0095-1137/06/$08.00⫹0 doi:10.1128/JCM.02039-05
Identification and Characterization of Variable-Number
Tandem-Repeat Markers for Typing of
Brucella
spp.†
Adrian M. Whatmore,* Stephen J. Shankster, Lorraine L. Perrett, Terry J. Murphy,
Simon D. Brew, Rachel E. Thirlwall, Sally J. Cutler, and Alastair P. MacMillan
Department of Statutory and Exotic Bacterial Diseases, Veterinary Laboratories Agency, Addlestone, Surrey KT15 3NB, United Kingdom
Received 29 September 2005/Returned for modification 2 December 2005/Accepted 10 March 2006
Members of the genus Brucella infect many domesticated and wild animals and cause serious zoonotic infection in humans. The availability of discriminatory molecular typing tools to inform and assist conven-tional epidemiological approaches would be invaluable in controlling these infections, but efforts have been hampered by the genetic homogeneity of the genus. We report here on a molecular subtyping system based on 21 variable-number tandem-repeat (VNTR) loci consisting of 13 previously unreported loci and 8 loci previ-ously reported elsewhere. This approach was applied to a collection of 121Brucellaisolates obtained worldwide and representing all six classically recognizedBrucellaspecies. The size of repeats selected for inclusion varied from 5 to 40 bp giving VNTR loci with a range of diversities. The number of alleles detected ranged from 2 to 21, and Simpson’s diversity index values ranged from 0.31 to 0.92. This assay divides the 121 isolates into 119 genotypes, and clustering analysis results in groups that, with minor exceptions, correspond to conventional species designations. Reflecting this, the use of six loci in isolation was shown to be sufficient to determine species designation. On the basis of the more variable loci, the assay could also discriminate isolates origi-nating from restricted geographical sources, indicating its potential as an epidemiological tool. Stability studies carried out in vivo and in vitro showed that VNTR profiles were sufficiently stable such that recovered strains could readily be identified as the input strain. The method described here shows great potential for further development and application to both epidemiological tracing ofBrucellatransmissions and in deter-mining relationships between isolates worldwide.
Brucellosis is a zoonotic disease of major public health, animal welfare, and economic significance worldwide. In
hu-mans, infection withBrucellacan lead to a chronic debilitating
infection; in domesticated animals, the main symptom is re-productive failure. Disease in humans usually reflects occupa-tional exposure or the consumption of unpasteurized dairy products. Brucellosis remains a major problem in many parts of the world, particularly Mediterranean regions, western Asia, and parts of Africa and Latin America (11), although in many developed countries it has been eradicated or severely cur-tailed by a combination of strict veterinary hygiene measures, monitoring programs, and improved food safety measures.
Brucellaspecies have also long been considered potential
bio-logical warfare agents, and in 1954Brucellabecame the first
biological agent to be treated as a weapon and field tested on animals under the old U.S. offensive biological weapons pro-gram. Recent history has raised awareness in this area (28), and the organism remains on the list of Centers for Disease Control and Prevention category B potential biological warfare agents (25).
ClassicalBrucellataxonomists developed a classification
sys-tem that recognized six species based on subtle phenotypic and
antigenic differences and host specificity:B. abortus(bovine),
B. melitensis(caprine and ovine),B. ovis(ovine),B. canis
(ca-nine),B. suis (porcine), and B. neotomae(desert wood rat).
Some of these species are classically divided into biovars. The development of discriminatory molecular tools for
identifica-tion and typing ofBrucellahas been problematic, reflecting the
lack of genetic polymorphism in Brucella. A high degree of
homology initially implied by DNA-DNA hybridization (30) has been confirmed by a variety of approaches, such as multi-locus enzyme electrophoresis (MLEE) (13) and 16S rRNA sequencing. These sequences were long considered highly con-served, and a recent systematic study has confirmed 100%
identity of the 16S rRNA sequences between all of theBrucella
spp. (14). While the development of genus-specific and, in some cases, species-specific PCR assays for identification has been possible (3, 4), typing tools of sufficient resolution to permit epidemiological tracing of outbreaks or which might be required to try to identify the source of a bioterror attack are still lacking.
In recent years the availability of microbial genome se-quences has revolutionized DNA fingerprinting by facilitat-ing the development of multilocus sequence-based typfacilitat-ing ap-proaches such as multilocus sequence typing (MLST) and multilocus VNTR analysis (MLVA). The genetic homogeneity ofBrucellaspp. implies that there will be insufficient sequence polymorphism in housekeeping genes to provide the resolution required to use MLST as a tool for epidemiological traceback. However, MLVA targets tandemly repeated DNA regions that are considered high-speed molecular clocks (27). The addition or deletion of repeat units reflecting either slipped strand nu-cleotide mispairing during replication or unequal crossover
* Corresponding author. Mailing address: Department of Statutory and Exotic Bacterial Diseases, Veterinary Laboratories Agency, Addlestone, Surrey KT15 3NB, United Kingdom. Phone: 44 1932 357311. Fax: 44 1932 357423. E-mail: [email protected].
† Supplemental material for this article may be found at http://jcm .asm.org/.
1982
on May 16, 2020 by guest
http://jcm.asm.org/
events results in a high rate of mutation at these loci. As a result, this approach has proven particularly useful as a tool for strain discrimination in bacterial species with little genomic
variation, notablyBacillus anthracis(17), Yersinia pestis (17),
Francisella tularensis (16), and various mycobacterial species
such asMycobacterium tuberculosis(22),Mycobacterium leprae
(15), andMycobacterium aviumsubsp.paratuberculosis(20). As
well as the potential for high resolution, MLVA has a number of other technical advantages, most notably its relative simplic-ity, fast turnaround time, amenability to high-throughput ap-proaches, applicability to nonviable and/or crude preparations, and the ability to easily compare digital results between labo-ratories.
None of the existing molecular tools provide adequate res-olution to confidently permit epidemiological traceback in the case of accidental import or deliberate release. However, the
completion of genome sequences for aBrucella suis(21) and a
Brucella melitensis(12) strain provided an opportunity to assess the presence of tandem repeats that might facilitate the devel-opment of an MLVA scheme. Initial analysis indicated the presence of many potentially useful regions of diversity in the
Brucellagenomes and, indeed, during the planning stages of the present study, an MLVA scheme that utilizes eight distinct copies of an octameric repeat (the “HOOF-Prints” assay) was described (5). Since our initial aim was to develop a high-resolution typing system, we have expanded on this existing scheme by assessing the performance of a number of addi-tional highly polymorphic markers, in conjunction with those previously described, in a diverse range of isolates representing
all currently recognizedBrucellaspecies. The identification of
these additional markers increases the potential discriminatory capacity of the existing HOOF-Prints assay and reduces the risk of unrelated isolates having identical profiles due to ho-moplasy. Furthermore, during the course of this work, it be-came clear that, although these highly discriminatory markers should facilitate a degree of epidemiological traceback impos-sible in the past, the rapid mutation rate at these loci could make it difficult to assign isolates to one of the classically recognized species. We therefore assessed a number of addi-tional tandem repeat loci with lower mutation rates for inclu-sion in a scheme that permits rapid speciation on the basis of apparently species-specific alleles or species-specific allelic profiles at these loci. We have thus examined VNTR markers
with a range of diversity levels in 121 Brucella isolates and
present an MLVA scheme that offers the potential of great discriminatory power and yet retains the capacity for isolate resolution at the taxonomic level.
MATERIALS AND METHODS
Isolates and template preparation.The details for all strains used in the present study are included in Table 1. The template for the VNTR PCR was either genomic DNA prepared as described previously (29) or a crude methanol extract prepared by harvesting growth into 66% (vol/vol) methanol. Where the biovar was determined in our laboratory, this was carried out according to standard procedures (1), although in some cases the biovar designations are those given by the strain providers.
Identification of tandem repeats.Tandem repeats were identified by using the genome sequences ofB. suis(21) andB. melitensis(12) and the Tandem Repeat Finder software (2). This program identified a large number of tandem repeats in addition to those described by Bricker et al. (5) and incorporated into the HOOF-Prints assay. A selection of these were assessed for their potential value in a typing scheme by designing PCR primers flanking the tandem repeats and
assessing the size diversity of resulting PCR products by agarose gel electro-phoresis across a panel ofBrucellaisolates. Thirteen novel repetitive loci were chosen for use in the assay described here to give, when used in conjunction with the existing eight HOOF-Prints loci, an assay examining variability at twenty-one loci.
Sequence verification.In order to verify the basis of repeat copy number variation, PCR products representing at least three alternative alleles (or both alleles in the case of loci with only two allelic states) were sequenced for all 13 novel VNTR markers. PCR products were purified by passage through QiaQuick PCR purification columns (QIAGEN) and sequenced by using forward and reverse primers and an ABI BigDye terminator cycle sequencing kit (Applied Biosystems). Amplicon sizes were used in conjunction with sequence data to predict repeat copy number for each locus and isolate.
