THE CONTRIBUTION OF FIBRINOGEN AND RED BLOOD CELLS TO ARTERIAL THROMBOSIS
Bethany Lynne Walton
“A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in the Department
of Pathology and Laboratory Medicine in the School of Medicine.”
Chapel Hill 2015
Approved by: Alisa S. Wolberg William B. Coleman
! !
© 2015
! !
ABSTRACT
Bethany Lynne Walton: The Contribution of Fibrinogen and Red Blood Cells to Arterial Thrombosis
(Under the direction of Alisa S. Wolberg)
Cardiovascular disease is the leading cause of death and disability worldwide. This dissertation explores the role of the clotting factor fibrinogen and red blood cells (RBCs) to arterial thrombosis.
Elevated plasma fibrinogen is associated with arterial thrombosis in humans and directly promotes thrombosis in mice, but the contribution of the γA/γ′ fibrinogen isoform to thrombosis is controversial. To determine if γA/γ′ is prothrombotic, we separated γA/γA and γA/γ′ from human plasma and determined the effects on in vitro clot formation and on in vivo
thrombus formation. Both γA/γA and γA/γ′ were cleaved by murine and human thrombin and were incorporated into murine and human clots. When γA/γA or γA/γ′ was spiked into plasma, γA/γA increased the fibrin formation rate to a greater extent than γA/γ′. In mice, compared to controls, γA/γA infusion shortened the time to carotid artery occlusion, whereas γA/γ′ infusion did not. Additionally, γA/γ′ infusion led to lower levels of plasma thrombin–
! !
! !
ACKNOWLEDGEMENTS
I would like to express my gratitude for the Wolberg Laboratory for serving as an excellent place to perform this dissertation work. Alisa has been a wonderful mentor to me and has helped guide me through every step of this research over the past 5 years. Thank you to both current and former lab members for keeping the lab a fun place to work everyday. I have also been fortunate to have the support of faculty, administration, and graduate students within the Pathology Department. Thanks to the members within the UNC Hemostasis and Thrombosis group and our many collaborators for their scientific input and experimental help. Thank you to my committee members, Drs. Alisa Wolberg, Bill Coleman, Brian Cooley, Nigel Key, and Leslie Parise, for their expertise to guide my research. Also, thank you to my funding sources including the UNC Program in Translational Research and the Integrative Vascular Biology program for helping provide the funds needed to complete this dissertation.
Thank you to all my supportive and caring friends in Chapel Hill for filling my life outside the lab with so many great memories. A special thanks to all the pub runners for hours upon hours of running and great conversation. Thanks to BAKTDL for being my best friends since elementary school. I would especially like to thank my amazing family
! !
TABLE OF CONTENTS
LIST OF TABLES……….x
LIST OF FIGURES………..xi
LIST OF ABBREVIATIONS AND SYMBOLS………...xiii
CHAPTER 1: INTRODUCTION: FIBRINOGEN AND RED BLOOD CELLS IN HEMOSTASIS AND THROMBOSIS………..…1
1.1 Introduction………..…1
1.2 Fibrinogen………....2
1.2.1 Fibrinogen structure, fibrin formation and fibrin mechanical properties………...…....2
1.2.2 Fibrinogen and arterial and venous thrombosis………...…….3
1.2.3 Abnormal fibrin structure and stability in thrombosis………..………....3
1.2.4 γ’ fibrinogen and arterial and venous thrombosis …………..………..…4
1.2.5 Fibrinogen interaction with cells and blood proteins………...….8
1.3 Red Blood Cells………...8
1.3.1 RBCs in circulation………...8
1.3.2 Causes of high hematocrit………...9
1.3.3 Hematocrit and arterial and venous thrombosis………9
1.3.4 RBCs mediate blood rheology………...……….13
1.3.5 RBC interaction with fibrinogen……… 13
! !
1.3.7 RBCs influence platelet reactivity………...………...16
1.3.8 RBCs and thrombin generation……….……..16
1.3.9 RBCs alter fibrin structure & stability………...………...19
1.4 Conclusions………....19
1.5 Focus of this Dissertation………...19
REFERENCES………....21
CHAPTER 2: THE FIBRINOGEN γA/γ’ ISOFROM DOES NOT PROMOTE ACUTE ARTERIAL THROMBOSIS IN MICE………32
2.1 Introduction………..……..32
2.2 Materials and Methods………...………34
2.2.1 Proteins and Materials……….…………34
2.2.2 Plasma Preparation……….……….35
2.2.3 Isolation of γA/γA and γA/γ’ fibrinogen...……….35
2.2.4 SDS-PAGE and western blotting………36
2.2.5 Clot formation with purified fibrin(ogen)………...36
2.2.6 Clot formation in plasma………36
2.2.7 Intravital microscopy………..36
2.2.8 FeCl3 thrombosis model……….…….37
2.2.9 Measurement of circulating TAT complexes……….38
2.2.10 Statistical Methods………38
2.3 Results………38
2.3.1 γA/γA fibrinogen increases the fibrin polymerization rate to a greater extent than γA/γ’ fibrinogen……….38
! !
