the KpnI V fragment relativetotheprobe.
6). pKEF-P4containsa55-bpdeletion intheoriL palindrome and an intact ICP8 gene (4, 45). On the basis of our
experience withd61, weanticipatedthatthe deletioninoriL
wouldbetransferredtothe viralgenomeconcomitantlywith therescueof the tsmutationby wild-type ICP8sequencesin
pKEF-P4. Of the 14
ts'
recombinants examined, one, ts47, wasfoundtocontainadeletion in theKpnIVfragment(Fig.3). Unlike the deletion in the parental plasmid pKEF-P4, however, the deletion in ts+7 was -150bp, rather than 55
bp.Two otherplaqueisolateswerepickedfrom the progeny
of the marker transfer experiment: ts*3, from 39°C plates,
and tsC1, from 34°C plates. The DNA restriction patterns
and Southern blots shown in Fig. 3 clearly reflect the
existence of three classesof theKptIVfragment: undeleted
(KOS, tsA24, tsC1, and
ts`3),
containing a -55-bp deletion(pKEF-P4 and d61), and containing a --150-bp deletion
(ts+7). Thus,tsC1 wasphenotypicallyandgenotypicallylike tsA24 (ts and
oriL'),
whereas ts+3 was phenotypically andgenotypically wildtype (ts+, oriL+)and ts+7 was
phenotyp-ically wild type
(ts')
and genotypically mutant (lackingoriL).
Replication efficiency of oriL deletion mutants. The two
independently derived oriL mutants, ts 7 and d61, were
capable of efficient replication in one-cycle growth
experi-ments in their respective permissive cells (Table 1). Thus,
burst sizes ofts+7 and d61 were similar to thoseof control
viruses KOS and d21 in thetwocelltypes.The differencesin
the burst sizes of d21 and d61 relative to KOS and ts+7 probablyreflect therequirementforcomplementation of the
former mutant by the resident wild-type ICP8 gene in
permissive U-47 cells. Consistent with the data shown in Table1,the plaquesizes of KOS and d21 were slightly larger than those of ts47 and d61 in Vero and U-47 cells, respec-tively.
Testsof originfunctionofisolated viral DNAfragments.To
determinewhether the deletion inoriL-associated sequences
of ts+7 had affected origin function, we carried out in vitro origin function tests similar to those describedby others (22, 39, 40. 45). The distinguishing feature of the procedure employed in this study wasthat uncloned, gel-purified viral DNA fragments were used directly after ligation to test plasmidsequences. thus avoiding the possibility of introduc-ing additional deletions durintroduc-ing cloning in bacteria. As ex-pected, pUC18 sequences were not amplified in CV-1 cells after mock infection (Fig. 4, lane 1) or superinfection with HSV-1 (lane 2). The requirement for HSV-1-associated factors supplied in trans for
o5is
and(riL function is seen in lanes 3 and4(ptkCAToris) and lanes 5 and 6 (pUC18 ligated to the wild-type o0iL-containing KpnI V fragment), respec-tively. By contrast, pUC18 sequences ligated to the KpnI V fragment of ts#7 were not amplified after superinfection with HSV-1 (lane 8), nor was pDX, the plasmid used in the construction of d61 (lane 10). These tests also confirmed previous reports (45) that the oriLdeletion present inpKEF-P4renders itincapable ofreplicating in experiments such as this (data not shown). These tests thus demonstrate that neither ts+7 nor pDX possesses an origin of viral DNA synthesis that can be driven by HSV-1 factors supplied in trans.