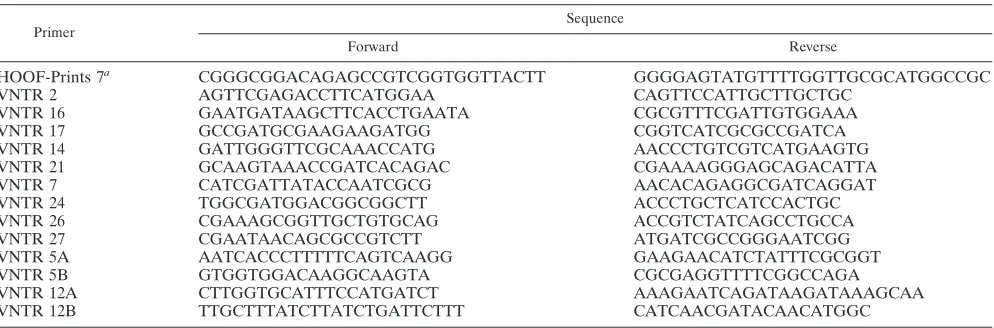
VNTR analysis.The primer pairs used for the amplification of all loci de-scribed for the first time here are shown in Table 2. Primers used for amplifica-tion of the HOOF-Prints loci were as described previously (5), with the excepamplifica-tion of the HOOF-Prints 7 primers that were redesigned since we found the originally reported primer set difficult to work with. PCR amplification was performed by using Fast StartTaqDNA polymerase (Roche). Each reaction mixture consisted of 2.5l of Fast Start 10⫻PCR buffer with MgCl2(20 mM), 3.125l of 2 mM deoxynucleoside triphosphates, 0.05l of 100M forward and reverse primer, and 0.125l of Fast StartTaqpolymerase in a final volume of 24.75l of water. As the PCR template, either 0.25l of purified genomic DNA or a crude methanol extract was used. Primer pairs consisted of one primer fluorescently labeled with NED, FAM, or HEX and one unlabeled primer. Initially, amplifi-cation was carried out one locus at a time but eventually amplifiamplifi-cation was multiplexed into sets of three loci using the three different dyes and reaction volumes were halved compared to the above. The PCR conditions were as follows: an initial step of 95°C for 5 min; followed by 95°C for 30 s, 55°C for 30 s, and 75°C for 1 min repeated for 30 cycles; followed by a 59-min incubation at 75°C. After PCR, the amplification products were routinely diluted 1:20 in water, and 1l was separated by capillary electrophoresis on an ABI377 genetic ana-lyzer. All samples were run with GeneScan 500 (ROX) size markers as an internal standard, and the bands were sized relative to these markers by using the GeneScan software. Sizes of the HOOF-Prints loci PCR products were consis-tent with those described by Bricker et al. (5) (with the exception of the modified HOOF-Prints 7 primers). To ensure the comparability of datasets, we used the same numbering system and adjusted the HOOF-Prints 7 allele designations such that those presented here are equivalent to those previously described. Newly described loci were given allele designations generally based on repeat number. However, in some cases, as described in Results, alleles were found to reflect changes within regions other than the selected tandem repeat, and in these cases different alleles therefore do not necessarily reflect the numbers of tandem repeats. A full list of the sizes of PCR products obtained and the corresponding allele designations is provided in the supplemental material.
Assessment of in vitro and in vivo stability.In order to assess the stability of VNTR profile on in vitro passage, cultures ofB. abortusUK18-03-211,B. meliten-sisF8/01-155, andB. suis01-5744 were maintained on serum dextrose agar slopes with the addition of 10% (vol/vol) horse serum for approximately 10 months. The cultures were transferred to fresh media at approximately three weekly intervals, at which point the population was sampled and subjected to VNTR analysis. In order to assess in vivo stability, samples obtained after experimental infection of pigs withB. suis01-5744 were utilized. After intraconjunctival inoculation, Bru-cellawas reisolated from the blood of six of the ten pigs initially infected by using the method of Casteneda (7). Blood culture was carried out twice weekly up to 56 days postinfection and weekly for a further 113 days. The number of occasions on whichBrucellaisolation was successful for an individual pig ranged from only a single bleed to up to nine bleeds. Crude methanol extracts of allBrucellastrains reisolated were prepared to assess the VNTR profiles of the recovered isolates relative to the input strain.
Computer analysis and tree construction.Genetic diversity was determined by using the Simpson’s diversity index (DI) (26) via the online tool V-DICE avail-able at the HPA website (http://www.hpa.org.uk/srmd/bioinformatics/tools/tools .htm). Values of this index can range from 0 (no diversity) to 1 (extreme diver-sity). Analysis of genetic relationships based on VNTR amplicon size profiles was performed by using the neighbor-joining tree algorithm in the PAUP4.0 beta version (Sinauer Associates, Inc., Sunderland, Mass.).
Sequence accession numbers.Sequences of the VNTR loci newly described in the present study that were determined in order to verify repeat numbers have been deposited in EMBL under accession numbers AM260641 to AM260683.
on May 16, 2020 by guest
http://jcm.asm.org/
TABLE 1. Characteristics ofBrucellastrains examined in this studya
Isolate Tree location Biovar Animal host Source
B.abortus544 1 Biovar 1 Reference strain NA
B.abortus86/8/59 2 Biovar 2 Reference strain NA
B.abortus870 3 Biovar 6 Reference strain NA
B.abortusF8/03-A 4 Biovar 1 NK New Zealand
B.abortusUK29/03 5 Biovar 1 Human United Kingdom
B.abortusF6/03 MS553 6 Biovar 1 Bovine Dubai
B.abortusUK8/01 7 Biovar 1 Human Eire
B.abortusUK34/99 8 Biovar 1 Human United Kingdom
B.abortusI12 9 Biovar 1 Bovine United Kingdom
B.abortusS19 Vaccine strain NA
B.abortusRB51 10 Vaccine strain NA
B.abortusUK25/01 11 Biovar 1 Human Eire
B.abortusF7/01-1476 12 Biovar 1 Ovine Kosovo
B.abortusF9/01-14 13 Biovar 1 Water buffalo Trinidad
B.abortusUK3/01 14 Biovar 1 Bovine United Kingdom
B.abortusUK14/02-6037 15 Biovar 2 Bovine Eire
B.abortusF11/02-V1 16 Biovar 1 Water buffalo Trinidad
B.abortus1 NK Bovine Sri Lanka
B.abortusUK11/02 17 Biovar 1 Human Eire
B.abortusCow4 18 Biovar 1 Bovine Cornwall, United Kingdom
B.abortusR51/03 19 Biovar 1 Bovine Scotland
B.abortusR8-22 20 Biovar 1 Bovine Scotland
B.abortusF6/04 04376 21 Biovar 1 Human New Zealand
B.abortus99-9971-135A 22 Former biovar 7❋ NK Mongolia
B.abortus99-9971-159B 23 Former biovar 7❋ NK Mongolia
B.abortus03/4923-239 24 Former biovar 7❋ Bovine Turkey
B.abortusF5/03-1 25 Biovar 3 Bovine Portugal
B.abortus62/5 26 NK Bovine Poland
B.abortus5/93 27 Biovar 2 Bovine Anglesey, United Kingdom
B.abortusF5/04-1 28 Biovar 1 Bovine Portugal
B.abortusF5/04-2 29 Biovar 1 Bovine Portugal
B.abortusF5/04-3 30 NK Bovine Portugal
B.abortusF5/04-4 31 Biovar 1 Bovine Portugal
B.abortusF5/04-5 32 Biovar 3 Bovine Portugal
B.abortusF5/04-7 33 Biovar 3 Bovine Portugal
B.abortusF5/04-8 34 Biovar 1 Bovine Portugal
B.abortusF5/04-9 35 Biovar 1 Bovine Portugal
B.abortusF5/04-10 36 Biovar 1 Bovine Portugal
B.abortusTulya 37 Biovar 3 Reference strain NA
B.abortus292 38 Biovar 4 Reference strain NA
B.abortusB3196 Biovar 5 Reference strain NA
B.abortusC68 39 Former biovar 9❋ Reference strain NA
B.abortus63/66 NK NK Poland
B.abortus63/294 40 NK NK Kenya
B.melitensis16M 41 Biovar 1 Reference strain NA
B.melitensis1BM1 42 Biovar 1 NK Northern Portugal
B.melitensis3BM1 43 Biovar 1 NK Northern Portugal
B.melitensis4BM1 44 Biovar 1 NK Northern Portugal
B.melitensisB115 45 Rough mutant Caprine Malta
B.melitensisF3/02 46 Biovar 2 Human NK
B.melitensis63/9 47 Biovar 2 Reference strain NA
B.melitensis65/155 48 Biovar 3 Ovine Mongolia
B.melitensis66/59 49 Biovar 3 Ovine India
B.melitensisUK7/01 50 Biovar 1 Human India
B.melitensisUK23/01 51 Biovar 3 Human United Kingdom (Greece derived)
B.melitensisF8/01-155 52 Biovar 3 Bovine Kosovo
B.melitensisF4/03-917 53 NK Human Greece
B.melitensisF8/03-D 54 Biovar 2 Human New Zealand
B.melitensisF6/03-D1127 55 Biovar 3 Gazelle Dubai
B.melitensis5BM3 56 Biovar 3 NK North Portugal
B.melitensis9BM3 57 Biovar 3 NK North Portugal
B.melitensis13BM3 58 Biovar 3 NK North Portugal
B.melitensis17BM3 59 Biovar 3 NK North Portugal
B.melitensisUK4/05 60 NK Human United Kingdom (Portugal derived)
B.melitensisF5/04-12 61 Biovar 3 Porcine Portugal
B.melitensisEther 62 Biovar 3 Reference strain NA
B.melitensisUK31/99 63 Biovar 1 Human NK
Continued on following page
on May 16, 2020 by guest
http://jcm.asm.org/
RESULTS