into murine thrombi in vivo……….47
2.3.3 Following FeCl3 injury, γA/γA, but not γA/γ’, fibrinogen shortens the time to artery occlusion……….49
2.3.4 Following FeCl3 injury mice infused with γA/γ’ fibrinogen have lower circulating TAT complexes than mice infused with γA/γA fibrinogen………51
2.4 Discussion………..53
REFERENCES………..………..59
CHAPTER 3: ELEVATED HEMATOCRIT PROMOTES ARTERIAL THROMBOSIS INDEPENDENT OF THROMBIN GENERATION………64
3.1 Introduction……….……64
3.2 Materials and Methods………66
3.2.1 Proteins and materials……….…66
3.2.2 Phlebotomy……….67
3.2.3 Reconstituted whole blood model……….…..67
3.2.4 Thrombin generation in whole blood……….….…68
3.2.5 Mouse model of elevated hematocrit………..…68
3.2.6 Blood smears………...…69
3.2.7 Flow cytometry………...69
3.2.8 Blood viscosity measurements………....70
3.2.9 FeCl3 model of arterial thrombosis………...…..…70
3.2.10 Measurement of circulating thrombin-antithrombin (TAT) complexes ………..…71
3.2.11 Histology………...…71
! !
3.3 Results……….72
3.3.1 The RBC increase in thrombin generation in whole blood is dependent on platelet concentration………72
3.3.2 RBC infusion raises hematocrit in mice without altering RBC morphology or PS-exposure………..75
3.3.3 Compared to controls, RBCHIGH mice have a faster time to arterial occlusion………...77
3.3.4 In vivo thrombin generation did not differ between control and RBCHIGH mice……….79
3.3.5 Thrombi from control and RBCHIGH mice do not differ in size or fibrin content……….…..…81
3.3.6 PF4 levels did not differ between control and RBCHIGH mice…………83
3.4 Discussion………..…….85
REFERENCES………..………..90
CHAPTER 4: SUMMARY AND FUTURE DIRECTIONS………..…95
4.1 Summary………....95
4.2 Future Directions……….…..95
! !
LIST OF TABLES
Table 1.1 Epidemiological studies are conflicted on the association
between hematocrit and arterial thrombosis………....11 Table 1.2 Epidemiological studies are conflicted on the
association between hematocrit and venous thrombosis……….……12 Table 2.1 Polymerization of purified fibrinogen isoforms by human
! !
LIST OF FIGURES
Figure 1.1 The fibrinogen γ chain undergoes alternative processing to
form the γA and γ’ isoforms………..…7 Figure 1.2 RBCs marginate platelets toward the vessel wall under
arterial shear………...…………..14 Figure 1.3 A portion of RBCs express PS on their cell
membrane.………18 Figure 2.1 Purified fibrinogen contains all three chains
(Aα, Bβ, and γA and/or γ’) at the expected molecular weights
and is equally cleaved by human and mouse thrombin …………...……...…40 Figure 2.2 Both γA/γA and γA/γ’ fibrinogen accelerate clotting in human
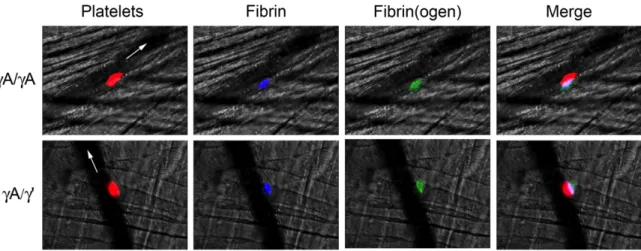
and mouse plasma………...…44 Figure 2.3 Intravital microscopy shows both γA/γA and γA/γ’ isoforms
are incorporated into murine thrombi………..……48 Figure 2.4 γA/ γA fibrinogen shortens the time to vessel occlusion after
arterial injury, but γA/γ’ does not………50 Figure 2.5 Following arterial injury, mice infused with γA/γ’ fibrinogen
have reduced circulating TAT complexes………...52 Figure 2.6 γA/γ’ fibrinogen binds thrombin, resulting in lower
thrombin-antithrombin levels and similar TTO as controls ………...55 Figure 3.1 The RBC effect on thrombin generation
is dependent on platelet concentration………...74 Figure 3.2 RBC transfusion raises hematocrit in recipient mice………...76 Figure 3.3 RBCHIGH mice have a shortened time to vessel occlusion following
FeCl3 injury to the carotid artery, ………..78 Figure 3.4 Thrombin-antithrombin complexes are similar in RBCHIGH mice
and controls following FeCl3 injury……….………80
Figure 3.5 Thrombi from control and RBCHIGH mice have similar
! !
Figure 4.1 Conceptual model showing influence of RBCs on thrombus
! !
LIST OF ABBREVIATIONS AND SYMBOLS
α Alpha
AU Arbitrary Units
β Beta
BSA Bovine Serum Albumin °C Degrees Celsius
CaCl2 Calcium Chloride
CAT Calibrated Automated Thrombography CGS Sodium citrate, Glucose, Sodium Chloride CHD Coronary Heart Disease
CVD Cardiovascular disease DVT Deep Vein Thrombosis
ELISA Enzyme-Linked Immunosorbent Assay FeCl3 Ferric Chloride
FXIII Factor XIII
γ Gamma
γ’ Gamma Prime
g Gram
H&E Hematoxylin and Eosin HBS HEPES Buffered Saline
hNPP Human Normal Pooled Plasma
Hz Hertz
! !