Sequencing of oriL deletions in d61 and ts+7. To further characterize the deletion in ts+7, we cloned the 309-bp RsaI-Ba,nHI fragment of the mutant in M13, sequenced it,
andcompared this sequence with that of thewild-type virus. The results of this analysis are shown in Fig. 5. Figure 5 presentsthe sequence of the 425-bp BstEII-BacmHIfragment of strain KOS (45), showingthe RsaI site at 114 to 118 bp, thelocation of the 144-bpinverted repeat, and the locations ofpertinent promoter regulatory sequences for the divergent
transcripts specifying the HSV major DNA-binding protein
and DNApolymerase (10, 15, 26, 41). The deletion in ts+7 is
148 bp in size and retains only one copy of the sequence CCAC found at positions 156 to 160 and 304to 308(Fig. 4). Thus, like other oriL-associated deletions described previ-ously, the ts 7 deletion appears to have occurred between shortrepeats (45). The deletion isasymmetric relative to the 144-bp inverted repeat, lacking 17 bp immediately to the right of the repeat and all but 13 bp of the 72-bp repeat
making up the left half of the palindrome. The deleted
sequences include the second distal signal upstream of the transcriptional start site of the major DNA-binding protein,
ICP8 (41), and may include as yet unidentified signals
upstream of the transcriptional start site and probably the
TABLE 1. Burst sizesof viruses containing intact ordeleted (IiL-associated sequences
Virus oril- ICP8 Burstsize"
sequences sequences (PFU/cell)
KOS Intact Intact 135
ts+7 Deleted Intact 96
d21 Intact Deleted 62
d61 Deleted Deleted 12
11Cells(106) wereinfectedat amultiplicityof 2.5 PFUJper cell(effective
multiplicity, IPFUpercell). washed, incubatedat37°Cfor 18h, harvested,
andassayed forinfectious virus. ICP8-deficient d21 and d61weretested in
ICP8-expressingU-47cells,andKOS andts-7weretested in Vero cells.
A
a.
a
a.%i*
hon November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.64.293.64.305.2]3532 POLVINO-BODNAR ET AL.
"ATA" sequence of the DNApolymerasegene at 350 to 354
bp (10).
On the other hand, it is unlikely that the deletion includes any elements critical for transcription of eitheressentialgeneasthemutantreplicates nearlyaswellasdoes
wild-type
virus(Table
1).Latency tests.
Having
established that ts+7 is replicationcompetent in cell
culture,
wenexttested therequirementfor functional oriL in the establishment of latency. For this purpose, groups of 7-week-old CD-1 mice were inoculatedwith 2 x 106 PFU per eye of either wild-type virus orts+7
aftercorneal scarification. Eye swabs andtrigeminalganglia
were
assayed
directly
on Vero cells for infectious virusduring
acute infection(day
3), and 12 ganglia from six survivingmiceper virusweretestedonday30bycocultiva-tion for reactivacocultiva-tion of latent virus. The results of thesetests
demonstratethatts+7 behavedinvivo inamannersimilar to
wild-type
virus (Table 2). Thus ts+7was nearlyaslethal forCD-1 miceaswas
wild-type
virus. Moreover, itreplicated
atthe site
of
inoculation and reachedtrigeminal ganglia,
asdemonstrated
by
the presence of virus in eye swabs andganglia
onday
3, andwasreactivated fromlatent infection asefficiently
aswaswild-type
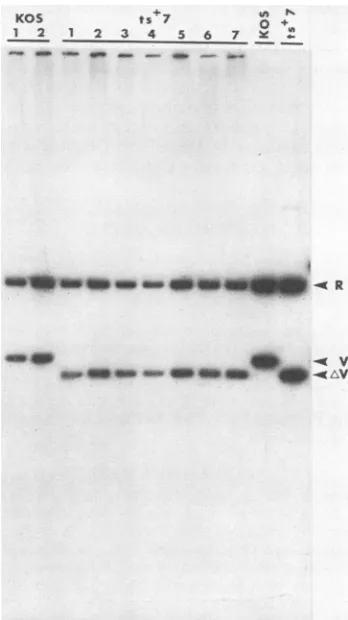
virus. The DNAs of 7 of 12 virusisolates reactivated from latent infection were tested by
Southern blot
analysis
for the presence of the deletioncharacteristic ofts+7
(Fig.