Identification and characterization of loci. Since, prior to the advent of MLVA, there were essentially no molecular tools that could provide useful discrimination beyond the level of biovar, the first aim of this study was to characterize highly discriminatory loci that would facilitate the development of
tools to permit epidemiological traceback of Brucella to the
[image:4.585.48.547.85.599.2]source of infection. During the planning stages of this work, the HOOF-Prints scheme, based on eight hypervariable octa-meric loci, was published by Bricker et al. (5). In order to further improve the discriminatory capacity of this scheme, to reduce the risk of match due to homoplasy, and to offer an
TABLE 1—Continued
Isolate Tree location Biovar Animal host Source
B.melitensis65/94 64 Biovar 3 Ovine France
B.melitensis80/82 65 Biovar 2 Human Former Yugoslavia
B.melitensisF12/01 Biovar 1 Ibex United Arab Emirates
B.melitensisR39/03-60 Biovar 1 Livestock Tanzania
B.melitensisUK19/04 66 Biovar 1 Human United Kingdom (Somalian/ Ethiopian origin)
B.melitensisUK21/04 67 Biovar 1 Human United Kingdom (Ethiopian origin)
B.melitensisUK22/04 68 Biovar 1 Human United Kingdom (unknown origin)
B.melitensisUK23/04 69 Biovar 1 Human United Kingdom (unknown origin)
B.melitensisUK25/04 70 Biovar 3 Human United Kingdom (unknown origin)
B.melitensisUK26/04 71 Biovar 1 Human United Kingdom (Somalia)
B.ovis63/290 72 NA Reference strain NA
B.ovisREO 73 NA NK France
B.ovis81/2 NA Ovine Germany
B.ovis63/96 74 NA Ovine Argentina
B.ovis81/8 75 NA Ovine Spain
B.ovisS92 76 NA Ovine France
B.ovis80/125 77 NA Ovine Australia
B.ovis79/69 78 NA Ovine New Zealand
B.ovis79/160 79 NA Ovine United States
B.ovisF10 D4/02 80 NA Ovine New Zealand
B.suis1330 81 Biovar 1 Reference strain NA
B.suis686 82 Biovar 3 Reference strain NA
B.suis64/24 83 Biovar 1 Porcine United States
B.suis63/202 Biovar 4 Reindeer Former Soviet Union
B.suis01-5744 84 Biovar 1 Porcine France
B.suis79/30 85 Biovar 4 Human Finland
B.suisF6/04 79/0349 Biovar 1 Human Tonga
B.suisF6/04 73/1616 86 Biovar 1 Human Tonga
B.suis40 87 Biovar 4 Reference strain NA
B.suis63/34 88 Biovar 3 NK United States
B.suis63/252 89 Biovar 4 Caribou United States
B.suisF1/04 90 Biovar 1 Human Holland
B.suis63/219 91 Biovar 4 Reindeer Former Soviet Union
B.suis63/198 92 Biovar 4 Reindeer Former Soviet Union
B.suisThomsen 93 Biovar 2 Reference strain NA
B.suis74/12 94 Biovar 2 N/K Denmark
B.suis79/194 Biovar 2 Hare Former Czechoslovakia
B.suisF12/02 95 Biovar 2 Hare Denmark
B.suisF13/02 Biovar 2 Hare Denmark
B.suis92/63 Biovar 2 Hare Slovenia
B.suisF5/03-2 96 Biovar 2 Porcine Portugal
B.suis32 97 Biovar 2 NK North Portugal
B.suis31 98 Biovar 2 NK Central Portugal
B.suis23 99 Biovar 2 NK South Portugal
B.suis513 100 Biovar 5 Reference strain NA
B.neotomae5K33 NA Reference strain NA
B.neotomae65/197 101 NA Desert wood rat United States
B.neotomae65/196 102 NA Desert wood rat United States
B.canisF7/02 103 NA Canine Germany
B.canisUK10/02 104 NA Canine United Kingdom
B.canisRM6/66 NA Reference strain NA
B.canis79/139 105 NA Canine United States
B.canis79/92 NA Canine Germany
B.canis79/85 NA Canine Peru
a
The corresponding allelic profile data are provided in the supplemental material. Where multiple allelic states were obviously present, the alternatives are shown. For the original HOOF-Prints loci, the allele naming system of Bricker et al. (5) was used, including the designation of alternative allele sizes for equivalent repeat numbers at HOOF-Prints 4, reflecting an upstream deletion in all other species relative toB.abortusandB.melitensis. Where no consistent amplification was obtained, a null allele (i.e., 0) was recorded. NA, not applicable; NK, not known.❋, strains related to pre-1988 designations (10).
on May 16, 2020 by guest
http://jcm.asm.org/
increased choice of loci that might be applicable in a future
standardized Brucella MLVA scheme, we identified further
loci and compared their diversity with the HOOF-Prints loci. Tandemly repeated loci were identified by using Tandem Re-peat Finder (2), and a number of loci were screened by PCR
for size variation across a panel ofBrucellaisolates (data not
shown).
The location and characteristics of all novel loci taken for-ward for full assessment in the present study are described in Table 3. The initial stages of the work focused on loci that
appeared variable within the classically identifiedBrucella
spe-cies and were therefore most likely to add substantial discrim-inatory power to an MLVA scheme. Seven such loci were included in the present study and, as might be expected, all represented short sequence repeats of either 5 bp (VNTR 16) or 8 bp (VNTR 2, VNTR 5A, VNTR 5B, VNTR 12A, VNTR 12B, and VNTR 17). Sequencing of a number of alleles at each of these loci confirmed that the size variation in VNTR PCR products usually reflected exact changes in the number of re-peats, and this information was used to predict the number of repeats present in remaining alleles. In the case of two of these repeats the situation was more complex. Sequencing of a rep-resentative of each of six different VNTR 17 alleles varying in size in 8-bp increments revealed that in addition to the tandem repeat described in Table 3, which varied from one to four copies, an additional octameric repeat varying from two to four copies is located approximately 30 bp downstream of this VNTR. Thus, in the case of VNTR 17, the combination of variation at both of these sites contributes to variation at this locus. In addition, sequencing of VNTR 16 alleles revealed that some strains harbor a 5-bp deletion immediately upstream of the pentameric repeats.
After assessment of the shorter sequence repeats, six VNTR loci comprising of slightly larger repeats were added to the scheme. The sizes of these repeats predicted by Tandem Re-peat Finder ranged from 13 bp (VNTR 21), 15 bp (VNTR27), 17 bp (VNTR 14), and 18 bp (VNTR 26 and VNTR 7) through to 40 bp (VNTR 24). Preliminary work suggested there was only very limited variation at these loci within a species, and thus it was predicted that variation at these loci between spe-cies might provide a useful taxonomic and phylogenetic
frame-work when used in conjunction with the shorter and apparently more variable repeats. Again, representatives of a number of allelic states were sequenced. Only two alternative allelic states were detected for VNTRs 14, 21, and 27 and these
corre-sponded to those predicted from theB. suisandB. melitensis
genome sequences. For VNTR 26 the designation of alleles was simple since, despite being a rather degenerate repeat, each allele was found to reflect an exact 18-bp increment in repeat number. The situation for the two remaining loci is more complex since allele sizes were not exact multiples of the sizes predicted in Table 3, and sequencing confirmed complex patterns of variation at these two loci that do not reflect exact changes in repeats. In the case of VNTR 7, whereas most alleles did reflect 18-bp increments, one allele reflected the presence of a 3-bp deletion in one copy of the repeat. VNTR 24 was found to represent a particularly complex structure with a series of alleles that did not reflect exact changes in the 40-bp repeat originally identified. Representative sequences of all of the loci with novel variants described here have been deposited in EMBL under accession numbers given in Materials and Methods.
Thus, in total, 13 novel loci were examined here in conjunc-tion with the existing 8 HOOF-Prints loci to give a 21-locus MLVA scheme. With the exception of two pairs of loci (VNTR 5A and 5B, and 12A and 12B), the novel loci are widely
distributed across theBrucellagenome (seven on chromosome
I and six on chromosome II). The two VNTR 12 loci represent octameric repeats that are separated by less than 40 bp but are highly diverse and appear to vary independently. Similarly, the two VNTR 5 loci are separated by approximately 60 bp but appear to vary at relatively high frequency independently.