ICAM Intercellular Adhesion Molecule IVC Inferior Vena Cava
K Thousand
KCl Potassium Chloride
L Liters
µL Microliter µM Micromolar
M Molar
mg Milligram
MgSO4 Magnesium Sulfate Min Minute
MI Myocardial infarction mL Milliliter
mM Millimolar
mNPP Murine Normal Pooled Plasma NaCl Sodium Chloride
nm Nanometer
! !
PRP Platelet-rich Plasma PS Phosphatidylserine PV Polycythemia Vera RBCs Red Blood Cells
RBCHIGH Mice with Elevated Hematocrit SCD Sickle Cell Disease
SD Standard Deviation SE Standard Error
SEM Standard Error of the Mean TAT Thrombin-antithrombin TF Tissue Factor
tPA Tissue-type Plasminogen Activator TTO Time to Occlusion
VTE Venous Thromboembolism WBC White Blood Cell
Chapter 1: Introduction: Fibrinogen and Red Blood Cells in Hemostasis and Thrombosis1
1.1 Introduction
Arterial thrombosis is a leading cause of death and disability worldwide. Arterial
thrombosis is usually initiated following rupture of an atherosclerotic plaque. This causes the
formation of thrombi that may become occlusive and cause ischemic damage to the
surrounding tissues. Intracardiac thrombosis may also occur due to atrial fibrillation or the
presence of a mechanical valve [1]. Arterial thrombi are usually termed “white-thrombi” due
to their high platelet count and efforts to understand the pathogenesis of arterial thrombosis
have mainly focused on platelets. However growing evidence suggests that the plasma
protein fibrin(ogen) and RBCs may also be involved in the development of arterial thrombi.
Venous thrombosis is initiated by endothelial dysfunction and inappropriate
expression of plasma and cellular procoagulant activity under low blood flow/stasis
(so-called Virchow’s Triad). The epidemiology, risk factors, and treatment of venous thrombosis
have been recently reviewed in [2]. However, the pathophysiologic mechanisms that
contribute to thrombus formation, composition, and stability are still poorly understood.
Clues may be found in the distinctive appearance of venous thrombi, which demonstrate
regions of high RBC and fibrin content (so-called “red thrombi”). Notably, RBCs can be
found between layers of fibrin in a “brick and mortar” construction, where they lose their
typical discoid shape and acquire a compressed morphology (so-called “polyhedrocytes”) [3].
These observations suggest RBCs and fibrin(ogen) interact during venous thrombosis, and
that thrombi undergo substantial consolidation during their maturation.
Herein, I will review the contributions of fibrinogen and RBCs to coagulation, and
provide evidence supporting their potential roles in both arterial and venous thrombosis.
1.2 Fibrinogen
1.2.1 Fibrinogen structure, fibrin formation, and fibrin mechanical properties.
The fibrinogen molecule consists of 2 sets each of 3 polypeptide chains (AαBβγ)2.
During coagulation, thrombin cleaves N-terminal peptides from the Aα- and Bβ-chains
promoting the formation of protofibrils and subsequently, fibrin fibers. Branching results in
the characteristic fibrin network seen in micrographs of purified and plasma clots.
Fibrinogen circulates at high concentrations (2-4 mg/mL) in plasma, and levels may increase
further during inflammation. The concentrations of thrombin and fibrinogen present during
clot formation influence fibrin network structure and stability. For example, clots formed in
the presence of high thrombin or fibrinogen concentrations have increased fibrin network
density and resistance to fibrinolysis compared to clots formed under normal conditions.
These processes have been previously reviewed [4, 5].
Crosslinked fibrin is also known for its ability to stabilize clots. This property is
determined at both micro- and macro-scales. Individual fibrin fibers have astounding
viscoelasticity. Crosslinked fibrin fibers can be stretched to 2.5-times their original length
of fibers from elongations up to 100% can occur within milliseconds [9]. Branchpoints within the fibrin network are surprisingly strong. When strained, individual fibers fail before branchpoints fail [10]. Thus, it is not surprising that fully-formed fibrin clots have similar extensibility and elasticity as individual fibers.
1.2.2 Fibrinogen and arterial and venous thrombosis.
Elevated total fibrinogen is correlated with increased arterial thrombosis [11-15] and increased venous thrombosis risk [16-20], and risk is concentration-dependent and present in both men and women. Studies using transgenic mice and murine infusion models have associated elevated fibrinogen with increased prothrombotic biomarkers (e.g., D-dimer) [21] and a shorter time to vessel occlusion and increased thrombus fibrin content [22]. Moreover, compared to control mice, thrombi in fibrinogen-infused mice also show increased resistance to fibrinolysis [22]. These findings suggest hyperfibrinogenemia is not merely a biomarker of thrombosis risk, but is causative in arterial and venous thrombosis etiology.