6).Compared
with twoisolatesderived
from mice inoculated withwild-type virus,
allsevenisolates
derived fromts+7-infected
mice exhibited the148-bpdeletion.We concludefromthesestudiesthatfunctionaloriL
is notrequiredfor the establishment orreactivationoflatent infection of mice.
DISCUSSION
The data
presented
herein demonstrate that oriL is notrequired
forgrowth
ofHSV
in vitroorfortheestablishmentand reactivation of latent infection inthe murineeye model.
These conclusions are based on observations made with
twodeletion mutantswhich
lack
afunctionaloriL.
The firstof the two mutants
isolated, d61,
contains mutations inICP8-coding
sequences,inoriL-associatedsequences, and inseveraladditional sites
(27).
Although unsuitable forbiolog-ical and molecular characterization because of its multiple
mutations,
thereplication
competence of d61 inICP8-expressing
cellsprovided
the initial clue that HSV-1 canreplicate
inthe absence ofafunctionaloriL. Construction ofthe second oriL deletion mutant, ts+7, provided the
neces-sarytooltoexaminethereplicationandlatencycompetence
of HSV-1
lacking
afunctionaloriL.
DNAsequenceanalysisandfunctionalcharacterization of
ts+7
revealed that the148-bp deletion (i)eliminates all but 11bp
on theright
end of the 144-bp oriL palindrome, (ii)occurred between
direct
repeats, (iii) renders ts+7 oriLunresponsive
to HSV-1 factors supplied in trans, (iv) has little effect on the growth properties of the mutant in vitro, and(v)
has no detectable effect on the ability of ts+7 toestablish, maintain,
or bereactivated from latent infection. It is notable that during the construction of ts+7, the148-bp
deletioninoriL-associated sequences was introducedby using
aplasmid,
pKEF-P4,thatcontains a 55-bp deletion(45), demonstrating
that additional sequences can be lost inmammaliancellsduringmarker transfer of such mutations to
the viralgenome.Of interestis theobservation that the large
deletion occurred between short direct repeats, as
previ-ously reported
for deletions generatedduring the cloning oforiL
inbacteria
(45). Whether the mechanism underlyingdeletion of sequences between shortrepeats applies equally
1 2 3 4 5 6 7 8 9 10 11 12 13 14
,T4-4 ~~ .,
. 4
4.0 li,~~~~~~~~~~~~~
0
.*pDXiptkCAToris
-pUC18
am
9o
I
a
4.*
MVY M V M V M V MVY
pUC18 ptkCAT- KOS ts' 7 pDX
oris
FIG. 4. Origin function test. Total cellular DNAs from CV-1
cells,transfectedwith the indicated DNAs(lanes 1 to10) andeither
mock infected(M)orinfected with KOS(V)orplasmidDNAs(lanes
11to14),weredigestedwithKpnIonly (lane 11)orwithKpnI plus
DpnI (all other lanes), electrophoresed in an agarose gel, and transferredtoanitrocellulosefilter.Thefilterwasthenprobedwith
32P-labeled pUC18. Lane 11containsamixture ofpDX, ptkCAT-oris, and pUC18. Lane 12contains pUC18 only, lane 13 contains
ptkCAToris only,andlane 14 shows pDXonly.
in bacterial and mammalian cells is unknown. Possible
mechanisms
leading
to deletions between short repeatsduring cloning
inbacteria have been discussedby Weller etal. (45).
Like all other deletedoriL clonessequencedto date (45) of
which the deletions lieat least partially withinthe inverted
repeat, that of ts+7 eliminated the entire palindrome.
Be-causenoneof these clones exhibitsorigin functionin assays
in
vitro,
thepalindrome
must contain elements requiredfororigin
function. To date, detailed analysis of the cis-acting elements necessary for origin function has not beenre-ported,
and hence the specific role of the palindrome inorigin
function isnot known. Distinct homologies between thehighly
conserved (22) HSV-1 and HSV-2 oriL andoris
palindromes
and papovavirus and adenovirus origins of DNAreplication
have been reported (18). Moreover, Elias et al.(personal
communication) have reported that selectedpalindromic
sequences of HSV-1 and HSV-2 oriL andoris
likely
constitute thebindingsiteforaviral protein present ininfected cells.