Comparative genetic diversity.The 21-locus MLVA scheme
outlined above was applied to 121Brucellaisolates consisting
of 44B. abortus, 33B. melitensis, 10 B. ovis, 25B. suis, 3 B.
neotomae, and 6B. canis isolates (Table 1 shows the strains examined [the equivalent raw data are provided in the supple-mental material]). These isolates include all species and biovar reference strains and a collection of isolates that represent a diverse range of biovars and geographical origins. In addition, a small number of isolates from more restricted geographical origins were included to assess diversity within such
popula-TABLE 2. Primers used for amplification of loci newly described in this study
Primer
Sequence
Forward Reverse
HOOF-Prints 7a CGGGCGGACAGAGCCGTCGGTGGTTACTT GGGGAGTATGTTTTGGTTGCGCATGGCCGC
VNTR 2 AGTTCGAGACCTTCATGGAA CAGTTCCATTGCTTGCTGC
VNTR 16 GAATGATAAGCTTCACCTGAATA CGCGTTTCGATTGTGGAAA
VNTR 17 GCCGATGCGAAGAAGATGG CGGTCATCGCGCCGATCA
VNTR 14 GATTGGGTTCGCAAACCATG AACCCTGTCGTCATGAAGTG
VNTR 21 GCAAGTAAACCGATCACAGAC CGAAAAGGGAGCAGACATTA
VNTR 7 CATCGATTATACCAATCGCG AACACAGAGGCGATCAGGAT
VNTR 24 TGGCGATGGACGGCGGCTT ACCCTGCTCATCCACTGC
VNTR 26 CGAAAGCGGTTGCTGTGCAG ACCGTCTATCAGCCTGCCA
VNTR 27 CGAATAACAGCGCCGTCTT ATGATCGCCGGGAATCGG
VNTR 5A AATCACCCTTTTTCAGTCAAGG GAAGAACATCTATTTCGCGGT
VNTR 5B GTGGTGGACAAGGCAAGTA CGCGAGGTTTTCGGCCAGA
VNTR 12A CTTGGTGCATTTCCATGATCT AAAGAATCAGATAAGATAAAGCAA
VNTR 12B TTGCTTTATCTTATCTGATTCTTT CATCAACGATACAACATGGC
a
The primer pair used to amplify HOOF-Prints 7 was modified from that originally described by Bricker et al. (5).
on May 16, 2020 by guest
http://jcm.asm.org/
[image:5.585.45.542.79.243.2]TABLE 3. Genomic location and characteristics of novel repetitive loci examined in the present study, as defined by Tandem Repeat Finder
Locus Repeat size (bp)
Location inB.suis 1330 genome
No. of units
Location inB.melitensis 16M genome
No. of units
Consensus repeat sequence (B.suis)
VNTR 2 8 Chromosome II (NC004311) 4.4 Chromosome II (NC003318) 2.4 TAAGGGAG 749707-749741 548709-548727
Overlapping stop codon of BRA0768 (glutamine synthetase family protein)
Overlapping stop codon of BMEII0523 (glutamine synthetase)
VNTR 7 18 Chromosome I (NC004310) 5.0 Chromosome I (NC003317) 3.0 CCGGAAGAGCCGCCGCCG 1756031-1756120 233447-233500
(hypothetical protein) (hypothetical membrane spanning protein)
VNTR 5A 8 Chromosome I (NC004310) 5.2 Chromosome I (NC003317) 4.6 GAATAGGG 736284-736328 1250148-1250187
Overlapping stop codon of BR0749
Overlapping stop codon of BMEI1204
(phosphatase, Ppx/GppA family) (exopolyphosphatase)
VNTR 5B 8 Chromosome I (NC004310) 4.2 Chromosome I (NC003317) 4.2 CCCTTACT 736392-736427 1250050-1250084
Overlapping stop codon of BR0750
Overlapping stop codon of BMEI1203
(RNase D) (RNase D)
VNTR 27 15 Chromosome I (NC004310) 3.0 Chromosome I (NC003317) 4.0 CCAATATTGAAATTG 752169-752213 1234292-1234351
Intergenic; upstream of BR0766 Intergenic; upstream of BMEI1190 (4-hydroxybenzoate octaprenyltransferase)
VNTR 26 18 Chromosome I (NC004310) 3.2 Chromosome I (NC003317) 4.2 CCNGCACAGAATGANTGG 850597-850653 1134980-1135054
Intragenic; within BR0878 Intragenic; within BMEI1088 (hypothetical protein) (soluble lytic murein
transglycosylase)
VNTR 16 5 Chromosome II (NC004311) 5.2 Chromosome II (NC003318) 2.2 GGCCG 954343-954368 344643-3446553
Intergenic; downstream of BRA0966
Intergenic; downstream of BMEII0332
(ribosomal protein S21) (ribosomal protein S21)
VNTR 21 13 Chromosome I (NC004310) 2.0 Chromosome I (NC003317) 1.0 TTGTTCATCCTGC 1965388-1965413 28455-28467
Overlapping stop codon of BR2042
Overlapping stop codon of BMEI0029
(L-sorbose dehydrogenase) (L-sorbose dehydrogenase)
VNTR 14 17 Chromosome II (NC004311) 2.0 Chromosome II (NC003318) 1.0 AAAGCGCAAAGATCACA 645465-645498 652250-652266
Intragenic; in BRA0662/663 Intragenic; in BMEII0616 (hypothetical protein; disrupted by
frameshift)
(hypothetical membrane-spanning protein)
VNTR 12A 8 Chromosome II (NC004311) 2.9 Chromosome II (NC003318) 1.9 GAGTAAGG 73008-73030 19069-19083
Overlapping stop codon of BRA0073
Overlapping stop codon of BMEII0021
(sugar isomerase KpsF/GutQ) (KpsF polysialic acid capsule expression protein)
VNTR 12B 8 Chromosome II (NC004311) 11.1 Chromosome II (NC003318) 6.1 TTCCCCTA 73070-73158 18981-19029
Overlapping stop codon of BRA0074
Overlapping stop codon of BMEII0020
(hypothetical protein) (nodulation protein NfeD, C-terminal only)
VNTR 24 40 Chromosome I (NC004310) 2.3 Chromosome I (NC003317) 3.3 GACAGGGATATAACCAAGAGG 2044647-2044739 2066414-2066545 CGTGAAGTGCCGTAGGCAC Overlapping stop codon of Overlapping stop codon of
BR2121 BMEI2006
(hypothetical protein) (hypothetical cytosolic protein) Immediately upstream of Immediately upstream of
BR2122 BMEI2005
(pheylalanyl-tRNA (pheylalanyl-tRNA synthetase, alpha subunit) synthetase, alpha subunit)
VNTR 17 8 Chromosome II (NC004311) 3.8 Chromosome II (NC003318) 4.8 AGTAAGGG 959110-959140 339863-339902
Intergenic; between Intergenic; between BRA0970/0971 BMEII0325/0326 (putative oxidoreductase/ (NADP transhydrogenase/
NADP transhydrogenase) homoserine dehydrogenase)
on May 16, 2020 by guest
http://jcm.asm.org/
tions. The discriminatory power of the technique is readily demonstrated by the fact that, of the 121 genotypic profiles
determined, 117 were unique. Only two pairs of strains (B.
neotomae65/196 and 65/197 and B. melitensis UK21/04 and UK26/04) gave identical profiles. With one exception, all loci could be amplified from all strains – we could not obtain a
reliable amplification product fromB. ovis isolates using the
HOOF-Prints 5 primers and thus treated this as a null allele for allB. ovisisolates.
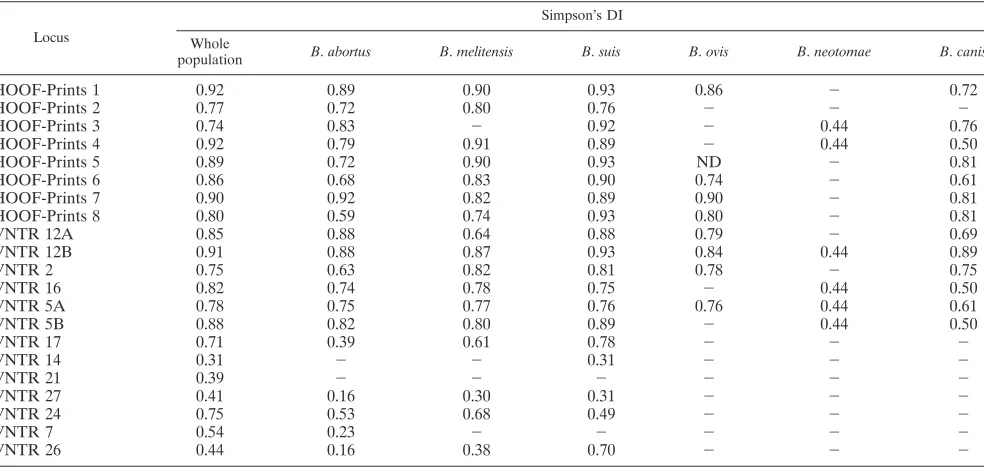
[image:7.585.97.485.68.312.2]The number of alleles present at each locus within the pop-ulation examined here ranged from 2 in the case of VNTR 14, VNTR 21, and VNTR 27 up to 21 in the case of HOOF-Prints
FIG. 1. Genetic diversity of the 21 VNTR loci examined in this study. The number of alleles at each locus is shown by the filled bars (left-hand scale). Simpson’s DI values for each VNTR calculated using a maximum of 121 isolates (isolates with multiple allelic states at a particular locus were excluded) are shown as diamonds (right-hand scale).