1.2.3 Abnormal fibrin structure and stability in thrombosis.
Several studies have reported abnormal fibrin structure and/or stability in both arterial and venous thrombosis, even when circulating fibrinogen levels are normal. For example, compared to controls, patients with a history of MI produced clots with shorter fibrin fibers with increased stiffness, and an increased resistance to lysis [23, 24]. Additionally,
fibrin network density, reduced permeability, and increased lysis times[27]. Interestingly, compared to plasma clots from patients with deep vein thrombosis, clots from patients that experienced pulmonary embolism are less compact and more susceptible to fibrinolysis [27, 28]. In total, these data suggest abnormal fibrin network structure and stability contribute to arterial and venous thrombosis.
1.2.4 Fibrinogen γ’-chain and arterial and venous thrombosis.
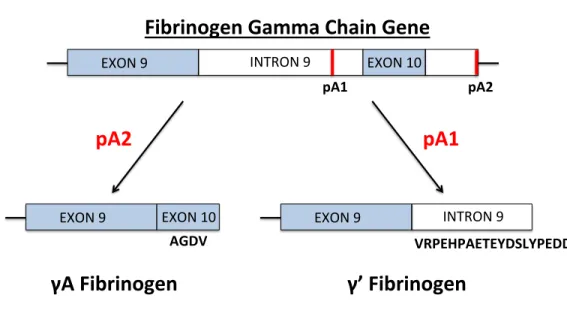
The fibrinogen γ-chain can undergo alternative splicing, leading to replacement of 4 C-terminal amino acids with a unique 20 amino acid sequence (γ’; Figure 1.1). The γ’-chain is present in 8-15% of fibrinogen molecules (as γA/γ’) in healthy individuals. The genes encoding the fibrinogen chains are co-regulated to maintain the level of fibrinogen in
circulation (reviewed in [29]). However, the levels of the γA- and γ’-chains are mediated by independent mechanisms that differentially regulate their expression. Expression of γ’-containing fibrinogen is disproportionally increased by interleukin-6-dependent
inflammatory responses [30], suggesting an independent relationship between the γ’-chain, inflammation, and thrombosis. Accordingly, although total fibrinogen levels are positively correlated with thrombosis risk, the fraction of circulating γ’-fibrinogen (γ’/total fibrinogen ratio) modulates risk independently of the total fibrinogen level. Notably, an elevated γ’-to-total fibrinogen ratio is associated with increased risk of arterial thrombosis in numerous epidemiological studies [31-36], suggesting γ’ may be driving arterial thrombosis.
Determining the operant mechanisms has been difficult because γA/γ’ fibrinogen has
both procoagulant and antithrombotic properties (reviewed in [39]). Briefly, compared to
γA/γA clots, clots that contain γ’ fibrinogen have a denser network of thin fibrin fibers,
reduced permeability, reduced plasminogen binding, and increased resistance to fibrinolysis.
The γ’-chain can also bind and sequester thrombin, protecting it from inactivation by
antithrombin. These properties are consistent with prothrombotic functions. However, γ’
fibrinogen also exhibits impaired polymerization. Recent studies have shown that a γ’
carboxyl-terminal peptide reduces plasma thrombin generation even in the presence of
anti-factor VIII antibody, suggesting γ’/thrombin interactions reduce anti-factor V activation [40]. By
reducing thrombin generation, this peptide also increases the sensitivity of coagulation to
activated protein C, thus augmenting endogenous anticoagulant mechanisms [41].
Studies to determine the contribution of the γ’-chain to thrombosis in vivo have
consistently demonstrated antithrombotic effects. Transgenic expression of the human
γ’-chain reduces venous thrombus volume in mice that are heterozygous for the factor V Leiden
mutation [42]. A peptide mimicking the γ’-chain C-terminus inhibits fibrin-rich thrombus
formation in a baboon model of thrombosis [43]. We recently infused mice with identical
levels of either γA/γA or γA/γ’ fibrinogen isolated from human plasma [44]. Compared to
controls, γA/γA infusion shortens the time to carotid artery occlusion, whereas γA/γ’ infusion
does not. Additionally, γA/γ’ infusion reduces levels of circulating thrombin-antithrombin
complexes. These data are consistent with the premise that the γ’-chain reduces thrombin
activity. By extension, these data implicate the γA-chain as the prothrombotic mediator in
Figure 1.1: The fibrinogen γ chain undergoes alternative processing to form the γA and γ’ isoforms. The fibrinogen γ-chain mRNA transcript may undergo splicing at two main polyadenylation sites. The γA-chain forms when polyadenylation (pA) occurs downstream of exon 10 (pA2), leading to translation of exon 10 and the formation of a γ-chain with 10 exons, 9 introns, and 411 amino acids (ending in AGDV). The γ’-chain forms when
polyadenylation occurs upstream of exon 10 (pA1), forming a γ-chain which includes intron 9. This results in the translation of an extra 20 amino from intron 9
(VRPEHPAETRYDSLYPEDDL), forming a γ-chain with 427 amino acids.