Whatever theirrole in the initiationof viral DNA
replica-tion,
oriLsequenceshave also been postulated to play a roleJ. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.327.557.62.389.2]20 30 40 50 60 70 GCCGATGAAC CCCGGCGGCT GGCAACGCGG GGTCCCTGCG AGAGGCACAG ATGCTTACGG CGGCTACTTG GGGCCGCCGA CCGTTGCGCC CCAGGGACGC TCTCCGTGTC TACGAATGCC
DBPt
80 gp
TCAGGTGCTC CGGCCGGGT
AGTCCACGAG GCCCGGCCCA
1;0 Rsa1120 130 140 150 160 170 180 190 200
TGCGGTTGGT ATATGTACAC TTTACCTGGG GGCGTGCCGG ACCGCCCCAG CCCCTCfCAC ACCCCGCGCG TCATCA0CCG GTGG0CGTGG CCGCTATTAT ACGCCAACCA TATACATGTG AAATGGACCC CCGCACGGCC TGGCGGGGTC CGGGA GGTG TGGGCGCCGC AGTAGTCGGC CACCCGCACC GGCGATAATA
ts 7
210 220 230 240 250 260 270 280 290 300
AAAAAAAGTG AGAACGCGAA GCGTTCACTfT99 CCTAA TAATATATAT ATTATTAGGA CAAAGTGCGA ACGCTTCGCG TTCT CATTT TTTTATAATA
TTTTTTTCAC TCTTGCGCTT CGCAA CTCAAA CAGGATT ATTATATATA TAATAATCCT GTTTCACGCT TGCGAAGCGC AAGA rGAAA AAAATATTAT
pKEF-P4 TpoI I pKEF-P4
10 320 330 3140 350 360~ 370 1--10039040
GCGCAC CCACCGGCTA CGTCACGCTC CTGTCGGCCG CCGGCGGTCC ATAAGCCCGG CCGGCCGGGC CGACGCGAAT AAACCGGGCC GCCGGCCGGG CGCC G GGTGGCCGAT GCAGTGCGAG GACAGCCGGC GGCCGCCAGG TATTCGGGCC GGCCGGCCCG GCTGCGCTTA TTTGGCCCGG CGGCCGGCCC
ts*7
4410 420
GCGCCGCGCA GCAGCTCGCC GCCCGG
CGCGGCCCGT CGTCGACCGG CGGGCC
FIG. 5. Sequence of the 425-bpBstEII-BamHIfragmentcontainingoriL.The basesin bold print (positions 176to319)correspondtothe
144-bp invertedrepeat(45). Thesequencesin boxesarethe short 4-to6-bprepeatsbetween which deletions ints+7andpKEF-P4occurred,
respectively.The location of theRsaIsite (position114to118)usedtoclonethe RsaI-BamHIfragment ofts+7intoM13mpl8 for sequencing
purposesis alsoshown. The solid lines beneath bases 106to121, 137to152, and176to190representtheproximal, first, andsecond distal
signals, respectively, of the ICP8promoter(41).The location of the ATA box in the ICP8promoteris indicated by the dotted line (108to114).
Thetranscriptionalstartsiteof the ICP8geneis shownatposition 90. The probable ATA box and transcriptionalstartsitesofthepolymerase
gene areshownatpositions350to354 and 370 and 379, respectively (10).