TABLE 4. Simpson’s DI for all loci examined in this study shown for the whole population and for each of the individual classically identifiedBrucellaspeciesa
Locus
Simpson’s DI
Whole
population B.abortus B.melitensis B.suis B.ovis B.neotomae B.canis
HOOF-Prints 1 0.92 0.89 0.90 0.93 0.86 ⫺ 0.72
HOOF-Prints 2 0.77 0.72 0.80 0.76 ⫺ ⫺ ⫺
HOOF-Prints 3 0.74 0.83 ⫺ 0.92 ⫺ 0.44 0.76
HOOF-Prints 4 0.92 0.79 0.91 0.89 ⫺ 0.44 0.50
HOOF-Prints 5 0.89 0.72 0.90 0.93 ND ⫺ 0.81
HOOF-Prints 6 0.86 0.68 0.83 0.90 0.74 ⫺ 0.61
HOOF-Prints 7 0.90 0.92 0.82 0.89 0.90 ⫺ 0.81
HOOF-Prints 8 0.80 0.59 0.74 0.93 0.80 ⫺ 0.81
VNTR 12A 0.85 0.88 0.64 0.88 0.79 ⫺ 0.69
VNTR 12B 0.91 0.88 0.87 0.93 0.84 0.44 0.89
VNTR 2 0.75 0.63 0.82 0.81 0.78 ⫺ 0.75
VNTR 16 0.82 0.74 0.78 0.75 ⫺ 0.44 0.50
VNTR 5A 0.78 0.75 0.77 0.76 0.76 0.44 0.61
VNTR 5B 0.88 0.82 0.80 0.89 ⫺ 0.44 0.50
VNTR 17 0.71 0.39 0.61 0.78 ⫺ ⫺ ⫺
VNTR 14 0.31 ⫺ ⫺ 0.31 ⫺ ⫺ ⫺
VNTR 21 0.39 ⫺ ⫺ ⫺ ⫺ ⫺ ⫺
VNTR 27 0.41 0.16 0.30 0.31 ⫺ ⫺ ⫺
VNTR 24 0.75 0.53 0.68 0.49 ⫺ ⫺ ⫺
VNTR 7 0.54 0.23 ⫺ ⫺ ⫺ ⫺ ⫺
VNTR 26 0.44 0.16 0.38 0.70 ⫺ ⫺ ⫺
aFor the whole population (maximum,n⫽121), strains with multiple allelic states for a particular locus were excluded from the calculation for that locus.⫺, no diversity; ND, not done (no consistent amplification).
on May 16, 2020 by guest
http://jcm.asm.org/
[image:7.585.47.539.474.708.2]7 (Fig. 1). Note that although HOOF-Prints 4 appears to have the highest number of alleles (27); this is somewhat deceptive
since a deletion within the PCR product inBrucella species
other thanB. abortusandB. melitensismeans that each repeat
number has two possible allelic states (shown, for example, as 1 or 1A in the supplemental material) at this locus. In order to assess the diversity at individual loci, the DI was calculated for
both the whole population and for all isolates of eachBrucella
species individually. Values of this index range from 0 in the case of no diversity up to 1 in the case of extreme diversity and reflect the number of alleles detected and the individual allele frequency. In general, there was a close relationship between the total number of alleles and DI with values ranging from
0.31 in the case of VNTR 14 to⬎0.90 in the case of
HOOF-Prints 1, 4, and 7 and VNTR 12B (Fig. 1). The overall diversity of the novel short sequence repeat (5 or 8 bp) VNTR loci selected for use in the present study (range, 0.71 to 0.91; mean, 0.81) was similar to that for the HOOF-Prints loci considering the same population (range, 0.74 to 0.92; mean, 0.85), indicat-ing similar resolvindicat-ing power. With the exception of VNTR 24, the six larger repeats had much lower numbers of alleles (two to four) and DI values (0.31 to 0.54). VNTR 24, although originally defined as a 40-bp repeat, had a number of complex alleles that did not reflect exact changes in repeat number and thus had a larger number of alleles (6) and a DI (0.75) within the range of the VNTR loci based on shorter sequence repeats (5 to 8 bp).
The DI values determined within each of the individual
species (Table 4) demonstrate that most of the VNTR consist-ing of short (5- to 8-bp) repeats are also variable within species.
There are some exceptions, notably in B. ovis. This species
appears to harbor less genetic diversity than the otherBrucella
spp., with no variation detected in 7 of 15 such VNTR loci. In
contrast, all 15 of these loci were found to be variable withinB.
abortus and B. suis, only HOOF-Prints 3 is invariant in B. melitensis, and only two loci (HOOF-Prints 2 and VNTR 17)
were found to be invariant in the small number of B. canis
isolates examined. In contrast, there was limited intraspecies diversity when we considered the six VNTRs representing longer sequence repeats in this population. VNTR 21 does not
vary within a species with one allele confined toB. melitensis,
while a second allele is found in all other species. The only
intraspecies variation for VNTR 14 is seen in the singleB. suis
biovar 5 isolate that shares an allele with representatives of all other species in contrast to the unique allele seen in all other
B. suisisolates. VNTR 7 showed intraspecies variation in only
threeB. abortusisolates. VNTR 27, VNTR 24, and VNTR 26
all reveal diversity in isolates ofB. abortus,B. melitensis, andB.
suis, although the DI values indicate that this is less extensive
than that seen using the 15 VNTR loci containing 5- to 8-bp sequence repeats.
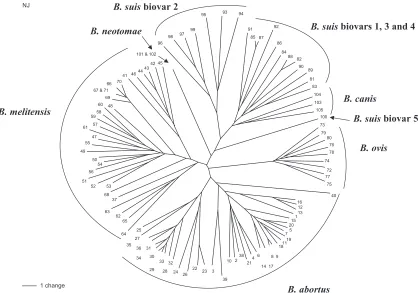
[image:8.585.84.502.69.362.2]Genetic relationships. The genetic relationships between 105 of the 121 isolates were determined by the construction of a neighbor-joining tree (Fig. 2). Sixteen of the original isolates included in Table 1 were found to possess multiple allelic states at one or more loci and were therefore excluded from this
FIG. 2. Neighbor-joining reconstruction of relationships between 105 of the 121 isolates examined in this study. Isolates that had multiple allelic states at one or more locus were excluded from this analysis. Designations of major clusters are shown, but in order to facilitate a clearer presentation the taxa were given numerical designations from 1 to 105. The corresponding isolate information is detailed in Table 1.
on May 16, 2020 by guest
http://jcm.asm.org/
analysis. A number of major clusters are immediately apparent
that correspond to the classically recognizedBrucellaspecies.
Thus, there are clusters consisting exclusively ofB. abortus,B.
ovis, andB. neotomaeisolates. Similarly, there is a large cluster
that, with the exception of a single isolate, theB. abortusbiovar
3 reference strain (Tulya), is comprised entirely ofB. melitensis
isolates. With the exception of theB. suisbiovar 5 reference
strain that appears to be distantly related to all other isolates,
B. suisisolates group together in a cluster that also containsB. canis.B. suisfurther subdivides into two clusters: one consist-ing exclusively of biovar 2 isolates and the other containconsist-ing biovar 1, 3, and 4 isolates.
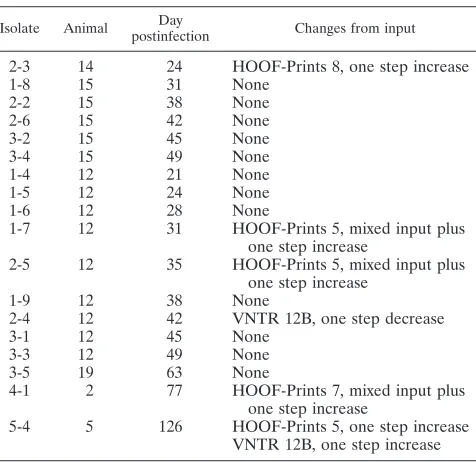
Stability of VNTR loci. In order to assess the stability of VNTR loci over time, changes to input strains were examined after both in vivo and in vitro passage. For the in vivo study
pigs were experimentally infected with B. suis 01-5744 and
frequent blood culture was carried out postinfection in an
attempt to reisolateBrucella. The VNTR profiles of both the
inoculum and any strains recovered postinfection were deter-mined (Table 5). Of six animals that became culture positive, isolates recovered from two of the animals at up to 63 days postinfection were identical to the input profile. Isolates from the four remaining animals showed minor changes in the VNTR profile. These changes were all either one-step in-creases or dein-creases in repeat number, often with a mixed profile such that the input allele was still visible, occurring at loci with high DI values. Thus, such changes were apparent at VNTR 12B, HOOF-Prints 5, and HOOF-Prints 7, all of which
have DI values of ⬃0.9, and a single change was noted at
HOOF-Prints 8 that, while having slightly lower diversity in the overall population, has the equal highest DI value recorded for
any population subset (0.93) forB. suisisolates. In order to
look at the effect of in vitro passage three strains were passaged
14 times over 270 days (Table 6). The VNTR profiles ofB. suis
andB. melitensisisolates remained unchanged, while the
pro-file of theB. abortusisolate showed only a one-step increase in
the VNTR 12B repeat number that became apparent at pas-sage 13 after 251 days of cultivation.