EXON%9% INTRON%9% EXON%10%
pA2$
EXON%9% EXON%10% AGDV$
pA1$
Fibrinogen$Gamma$Chain$Gene$
γA$Fibrinogen$
pA2$
VRPEHPAETEYDSLYPEDDL$ EXON%9% INTRON%9%
pA1$
1.2.5 Fibrin(ogen) interactions with cells and blood proteins.
Most studies of fibrin(ogen) function have used purified systems or plasmas. These studies have identified binding sites on fibrin(ogen) for soluble proteins involved in clot formation, stabilization, and fibrinolysis, including thrombin, FXIII, fibronectin, tissue-type plasminogen activator (tPA), plasminogen, and plasmin [45-49]. Notably, however,
fibrin(ogen) also interacts with cells and these interactions may contribute to the
incorporation of cells into venous thrombi. For example, fibrin(ogen) contains recognition sequences for integrins including αMβ2, αIIbβ3, αVβ3, which mediate fibrin(ogen)
interactions with leukocytes, platelets, and endothelial cells, respectively [50]. These interactions modulate leukocyte function, platelet aggregation and clot retraction, and may anchor thrombi to the endothelium. Fibrin(ogen) also binds to RBCs, which influences both the erythrocyte sedimentation rate and blood viscosity (discussed below) [51-53].
1.3 Red Blood Cells
1.3.1 RBCs in circulation.
1.3.2 Causes of high hematocrit.
The normal range of RBCs in blood (hematocrit) is 41-46% in men and 36-44% in women, and can be influenced by a number of physiologic and pathologic situations. In high altitude, the bone marrow increases RBC production to compensate for decreased oxygen saturation [62]. RBC levels may also increase in disease states such as polycythemia vera (PV) [63] and as a result of increased erythropoietin, either through exogenous erythropoietin use or abnormal erythropoietin production by certain types of tumors [64]. Conditions associated with hypoxia, such as smoking, lung disease, and heart disease, are also associated with increased RBC production [64].
1.3.3 Hematocrit in arterial and venous thrombosis.
Thrombosis is a common complication in patients with PV, with arterial thrombosis including MI and cerebrovascular events making up the majority of all thrombotic events in these patients [65]. The Cytoreductive Therapy in Polycythemia Vera (CYTO-PV), a large-scale, multicenter, prospective, randomized clinical trial compared maintaining hematocrit <45% or between 45-50% in patients with PV [66]. Compared to maintaining a hematocrit <45%, maintaining hematocrit in the higher range was associated with four times the rate of death from CVD and major thrombosis suggesting hematocrit is a cause for thrombosis in PV. However, this study did not control for patients taking hydroxyurea which complicates the interpretation of these results.
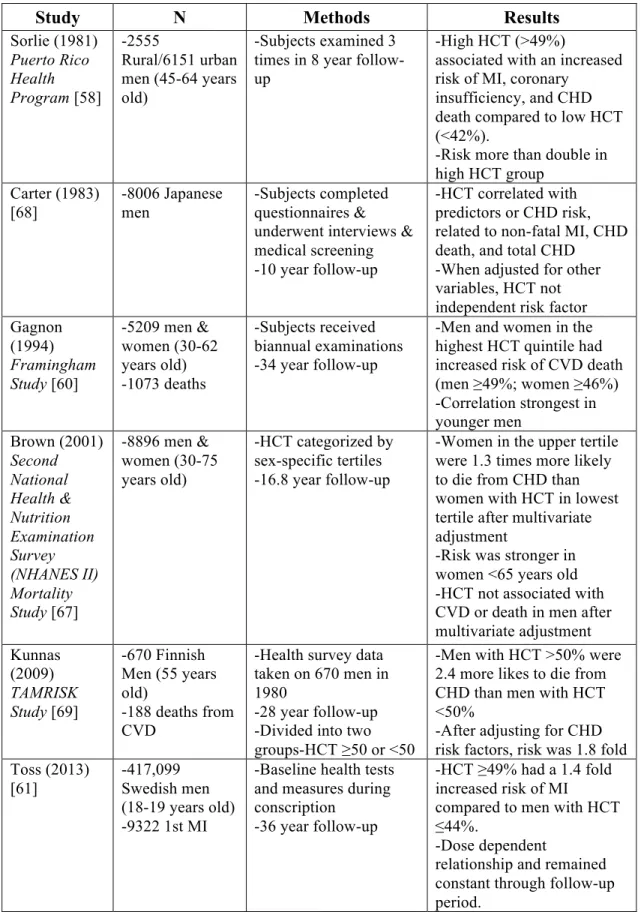
Table 1.1 Epidemiological studies are conflicted on the association between hematocrit and arterial thrombosis
Study N Methods Results
Sorlie (1981) Puerto Rico Health Program [58] -2555 Rural/6151 urban men (45-64 years old)
-Subjects examined 3 times in 8 year follow-up
-High HCT (>49%)
associated with an increased risk of MI, coronary
insufficiency, and CHD death compared to low HCT (<42%).