in the transcriptional regulation of the two essential genes flanking oriL: those encoding the major DNA-binding
pro-tein, ICP8, and DNA polymerase (15). The second distal
signal of ICP8(41)(and possibly regulatory elements of the
polymerase gene [10]) is absent in ts+7, yet the virus
replicates nearly as efficiently as wild-type virus. As the
deletion in ts+7 is among the larger of the HSV-1 oriL
deletions tobe sequencedand thefirsttobeintroduced into
theviralgenomewith littleeffect on replication competence,
the role of palindromic sequences per se in regulation of
ICP8 andpol transcription during productive infection
re-mains unclear. The measurement ofactual levels of
tran-scription of these two genes in ts+7-infected cells is in
progress. Although site-directed mutagenesis will be
re-TABLE 2. Resultsoflatency testsin CD-1 mice
Virus titer inspecimens
Virus Lethalitya Eye Latent
ViruLethalit
swabs"
Gangliac infectiondWildtype 9/26 (35) 1.2 x 102 1.5 x 104 12/12
3.7 x 102 2.2 x 104
4.2x 102 4.7 x 104
1.8 x 103 5.4 x 104
tsI7 3/10(30) 1.0 x 101 2.3 x 104 10/12
3.1 X 102 2.9 x 104
7.1 x 102 5.8 x 104
1.1X 1013 6.2 x104
aLethalityispresentedastheratio of the number of dead animalstothe
number of animals inoculatedwith2x 106PFUper eye.
bEye swabsweretaken from fourmiceonday3postinoculation. Swabs from both eyes of eachmouse werepooledandassayedonVerocells.Titers aretotalPFU ineye swabsuspensions.
cTwo miceweresacrificedonday3postinoculation,andindividualganglia were homogenized and plated directly on Verocells. Titers arePFU per ganglion.
dOn day 30 postinoculation, ganglia were removed from six mice (12 ganglia) and assayed for reactivatable virusbycocultivationwith Verocells.
Noinfectious viruswasdetectedin sixtrigeminalgangliaofmice inoculated withwild-typevirus, removedonday 30,homogenized,andplateddirectly.
KOS ts 7
1 2 12 3 4 5 6
.p -
-In N 0 +
7 X
4e-Om __m am_ _ _ _ R
w v
FIG. 6. Southern blot of DNAs of virus isolatesobtained from
trigeminal gangliaofmiceinoculated withwild-type virusorts+7.
Two isolates from KOS-infected mice were compared with seven
isolates from ts+7-infected mice. Total DNAs from cells infected with each isolatewerecleaved withBamHI, electrophoresedon an agarosegel, and transferred toa nitrocellulosefilter, andthe filter wasprobedwith theKpnIV fragmentwhich hybridizestoboththe BamHIR and Vfragments.ControlKOS andts+7DNAsareshown
in theright-handtwolanes. 10
ACCACGGGGT TCCTGCCCCA
100 GCGTCTGATA CGCAGACTAT
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.612.58.553.67.242.2] [image:6.612.345.519.322.632.2] [image:6.612.55.295.496.620.2]3534 POLVINO-BODNAR ET AL.
quiired
to elucidate thespecific
roles oforiL
sequences inorigin function and transcriptional regulation, theavailability
ofanisogenicseries of sequenced deletion mutationsinoriL should prove useful, with the unfortunate caveat that their
introduction into the viral genome by marker transfer may
result in further sequence loss.
The data
presented
herein demonstrate thatoriL
is notrequired for virus replication in vitro, implying that two
copies of
oris
are sufficient for this purpose. In a recentreport, Longnecker and Roizman reported the viability of a
mutant of HSV-1 strain F, R7023, that contains oriL and only one copy of
oris
(23). The viability ofthis mutantdemon-stratesthat the two remaining origins (one copy of
oris
andoriL) are sufficient for virus replication. The observations
that viable
mut4nts
of HSV-1 may lackeither oriL or one oftwo copies of
oris
raise interesting questions regarding therole(s)of the two types of origin in thebiology of HSV-1. (i)
Iseither type of origin preferentially used during the lytic or
latent modes of infection, and (ii) why have both been
conserved in the HSV-1 and HSV-2 genomes?
With regard tothe former question, we have demonstrated
the replication and latency competence of
ts+7
invivo,
implyingthat anintact oriL isnot requiredfor the
establish-ment, maintenance, or reactivation of latency. The only
other virus for which a latency-specific origin has been
suggested is Epstein-Barr virus, in which oriP has been
shown to function during latent infection (42, 47). In this
system, however, no origins other than oriP have yet been
identified (although they may exist), and it has not been
shown thatoriP does not also function during lytic infection.