DISCUSSION
The aim of this study was to identify and characterize VNTR markers that, when used in combination with loci previously described, provide the robust high-resolution typing tool that
Brucellaepidemiologists have lacked to date to facilitate epi-demiological traceback. Furthermore, by incorporation of more stable markers, it was deemed possible to develop a tool
that can simultaneously address issues of Brucella taxonomy
and phylogeny. We report on 13 repetitive loci not previously
described inBrucellaspp. that further progress toward both of
these goals. The novel loci are distributed throughout the ge-nome, with seven located on chromosome I and six located on chromosome II. Two sets of repeats (5A/B and 12A/B) were located very close to each other (ca. 40 to 60 bp apart) and were originally considered as single loci. However, eventually primers were designed such that these repeat tracts are con-sidered separately as, for a number of reasons, this approach enhances discriminatory capacity. It enables maximum infor-mation to be gathered from the extensive diversity seen at all four of these loci rather than just considering these sites as two loci. It reduces the risk of convergence to the same allele that could result from treating both sets of repeats as a single locus. Furthermore, should there be multiple alleles at one of the repetitive tracts, only data from one locus is lost and not from both as is the case if the tracts are considered together as a single locus. As with the original HOOF-Prints loci, the 13 novel loci are located variously either intragenically, intergeni-cally, or overlapping the predicted stop codons of putative proteins (Table 3).
The use of VNTRs to examine phylogeny and taxonomy in bacteria is open to criticism due to concerns about rapid evo-lution and potential homoplasy that could result in misleading
conclusions. However, in homogeneous organisms such as
[image:9.585.44.282.96.327.2]Bru-cella, where limited diversity is detectable by other approaches, the use of VNTRs offers a viable alternative to more tradi-tional approaches. With minor exceptions, analysis based on the 21-locus MLVA scheme divided the organisms in the present study into clusters corresponding to their taxonomic designations (Fig. 2). Application of the identical analysis to the same population with only the original 8 HOOF-Prints loci fails to group all isolates of the traditionally recognized species
TABLE 5. Stability of VNTR markers after in vivo passage: changes after experimental infection of pigs
withB.suis01-5744a
Isolate Animal Day
postinfection Changes from input
2-3 14 24 HOOF-Prints 8, one step increase
1-8 15 31 None
2-2 15 38 None
2-6 15 42 None
3-2 15 45 None
3-4 15 49 None
1-4 12 21 None
1-5 12 24 None
1-6 12 28 None
1-7 12 31 HOOF-Prints 5, mixed input plus one step increase
2-5 12 35 HOOF-Prints 5, mixed input plus one step increase
1-9 12 38 None
2-4 12 42 VNTR 12B, one step decrease
3-1 12 45 None
3-3 12 49 None
3-5 19 63 None
4-1 2 77 HOOF-Prints 7, mixed input plus one step increase
5-4 5 126 HOOF-Prints 5, one step increase VNTR 12B, one step increase
[image:9.585.300.542.99.165.2]aChanges from the input profile are shown.
TABLE 6. Stability of VNTR markers after in vitro passage: changes after in vitro passage ofB.suis01-5744,
B.melitensisF8/01-155, andB.abortusUK18-03-211a
Strain Passage no.
Time
(days) Changes from input
B.abortus 1-14 270 VNTR 12B, one-step increase from passage 13 (day 251)
B.suis 1-14 270 None
B.melitensis 1-14 270 None aChanges from the input profile are shown.
on May 16, 2020 by guest
http://jcm.asm.org/
together, while use of all 15 short-sequence repeat-containing loci (5 to 8 bp), although an improvement on the latter, also fails to completely resolve all of the species groups (data not shown). One of the minor exceptions mentioned above is the
B. abortusbiovar 3 reference strain (Tulya) that falls within the
B. melitensis cluster. Previous studies have noted that strain Tulya is genetically distinct from field biovar 3 strains (18), and
otherB. abortusbiovar 3 strains examined in the present study
did cluster withB. abortus. Thus, the status of this reference
strain and its relationship to otherB. abortusbiovar 3 strains
requires further investigation. Except for this one strain, all
isolates ofB. melitensis,B. abortus,B. ovis, andB. neotomaefall
into distinct clusters that reflect their traditional taxonomic
status.B. canisfalls on a separate branch within the B. suis
cluster that is most closely related toB. suisbiovar 1, 3, and 4
isolates.B. suisbiovar 2 isolates form a separate branch, while
biovar 5 isolates are only distantly related to other B. suis
isolates. The close association ofB. caniswithB. suishas been
noted in a number of studies using methods including ampli-fied fragment length polymorphism (AFLP) (29),
chromo-somal maps (23),omp2profiling (8, 9), MLEE (13), and
in-sertion sequence typing (19). Similarly, studies using AFLP
and MLEE have indicated thatB. suisbiovar 5 is distinct from
otherB. suisisolates (13, 29). Although the species generally
separated well into distinct genetic entities, there was less evidence of many of the biovars corresponding to distinct
ge-netic groups. Thus, in the case ofB. suis, although biovar 2 and
5 isolates separate well as described above, isolates of biovars 1, 3, and 4 were not clearly separated. Similarly, there was no
evidence of separation of the isolates ofB. melitensisbiovars 1,
2, and 3, and there were too few representatives of most of the
B. abortusbiovars to reach any conclusions. Genetic relation-ships within biovars as determined by this approach will be addressed elsewhere using much larger numbers of suitably selected isolates.
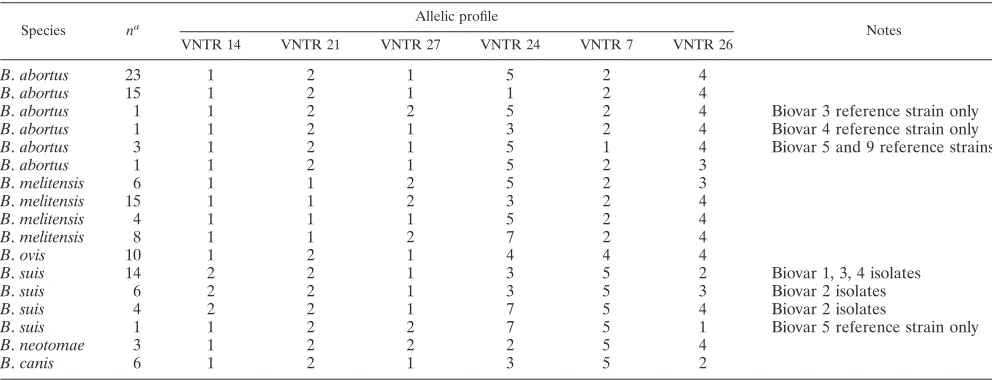
Data gathered in the present study indicate that the use of the six novel loci based on slightly longer sequence repeats (VNTRs 7, 14, 21, 24, 26, and 27) in isolation provide an assay
capable of speciation of all Brucella isolates. The potential
outcomes of such an assay are illustrated in Table 7. On the basis of alleles at these loci, the 121 isolates examined here divide into 17 distinct genotypes, all of which are associated
exclusively with only one of the traditionally recognized
Bru-cellaspecies (6B. abortus, 4B. melitensis, 1B. ovis, 4B. suis, 1
B. neotomae, and 1B. canis). Thus, although it must first be validated by using a much larger collection of isolates, this assay would represent by far the most straightforward method
currently available to speciateBrucellaisolates. Existing assays
that can speciateBrucellafail to distinguish all species or
bio-vars of species (e.g., AMOS PCR [4]) and/or involve complex technical procedures and genomic DNA preparation (e.g.,
IS711fingerprinting or AFLP [19, 29]). As discussed below, the
use of MLVA has considerable technical advantages and could easily be run with an agarose gel-based approach, overcoming the need for expensive apparatus and specialized reagents.
As a tool for epidemiological traceback, MLVA clearly of-fers a remarkable discrimination compared to any of the con-ventional molecular typing techniques previously applied to this organism that generally fail to provide much discrimina-tion below the biovar level. In contrast, the 121 strains exam-ined in the present study represent 119 distinct genotypes.