-Risk more than double in high HCT group
Carter (1983) [68] -8006 Japanese men -Subjects completed questionnaires & underwent interviews & medical screening -10 year follow-up
-HCT correlated with predictors or CHD risk, related to non-fatal MI, CHD death, and total CHD
-When adjusted for other variables, HCT not independent risk factor Gagnon
(1994) Framingham Study [60]
-5209 men & women (30-62 years old) -1073 deaths
-Subjects received biannual examinations -34 year follow-up
-Men and women in the highest HCT quintile had increased risk of CVD death (men ≥49%; women ≥46%) -Correlation strongest in younger men Brown (2001) Second National Health & Nutrition Examination Survey (NHANES II) Mortality Study [67]
-8896 men & women (30-75 years old)
-HCT categorized by sex-specific tertiles -16.8 year follow-up
-Women in the upper tertile were 1.3 times more likely to die from CHD than women with HCT in lowest tertile after multivariate adjustment
-Risk was stronger in women <65 years old -HCT not associated with CVD or death in men after multivariate adjustment Kunnas (2009) TAMRISK Study [69] -670 Finnish Men (55 years old)
-188 deaths from CVD
-Health survey data taken on 670 men in 1980
-28 year follow-up -Divided into two groups-HCT ≥50 or <50
-Men with HCT >50% were 2.4 more likes to die from CHD than men with HCT <50%
-After adjusting for CHD risk factors, risk was 1.8 fold Toss (2013)
[61]
-417,099 Swedish men (18-19 years old) -9322 1st MI
-Baseline health tests and measures during conscription
-36 year follow-up
-HCT ≥49% had a 1.4 fold increased risk of MI
compared to men with HCT ≤44%.
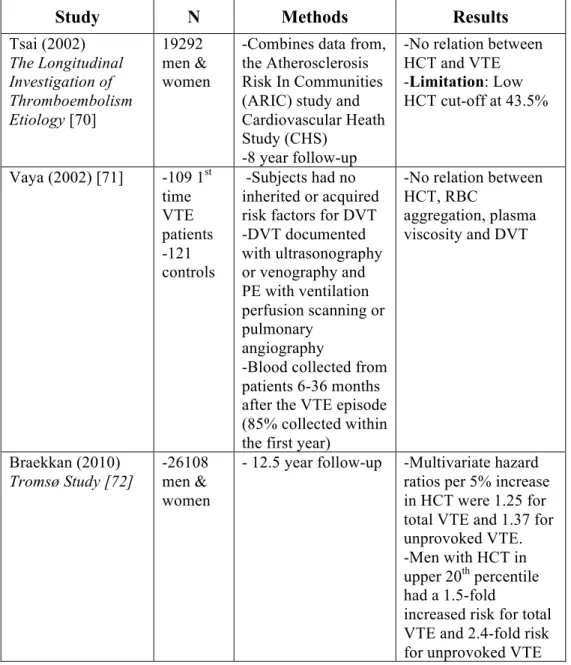
Table 1.2 Epidemiological studies are conflicted on the association between hematocrit and venous thrombosis
Study N Methods Results
Tsai (2002) The Longitudinal Investigation of Thromboembolism Etiology [70]
19292 men & women
-Combines data from, the Atherosclerosis Risk In Communities (ARIC) study and Cardiovascular Heath Study (CHS)
-8 year follow-up
-No relation between HCT and VTE -Limitation: Low HCT cut-off at 43.5%
Vaya (2002) [71] -109 1st time VTE patients -121 controls
-Subjects had no inherited or acquired risk factors for DVT -DVT documented with ultrasonography or venography and PE with ventilation perfusion scanning or pulmonary
angiography
-Blood collected from patients 6-36 months after the VTE episode (85% collected within the first year)
-No relation between HCT, RBC
aggregation, plasma viscosity and DVT
Braekkan (2010) Tromsø Study [72]
-26108 men & women
- 12.5 year follow-up -Multivariate hazard ratios per 5% increase in HCT were 1.25 for total VTE and 1.37 for unprovoked VTE. -Men with HCT in upper 20th percentile had a 1.5-fold
1.3.4 RBCs mediate blood rheology.
Hemorheology is the study of how flowing blood influences hemostasis. RBCs are the major determinant of blood rheology because of their prevalence, size, deformability, and ability to undergo reversible aggregation. Under high shear in the arterial circulation,
(typically 500-1500 s-1), RBCs promote platelet flux toward the vessel wall (so-called platelet margination), which increases the frequency of platelet-endothelial cell interactions, and platelet-platelet interactions which promotes platelet adhesion, activation, and
aggregation (Figure 1.2) [73]. However, under low shear in the venous circulation (typically 10-100 s-1), RBCs increase blood viscosity via their tendency to aggregate (rouleaux
formation). Increased blood viscosity is a risk factor for arterial and venous thrombosis [74-76]. Notably, RBC aggregation is mediated by plasma proteins including fibrinogen [51-53]. Consequently, inflammatory processes that increase fibrinogen levels also increase blood viscosity. These effects have been implicated in the association between elevated hematocrit and hyperfibrinogenemia with thrombosis. However, it remains unclear whether this
Figure 1.2: RBCs marginate platelets toward the vessel wall under arterial shear. RBCs dominate the rheology of blood due to their high number, large size, and deformability. Under the high shear rates present in arteries, RBCs move toward the center of the blood vessel. This marginates platelets towards the arterial walls, which promotes increased platelet-platelet interactions and platelet-endothelial interactions.