Some insightinto the secondquestion mightbegained by
comparisonofHSVwithotherherpesviruses.Inthis regard,
it is notablethat the genome of varicella-zoster virus, a virus
which shares extensive biological and DNA structural
simi-larities with HSV, contains a diploid
oris
but lacks an oriLequivalent (39).
ACKNOWLEDGMENTS
WethankN.DeLuca forvaluable discussions, M. Bush, D. Coen, J. Jacobson, D. Knipe, K. Tyler, E. Villareal, and D. Yager for assistance in latency tests of ts+7, and M. Cook for manuscript preparation.
This investigation was supported by Public Health Service
Pro-gramProject grant no. CA21082 from the National Cancer Institute and grant no. A124010 from the National Institute of Allergy and Infectious Diseases. M.P.-B. is supported by a Special Fellowship fromthe Leukemia SocietyofAmerica. P.K.O. was supported by Postdoctoral Fellowship no. DRG-840 from the Damon Runyon-WalterWinchell Cancer Fund.
LITERATURE CITED
1. Ben-Porat,T.1982.Organization and replication of herpesvirus DNA, p. 147-172. In A. S. Kaplan (ed.), Organization and replicationofviral DNA. CRC Press, Inc., Boca Raton, Fla.
2. Cuifo, D.M., and G. S. Hayward. 1981. Tandem repeat
defec-tive DNA fromthe L segment of the HSV genome, p. 107-128.
In Y. Becker (ed.), Herpesvirus DNA. Martinus Nijhoff, The Hague, The Netherlands.
3. Deiss, L. P., and N. Frenkel. 1986. Herpes simplex virus amplicon: cleavage of concatemeric DNA is linked to packaging
and involves amplification of the terminally reiterated a se-quence.J. Virol. 57:933-941.
4. DeLuca, N., D. Bzik, V. C. Bond, S. Person, and W. Snipes. 1982. Nucleotide sequences of herpes simplex virus type 1 (HSV-1) affecting virus entry, cell fusion, and production of glycoprotein gB (VP7). Virology 122:411-423.
5. DeLuca, N. A., M. A. Courtney, and P. A. Schaffer. 1984.
Temperature-sensitive mutants in herpes simplex virus type 1
ICP4 permissive for early gene expression. J. Virol. 52:767-776. 6. DeLuca, N. A., and P. A. Schaffer. 1985. Activation of immedi-ate-early, early, and late promoters by temperature-sensitive
and wild-type forms of herpes simplex virus type 1 protein
ICP4. Mol. Cell. Biol. 5:1997-2008.
7. Frenkel, N.1981.Defectiveinterferingherpesviruses,p. 91-120. In A. J. Nahmias, W. R. Dowdle, and R. F. Schinazi(ed.), The human herpesviruses. Elsevier, New York.
8. Frenkel, N.,
H.
Locker, and D. A.Vlazny.
1980. Studies of defective herpes simplex viruses. Ann. N.Y. Acad. Sci. 354:347-370.9. Friedmann, A., J. Shlomai, and Y. Becker. 1977. Electron microscopy of herpes simplex virus DNA molecules isolated from infected cells by centrifugation in CsCl density gradients.
J. Gen. Virol. 34:507-522.
10. Gibbs, J. S., H. C. Chiou, J. D. Hall, D. W. Mount, M. J.
Retondo, S. K. Weller, and D. M. Coen. 1985. Sequence and mapping
analyses
of the herpes simplex virus DNA polymerase gene predict a C-terminal substrate binding domain. Proc. Natl. Acad. Sci. USA 82:7969-7973.11. Goldin, A. L., R. M. Sandri-Goldin, M. Levine, and J. C.