EveryB. abortusbiovar 1 isolate examined in the present study
represented a different genotype. A further indication of the
power of the technique is that all isolates ofB. ovis, a species
long considered to harbor little diversity (24), could be sepa-rated. However, the lack of variation in 13 of 21 loci does
support the suggestion that, among theBrucellaspecies,B. ovis
is particularly highly conserved. Although 13 new loci are de-scribed here, it is the 7 based on short sequence repeats of 5 to 8 bp that are most useful in developing a genotyping tool that can be used to complement conventional epidemiological in-vestigations. Bricker et al. (6) have recently highlighted a
num-ber of inconsistencies between theB. abortusbiovar and the
[image:10.585.43.539.89.279.2]HOOF-Prints genotype. One possible explanation for this is homoplasy resulting from convergent evolution to an identical genotype among “unrelated” strains, and Bricker et al. (6)
TABLE 7. Species-specific allelic profiles generated for 121Brucellaisolates using the six longer VNTR loci demonstrating that use of these loci in isolation provides an assay that should place an isolate within one of the classically recognizedBrucellaspecies
Species na Allelic profile Notes
VNTR 14 VNTR 21 VNTR 27 VNTR 24 VNTR 7 VNTR 26
B.abortus 23 1 2 1 5 2 4
B.abortus 15 1 2 1 1 2 4
B.abortus 1 1 2 2 5 2 4 Biovar 3 reference strain only
B.abortus 1 1 2 1 3 2 4 Biovar 4 reference strain only
B.abortus 3 1 2 1 5 1 4 Biovar 5 and 9 reference strains
B.abortus 1 1 2 1 5 2 3
B.melitensis 6 1 1 2 5 2 3
B.melitensis 15 1 1 2 3 2 4
B.melitensis 4 1 1 1 5 2 4
B.melitensis 8 1 1 2 7 2 4
B.ovis 10 1 2 1 4 4 4
B.suis 14 2 2 1 3 5 2 Biovar 1, 3, 4 isolates
B.suis 6 2 2 1 3 5 3 Biovar 2 isolates
B.suis 4 2 2 1 7 5 4 Biovar 2 isolates
B.suis 1 1 2 2 7 5 1 Biovar 5 reference strain only
B.neotomae 3 1 2 2 2 5 4
B.canis 6 1 2 1 3 5 2
a
n⫽number of isolates of 121 representing this genotype.
on May 16, 2020 by guest
http://jcm.asm.org/
suggested that additional polymorphic loci should be included in the scheme in order to reduce the likelihood of this. We report here on seven such polymorphic loci that, as demon-strated in Fig. 1, display levels of diversity within the same range as the original HOOF-Prints loci. Although we will de-scribe in detail the use of this scheme in epidemiological trace-back in future studies, the incorporation of small numbers of isolates from restricted geographical locations does highlight the potential of this tool in assisting conventional epidemio-logical approaches. Thus, there are small groups of isolates of
B. abortus(isolate tree references in Fig. 2 of 28 to 36) B. melitensisbiovar 1 (42 to 44),B. melitensisbiovar 3 (56 to 59), andB. suisbiovar 2 (97 to 99) from Portugal included in the present study. These four groups all cluster together in Fig. 2,
with the exception of one of theB. melitensisbiovar 3 isolates
(i.e., isolate 56). However, isolates within each of these clusters still showed diversity at some of the most variable loci. Thus, the loci with the highest DI values (HOOF-Prints 1, 4, 5, and 7 and VNTR 12B) were variable within all four of these
groups. A substantial number of theB. abortusbiovar 1 isolates
included in this study originated from Eire or Northern Ire-land, and all of these isolates also cluster together, along with isolates from the United Kingdom that are known or suspected to reflect importation from these locations (isolate tree refer-ences 5, 7 to 9, 11, 14, 15, and 17 to 20). This cluster is separate
from all other isolates, such as the PortugueseB. abortusbiovar
1 cluster, but again all of the isolates within the cluster are distinguishable from each other, such that a broad geograph-ical origin of an isolate might be predicted from the MLVA profile, but the technique is sufficiently powerful to further differentiate isolates within such a restricted locality. Further examples of the potential of MLVA to assist with epidemio-logical traceback include an isolate from a case of human brucellosis in the United Kingdom (isolate tree reference 60). In this case the isolate profile most closely matches isolates of
B. melitensisfrom Portugal (see Fig. 2, isolate tree references 57 to 59) and, indeed, upon investigation the patient reported a history of recent travel to Portugal. There is also a
well-separated cluster ofB. melitensisisolates from human
brucel-losis in the United Kingdom (isolate tree references 66, 67, and 69 to 71). Where patient histories were available all of these isolates were associated with travel to Eritrea or Somalia.
These isolates are separated from otherB. melitensisisolates
even on the basis of variation in the six loci that facilitate speciation (Table 7). On this basis they share a profile with only a single isolate from livestock in Tanzania R39/03-60 (this isolate was excluded from Fig. 2 due to the presence of a multiple allele at one locus). All of these examples highlight
the potential power of international database ofBrucellastrain
genotypes against which new isolates could be compared.
The technical advantages of MLVA versus existingBrucella
typing schemes are overwhelming. In addition to the huge improvement in discriminatory power, the technique is tech-nically undemanding, and the numerical format means that data are unambiguous and readily comparable between
labo-ratories. All Brucella isolates appear to be typeable by this
approach, including the unclassified marine mammal isolates (data not shown). Only 1 of the 21 loci examined could not be
amplified from all strains, and this problem was confined toB.
ovis(HOOF-Prints 5). One problem was the occasional
iden-tification of multiple alleles among the most genetically vari-able loci. This has been reported previously (6), where the authors chose to report the major peak as the allele designa-tion. Since in our experience the “secondary” peaks were often of identical or similar intensity, we took the approach here of reporting multiple alleles where a second peak had an intensity
of⬎1/3 of that of the major peak. Bricker et al. (6) reported
that this was a particular problem with reference isolates; we also found a particular problem with reference isolates and vaccine strains that have been extensively subcultured. Less frequently passaged recent field isolates seem less problematic. Provided there are sufficient discriminatory loci included in a scheme, the loss of data from a single locus due to the presence of multiple alleles should not be problematic. Alternatively, both possible genotypes could be considered in analysis. Fur-ther advantages of an MLVA approach include rapid turn-around time, ease of automation, and the ability to use crude material or nonviable samples; these characteristics offer huge
advantages for a hazardous organism such asBrucella. As a
PCR-based method, it is also theoretically possible to type directly from infected tissue or other materials without prior culture.
A successful typing method needs to clearly differentiate unrelated isolates but to demonstrate the relationships of all organisms isolated from individuals infected through the same source. In order to formally confirm the stability of isolates obtained from a single source of infection, we examined the stability of isolates after both in vivo and in vitro passages. In both cases minor changes were seen, but they always repre-sented one-step changes in no more than two of the most variable loci such that the strain was clearly recognizable as the input strain. After in vitro cultivation over 14 passages and some 270 days, only a single-step change in a single strain was seen toward the end of this time course. Surprisingly, this change appeared to be absolute with no intermediate mixed genotype detected. We assume that the variant was better adapted to growth on artificial media than the parent strain and rapidly outgrew the original parent strain such that this was no longer detected. However, the length and number of serial passages undertaken far exceeds the in vitro cultivation that clinical isolates would undergo prior to routine MLVA testing and indicates that in vitro cultivation does not lead to significant changes in MLVA profile. After in vivo cultivation, isolates from four of six infected animals showed some minor changes that were always single-step changes and occurred at no more than two loci. Table 5 shows that in one animal, animal 12, one-step changes apparent at one time were not always maintained. We suspect that this reflects the fact that theBrucellaculture method includes an enrichment step and thus the VNTR profiles, though population based, may actu-ally be derived from the enriched clonal descendants of a very small number of bacteria. Thus, the most obvious explanation for the results from animal 12 is that by chance on some occasions the profile resembled the input strain and by chance on other occasions we isolated organisms originating from a clone(s) that had one-step mutations at a rapidly evolving locus either alone or in conjunction with clones representing the initial input strain. Again, all output strains would be recog-nized as identical or very closely related to input strains par-ticularly if ordered characters were considered in the analysis
on May 16, 2020 by guest
http://jcm.asm.org/
rather than considering each different allele at a locus as equally unrelated.
The increasing movements of humans and livestock and the ongoing threat of bioterrorism highlights the needs for inter-national epidemiological surveillance tools able to monitor
pathogens such asBrucellaat global levels and to track
trans-mission routes. In order to facilitate the development of such an approach based on VNTR markers, we have characterized 13 previously unreported VNTR markers that, together with the 8 previously reported markers (5), increase the potential pool of loci that could be used in such a scheme. The loci selected clearly have different rates of evolution, giving flexi-bility to use all markers or use selected markers matching the nature of the population being examined. Thus, while some of the VNTRs (i.e., those based on longer sequence repeats) are not likely to be useful in establishing transmission routes, they are better informers of evolutionary scenarios and useful for establishing phylogeny across a diverse isolate collection. How-ever, for tracking in areas of endemicity there will be little variation among these markers of relatively low diversity and, consequently, the use of more diverse markers will be more informative. The use of all 21 of the loci described here results in a scheme with potentially huge discriminatory capacity that can resolve isolates at a local level but also includes more stable markers, likely to be intrinsically less prone to ho-moplasy, that give a resolution at the taxonomic level that is difficult to achieve using highly variable loci alone. The most pressing requirement now is for the development of an
inter-national database ofBrucellastrain profiles against which new
isolates can be compared. In order to make the most of the power of MLVA, it is crucial that a common set of loci are agreed upon at this early stage in the development of the
technique forBrucella and that all, or a subset, of these are
used internationally such that all data are comparable between laboratories. We hope that the data presented in the manuscript on both novel and previously identified loci will facilitate the rational selection of the most appropriate loci for such a scheme.