RBC$ Platelets$
Blood$Flow$
Smooth$Muscle$Cell$ Endothelial$Cell$
1.3.5 RBCs interact with fibrin(ogen).
RBCs interact specifically with fibrin(ogen) and the fibrinogen motif that mediates
RBC interactions involve fibrinogen Aα-chain residues 207-303 [51]. Two potential RBC
receptors have been implicated in this interaction. Fibrinogen-RBC interactions can be
inhibited by the integrin-blocking molecule eptifibatide and are not supported by RBCs
lacking β3 isolated from patients with Glanzmann thrombasthenia [77], implicating β3 or a
β3-like molecule on the RBC surface. However, that study did not rule out the possibility that RBC-bound platelets mediate this interaction [77]. Fibrinogen-RBC interactions can also be
blocked with an antibody against the integrin-associated protein CD47 [78]. Since CD47 was
originally identified for its interaction with αvβ3, αIIbβ3, and α2β1 integrins, it is possible
that the RBC binding site comprises a complex with both of these molecules. It is
interesting to speculate that that blocked fibrin(ogen)-RBC interactions may reduce whole
blood viscosity and thus thrombosis risk.
1.3.6 RBCs interact with cells.
RBCs can interact with leukocytes, platelets, and endothelial cells. For example,
RBC ICAM-4 can bind leukocyte β1 and β2 integrins [79, 80] and platelet αIIbβ3 [81]. RBC
ICAM-4 also interacts with integrin αv [82]. RBCs are the first cells to adhere to ferric
chloride (FeCl3)-treated, intact arterial endothelium, prior to the arrival of platelets [83]. This
interaction is not dependent on von Willebrand factor or GPIbα. However, the molecular
receptors on RBCs and the endothelium that mediate this interaction have not been identified
[84], suggesting stage-specific receptors may decorate RBCs during differentiation and
further refine these interactions.
1.3.7 RBCs influence platelet reactivity.
RBCs have been shown to alter the biochemical responsiveness and functional
responsiveness of activated platelets [85]. Silvain et al. [86] showed that RBCs increase
ADP-induced platelet activation and aggregation in vitro in blood from healthy volunteers.
The same group went on to show that patients who received RBC transfusions displayed
increased ADP-induced platelet reactivity [87]. Moreover, it was specifically shown that
RBCs amplify platelet activation and degranulation by increasing platelet serotonin release,
increasing enzymatic ADP removal, and inhibiting proteases [85]. The increase in platelet
reactivity by RBCs could not be decreased by aspirin, suggesting that RBC level influences
therapeutic effect of aspirin. RBCs have also been shown to enhance the activation of αIIbβ3
and P-selectin on platelets, suggesting RBCs increase platelet activation and aggregation [88].
In sum, these studies suggest that RBCs contribute to thrombus formation by increasing
platelet reactivity.
1.3.8 RBCs support thrombin generation.
A small percentage (~0.5%) of RBCs circulate with exposed phosphotidylserine (PS)
on their outer membranes [89], suggesting RBCs can assemble prothrombinase complexes
and support thrombin generation (Figure 1.3). Interestingly, although levels of both
PS-positive RBCs and platelets are elevated in patients with sickle cell disease (SCD) genotypes,
including F1.2 and D-dimer [89]. This finding suggests PS-positive RBCs are the primary cell responsible for thrombophilia in SCD. In vitro studies support this premise; when added to platelet-poor plasma, RBCs shorten the lag time and increase the peak of thrombin
generation similar to that seen with platelets [90, 91], although in contrast to platelets, thrombin generation on RBCs occurs through the meizothrombin pathway [92]. RBCs can
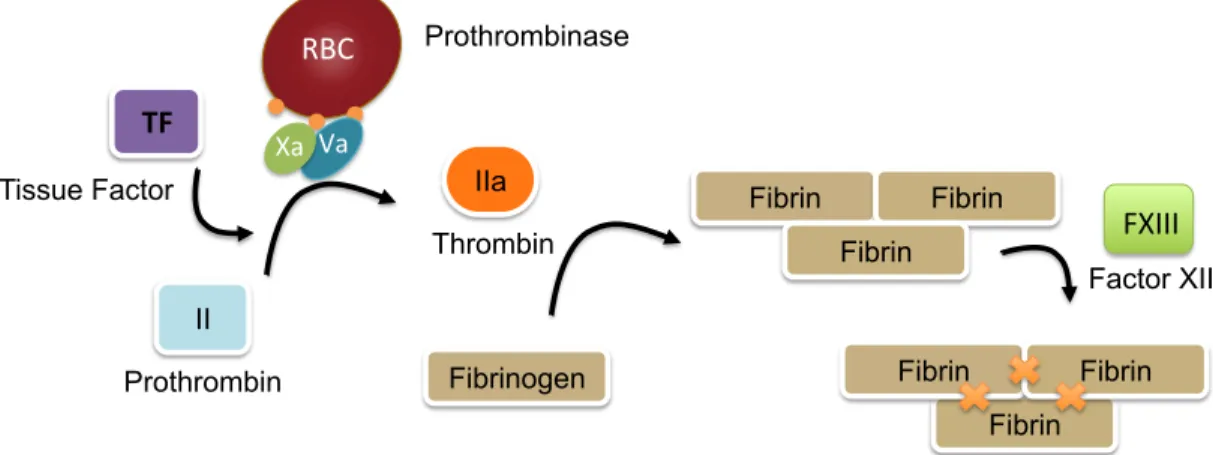
Figure 1.3: A portion of RBCs express PS on their cell membrane. A small percentage (0.5%) of RBCs express PS exposure on their cell surface. This surface allows for
prothrombinase complex assembly, prothrombin conversion to thrombin, and fibrin formation. Fibrin can then be crosslinked by the transglutaminase factor XIII, to form the structural support for a clot.