Glorioso. 1981. Cloning of herpes simplex virus type 1
se-quences representing the whole genome. J. Virol. 38:50-58. 12. Gray, C. P., and H. C.Kaerner. 1984. Sequence of the putative
origin of replication in the UL region of herpes simplex virus type 1 ANG DNA. J. Gen. Virol. 65:2109-2119.
13. Grunstein, M., and D. Hogness. 1975. Colony hybridization: a method for the isolation of cloned DNAs that contain a specific
gene. Proc. Natl. Acad. Sci. USA 72:3961-3965.
14. Hirsch, I., G. Cabral, M. Patterson, and N. Biswal. 1977. Studies
on the intracellular replicating DNA of herpes simplex virus type 1. Virology 81:48-61.
15. Holland, L. E., R. M. Sandri-Goldin, A. L. Goldin, J. C. Glorioso, and M. Levine. 1984. Transcriptional and genetic analyses of the herpes simplex virus type 1 genome: coordinates 0.29 to 0.45. J. Virol. 49:947-959.
16. Kaerner, H. C., I. B.Maichle,A. Ott, and C. H.
Schroder.
1979. Origin of two different classes of defective HSV-1 Angelotti DNA. Nucleic Acids Res. 6:1467-1478.17. Kaerner, H. C., A. Ott-Hartmann, R.Schatten, C. H.
Schroder,
and C. P. Gray. 1981. Amplification of a short nucleotide sequence in the repeat units of defective herpes simplex virus
type 1 Angelotti DNA. J. Virol. 39:75-81.
18. Knopf, C. W., B. Spies, and H. C. Kaerner. 1986. The DNA replication origins of herpes simplex virus type 1 strain Angelotti. Nucleic Acids Res. 14:8655-8667.
19. Knopf, C. W., G. Strauss, A. Ott-Harman, R. Schatten, and H. C. Kaerner. 1983. Herpes simplex virus defective genomes: structure of HSV-1 ANG defective DNA of classII and encoded polypeptides. J. Gen. Virol. 64:2455-2470.
20. Lacks, S., and B. Greenberg. 1977. Complementary specificity of restriction endonucleases of Diplococcus pneumoniae with respect to DNA methylation. J. Mol. Biol. 114:153-168. 21. Lee, C. K., and D. M. Knipe. 1983. Thermolabile in vivo
DNA-binding activity associated with a protein encoded by mutants of herpes simplex virus type 1. J. Virol. 46:909-919.
22. Lockshon, D., and D. A. Galloway. 1986. Cloning and
charac-terization oforiL2,a large palindromic DNA replication origin of herpes simplex virus type 2. J. Virol. 58:513-521.
23. Longnecker, R., and B. Roizman. 1986. Generation of an
invert-ing herpes simplex virus 1 mutant lackinvert-ing the L-S junction a sequences, an origin of DNA synthesis, and several genes including those specifying glycoprotein E and the
a47
gene. J. Virol. 58:583-591.24. Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular
cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
25. Mocarski, E. S., and B. Roizman. 1982. Herpesvirus-dependent amplification and inversion of cell-associated viral thymidine
kinase gene flanked by viral a sequences and linked to an origin of viral DNA replication. Proc. Natl. Acad. Sci. USA
79:5626-5630.
26. Morse, L. S., L. Pereira, B. Roizman, and P. A. Schaffer. 1978. J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
Anatomyof herpes simplex virus (HSV) DNA. X. Mapping of viral genes by analysis of polypeptides and functions specified by HSV-1 x HSV-2 recombinants. J. Virol. 26:389-410. 27. Orberg, P. K., and P. A. Schaffer. 1987. Expression of herpes
simplex virus type 1 major DNA-binding protein, ICP8, in transformed cell lines: complementation of deletion mutants and inhibition ofwild-type virus. J. Virol. 61:1136-1146.
28. Quinn, J. P., and D. J. McGeoch. 1985. DNA sequence of the region in the genome of herpes simplex virus type 1 containing the genes for DNA polymerase and DNA binding protein. Nucleic Acids Res. 13:8143-8163.