ACKNOWLEDGMENTS
We gratefully acknowledge the assistance of Betsy Bricker, U.S. Department of Agriculture, Ames, Iowa, in setting up the HOOF-Prints assay. We also gratefully acknowledge the role of coworkers in theBrucellafield who isolated and submittedBrucellastrains to the VLA over many years, providing an invaluable resource for the opti-mization of this technique.
All research for this study was funded by the United Kingdom Department of Environment, Food, and Rural Affairs (DEFRA).
REFERENCES
1.Alton, G. G., L. M. Jones, R. D. Angus, and J. M. Verger.1988. Techniques for the brucellosis laboratory. INRA, Paris, France.
2.Benson, G.1999. Tandem repeats finder: a program to analyze DNA se-quences. Nucleic Acids Res.27:573–580.
3.Bricker, B. J.2002. PCR as a diagnostic tool for brucellosis. Vet. Microbiol.
90:435–446.
4.Bricker, B. J., and S. M. Halling.1994. Differentiation ofBrucella abortus bv. 1, 2 and 4,Brucella melitensis,Brucella ovis, andBrucella suisbv. 1 by PCR. J. Clin. Microbiol.32:2660–2666.
5.Bricker, B. J., D. R. Ewalt, and S. M. Halling.2003.Brucella‘HOOF-Prints’: strain typing by multi-locus analysis of variable number tandem repeats (VNTRs). BMC Microbiol.3:15.
6.Bricker, B. J., and D. R. Ewalt.2005. Evaluation of the HOOF-Print assay for typingBrucella abortusstrains isolated from cattle in the United States: results with four performance criteria. BMC Microbiol.5:37.
7.Casteneda, M. R.1947. A practical method for routine blood cultures in brucellosis. Proc. Soc. Exp. Biol. Med. N. Y.64:114–115.
8.Cloeckaert, A., J. M. Verger, M. Garyon, and O. Gre´pinet.1995. Restriction site polymorphism of the genes encoding the major 25 and 36-kDa outer-membrane proteins ofBrucella. Microbiol.141:2111–2121.
9.Cloeckaert, A., J. M. Verger, M. Garyon, and N. Vizcaı´no.1996. Molecular and immunological characterisation of the major outer membrane proteins ofBrucella. FEMS Microbiol. Lett.145:1–8.
10.Corbel, M. J.1988. International Committee on Systematic Bacteriology Subcommittee on the Taxonomy ofBrucella.Report of the meeting, 5 Sep-tember 1986, Manchester, England. Int. J. Syst. Bacteriol.38:450–452. 11.Corbel, M. J.1997. Brucellosis: an overview. Emerg. Infect. Dis.3:213–221. 12.DelVecchio, V. G., V. Kapatral, R. J. Redkar, G. Patra, C. Mujer, T. Los, N. Ivanova, I. Anderson, A. Bhattacharyya, A. Lykidis, G. Reznik, L. Jablonski, N. Larsen, M. D’Souza, A. Bernal, M. Mazur, E. Goltsman, E. Selkov, P. H. Elzer, S. Hagius, D. O’Callaghan, J. J. Letesson, R. Haselkorn, N. Kyrpides, and R. Overbeek.2002. The genome sequence of the facultative intracellular pathogenBrucella melitensis. Proc. Natl. Acad. Sci. USA99:443–448. 13.Gandara, B., A. L. Merino, M. A. Rogel, and E. Martinez-Romero.2001.
Limited genetic diversity ofBrucellaspp. J. Clin. Microbiol.39:235–240. 14.Gee, J. E., B. K. De, P. N. Levett, A. M. Whitney, R. T. Novak, and T. Popovic.
2004. Use of 16S rRNA gene sequencing for rapid confirmatory identifica-tion ofBrucellaisolates. J. Clin. Microbiol.42:3649–3654.
15.Groathouse, N. A., B. Rivoire, H. Kim, H. Lee, S. N. Cho, P. J. Brennan, and V. D. Vissa.2004. Multiple polymorphic loci for molecular typing of strains ofMycobacterium leprae. J. Clin. Microbiol.42:1666–1672.
16.Johansson, A., J. Farlow, P. Larsson, M. Dukerich, E. Chambers, M. Bystrom, J. Fox, M. Chu, M. Forsman, A. Sjostedt, and P. Keim.2004. Worldwide genetic relationships amongFrancisella tularensisisolates deter-mined by multiple-locus variable-number tandem repeat analysis. J. Bacte-riol.186:5808–5818.
17.Le Fleche, P., Y. Hauck, L. Onteniente, A. Prieur, F. Denoeud, V. Ramisse, P. Sylvestre, G. Benson, F. Ramisse, and G. Vergnaud.2001. A tandem repeats database for bacterial genomes: application to the genotyping of Yersinia pestisandBacillus anthracis. BMC Microbiol.1:2.
18.Ocampo-Sosa, A. A., J. Aguero-Balbin, and J. M. Garcia-Lobo.2005. De-velopment of a new PCR assay to identifyBrucella abortusbiovars 5, 6, and 9 and the new subgroup 3b of biovar 3. Vet. Microbiol.110:41–51. 19.Ouahrani, S., S. Michaux, J. Sri Widada, G. Bourg, R. Tournebize, M.
Ramuz, and J. P. Liautard.1993. Identification and sequence analysis of IS6501, an insertion sequence inBrucellaspp.: relationship between genomic structure and the number of IS6501 copies. J. Gen. Microbiol.139:3265–3273. 20.Overduin, P., L. Schouls, P. Roholl, A. van der Zanden, N. Mahmmod, A. Herrewegh, and D. van Soolingen.2004. Use of multilocus variable-number tandem-repeat analysis for typingMycobacterium aviumsubsp. paratubercu-losis. J. Clin. Microbiol.42:5022–5028.
21.Paulsen, I. T., R. Seshadri, K. E. Nelson, J. A. Eisen, J. F. Heidelberg, T. D. Read, R. J. Dodson, L. Umayam, L. M. Brinkac, M. J. Beanan, S. C. Daugherty, R. T. Deboy, A. S. Durkin, J. F. Kolonay, R. Madupu, W. C. Nelson, B. Ayodeji, M. Kraul, J. Shetty, J. Malek, S. E. Van Aken, S. Riedmuller, H. Tettelin, S. R. Gill, O. White, S. L. Salzberg, D. L. Hoover, L. E. Lindler, S. M. Halling, S. M. Boyle, and C. M. Fraser.2002. The Brucella suisgenome reveals fundamental similarities between animal and plant pathogens and symbionts. Proc. Natl. Acad. Sci. USA99:13148–13153. 22.Mazars, E., S. Lesjean, A. L. Banuls, M. Gilbert, V. Vincent, B. Gicquel, M. Tibayrenc, C. Locht, and P. Supply.2001. High-resolution minisatellite-based typing as a portable approach to global analysis ofMycobacterium tuberculosis molecular epidemiology. Proc. Natl. Acad. Sci. USA98:1901–1906. 23.Michaux-Charachon, S., G. Bourg, E. Jumas-Bilak, P. Guigue-Talet, A.
Allardet-Servent, D. O’Callaghan, and M. Ramuz.1997. Genome structure and phylogeny in the genusBrucella. J. Bacteriol.179:3244–3249. 24.Ridler, A. L., M. J. Leyland, S. G. Fenwick, and D. M. West.2005.
Demon-stration of polymorphism amongBrucella ovisfield isolates by pulsed-field gel electrophoresis. Vet. Microbiol.108:69–74.
25.Rotz, L. D., A. S. Khan, S. R. Lillibridge, S. M. Ostroff, and J. M. Hughes.
2002. Public health assessment of potential biological terrorism agents. Emerg. Infect. Dis.8:225–230.
26.Simpson, E. H.1949. Measurement of diversity. Nature163:688. 27.van Belkum, A.1999. The role of short sequence repeats in epidemiologic
typing. Curr. Opin. Microbiol.2:306–311.
28.Whatmore, A. M., T. J. Murphy, S. J. Cutler, and A. P. MacMillan.2004. An assessment of the potential of amplified-fragment length polymorphism (AFLP) for use in identification and typing ofBrucellaisolates, p. 95–99.In P. H. Elzer and K. T. Metodiev (ed.), Risk infections and possibilities for biomedical terrorism. NATO Science Series, IOS Press, Amsterdam, The Netherlands.
29.Whatmore, A. M., T. J. Murphy, S. Shankster, E. Young, S. J. Cutler, and A. P. MacMillan.2005. Use of amplified fragment length polymorphism to identify and typeBrucellaisolates of medical and veterinary interest. J. Clin. Microbiol.43:761–769.
30.Verger, J. M., F. Grimont, P. A. D. Grimont, and M. Grayon.1985.Brucella, a monospecific genus as shown by deoxyribonucleic acid hybridization. Int. J. Syst. Bacteriol.35:292–295.