29. Sacks, W. R., C. C.Greene, D. P. Aschman,and P. A. Schaffer.
1985. Herpes simplex virus type1 ICP27 isanessential regula-toryprotein. J. Virol. 55:796-805.
30. Sanger, F., S. Nicklen, and A. Coulsen. 1977. DNAsequencing with chainterminating inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
31. Schaffer, P. A., V. C. Carter, and M. C. Timbury. 1978.
Collaborativecomplementation study of temperature-sensitive
mutants of herpes simplex virus types 1 and 2. J. Virol. 27:490-504.
32. Smith, H. 0. 1980. Recovery of DNA from gels. Methods Enzymol. 65:371-380.
33. Southern, E. M. 1975. Detection ofspecific sequences among
DNAfragments separated by gel electrophoresis. J. Mol. Biol.
98:503-517.
34. Southern, P.J.,and P.Berg. 1982.Transformation of
mamma-lian cells to antibiotic resistance with a bacterial gene under controlof the SV40 early region promoter. J. Mol. Appl. Genet. 1:327-341.
35. Spaete, R.R.,and N. Frenkel. 1985.The herpessimplex virus amplicon: analyses of cis-acting replication functions. Proc.
Natl. Acad. Sci. USA 82:694-698.
36. Spear,P.G.,and B. Roizman.1980.Herpessimplexviruses, p. 615-745. In J. Tooze (ed.), DNA tumor viruses. Cold Spring HarborLaboratory, ColdSpring Harbor,N.Y.
37. Stow,N. D. 1982. Localization ofanorigin ofDNAreplication
within theTRsIIRs repeatedregion of the herpes simplex virus type 1 genome. EMBO J. 1:863-867.
38. Stow, N. D. 1985. Mutagenesis of a herpes simplex virus origin of DNA replication and its effect on viral interference. J. Gen. Virol. 66:31-42.
39. Stow,N.D., andA. Davison. 1986. Identification ofa varicella-zoster virus origin of DNA replication and its activation by herpes simplex virus type 1 gene products. J. Gen. Virol.
67:1613-1623.
40. Stow, N., and E. C. McMonagle. 1983. Characterization ofthe
TRsIIRsorigin of DNA replication of herpes simplex virus type
1. Virology 130:427-438.
41. Su, L., and D. M.Knipe. 1987. Mapping of thetranscriptional initiation site of the herpes simplex virus type 1 ICP8 gene in infected and transfected cells. J. Virol. 61:615-620.
42. Sugden, B., K. Marsh, and J. Yates. 1985. A vector that
replicates as a plasmid and can be efficiently selected in
B-lymphoblasts transformed by Epstein-Barr virus. Mol. Cell. Biol. 5:410-413.
43. Vlazny, D. A., and N. Frenkel. 1981. Replication of herpes simplex virus DNA: localization of replication signals within defective virus genomes. Proc. Natl.Acad. Sci.USA 78:742-746.
44. Weller, S. K., K. J. Lee, D. J. Sabourin, and P. A. Schaffer.
1983.Genetic analysis oftemperature-sensitive mutantswhich define the gene for the major herpes simplex virus type 1
DNA-bindingprotein. J.Virol.45:354-366.
45. Weller,S.K.,A.Spadaro, J.E.Schaffer,A. W.Murray,A. M.
Maxam, and P. A. Schaffer. 1985. Cloning, sequencing, and functionalanalysis of oriL,aherpessimplex virus type1origin ofDNAsynthesis. Mol. Cell. Biol. 5:930-942.
46. Yanisch-Perron, C., J. Vierira,andJ. Messing. 1985.Improved
M13 phage cloning vectors and host strains: nucleotide
se-quences ofthe M13mpl8 and pUC19 vectors. Gene
33:103-119.
47. Yates, J. L.,N.Warren,and B.Sugden.1985.Stablereplication ofplasmids derived fromEpstein-Barr virus in various
mamma-liancells. Nature(London) 313:812-815.