II
Prothrombin
IIa
Thrombin Xa# Va#
Prothrombinase
Fibrinogen
Fibrin Fibrin
Fibrin TF#
Tissue Factor
Fibrin Fibrin Fibrin RBC#
1.3.9 RBCs alter fibrin structure and stability.
RBCs alter fibrin network structure [96, 97] and reduce fibrin network permeability [98]. RBCs also suppress plasmin generation and reduce clot dissolution [97]. In the
presence of the substantial contractile forces induced by platelets during clot retraction [99, 100], RBCs are dramatically compressed, which further reduces clot permeability and
restricts access of fibrinolytic enzymes to the clot [3, 101]. Importantly, this phenomenon was noted in thrombi harvested from the arterial vasculature [3] but is also likely to have a significant impact on venous thrombosis, since these thrombi contain platelets and large
numbers of RBCs. These data suggest reducing RBC content in thrombi may increase clot dissolution, thus reducing thrombosis.
1.4 Conclusions
Both fibrinogen and RBCs are essential components of blood and major players in
coagulation. Continued studies are needed to delineate the pathophysiologic mechanisms that mediate the roles of both fibrinogen and RBCs in thrombosis. Specifically, identifying
the specific fibrinogen isoform that promotes thrombosis and knowledge of the role of elevated hematocrit in thrombosis will provide new insight into mechanisms that drive abnormal clot formation. Additionally, identification of the RBC-receptor that binds
fibrinogen may provide new therapeutic targets to prevent thrombosis.
1.5 Focus of this dissertation
total fibrinogen (combined γA/γA and γA/γ’) and previous work on RBCs in arterial
thrombosis has been performed in murine models with co-morbidities. Therefore, this
dissertation will specifically focus on i) the effect of the γA/γA and γA/γ’ individually to
arterial thrombosis, and ii) the influence of elevated hematocrit to arterial thrombosis in a
RBCs transfusion model of elevated hematocrit. Knowledge on how fibrinogen isoforms and
RBCs contribute to thrombosis may help elucidate mechanisms involved in the formation of
arterial thrombi, reveal biomarkers to predict thrombosis, and provide clues into the best
REFERENCES
1 Previtali E, Bucciarelli P, Passamonti SM, Martinelli I. Risk factors for venous and arterial thrombosis. Blood Transfus. 2011; 9: 120-38.
2 Wolberg AS, Rosendaal FR, Weitz JI, Jaffer IH, Agnelli G, Baglin T, Mackman N. Venous Thrombosis. Nat Rev Dis Pri, In Press. 2015.
3 Cines DB, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov RI, Rauova L, Lowery TJ, Weisel JW. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. 2013.
4 Lord ST. Molecular mechanisms affecting fibrin structure and stability. Arterioscler Thromb Vasc Biol. 2011; 31: 494-9.
5 Wolberg AS. Thrombin generation and fibrin clot structure. Blood Rev. 2007; 21: 131-42.
6 Collet JP, Shuman H, Ledger RE, Lee S, Weisel JW. The elasticity of an individual fibrin fiber in a clot. Proc Natl Acad Sci U S A. 2005; 102: 9133-7.
7 Liu W, Carlisle CR, Sparks EA, Guthold M. The mechanical properties of single fibrin fibers. J Thromb Haemost. 2010; 8: 1030-6.
8 Liu W, Jawerth LM, Sparks EA, Falvo MR, Hantgan RR, Superfine R, Lord ST, Guthold M. Fibrin fibers have extraordinary extensibility and elasticity. Science. 2006; 313: 634.
9 Hudson NE, Ding F, Bucay I, O'Brien ET, 3rd, Gorkun OV, Superfine R, Lord ST, Dokholyan NV, Falvo MR. Submillisecond elastic recoil reveals molecular origins of fibrin fiber mechanics. Biophys J. 2013; 104: 2671-80.
11 Lindahl B, Toss H, Siegbahn A, Venge P, Wallentin L. Markers of myocardial damage and inflammation in relation to long-term mortality in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. N Engl J Med. 2000; 343: 1139-47.
12 Wilhelmsen L, Svardsudd K, Korsan-Bengtsen K, Larsson B, Welin L, Tibblin G. Fibrinogen as a risk factor for stroke and myocardial infarction. N Engl J Med. 1984; 311: 501-5.
13 Acevedo M, Pearce GL, Kottke-Marchant K, Sprecher DL. Elevated fibrinogen and homocysteine levels enhance the risk of mortality in patients from a high-risk preventive cardiology clinic. Arterioscler Thromb Vasc Biol. 2002; 22: 1042-5.
14 Kannel WB, Wolf PA, Castelli WP, D'Agostino RB. Fibrinogen and risk of cardiovascular disease. The Framingham Study. JAMA. 1987; 258: 1183-6.
15 Toss H, Lindahl B, Siegbahn A, Wallentin L. Prognostic influence of increased fibrinogen and C-reactive protein levels in unstable coronary artery disease. FRISC Study Group. Fragmin during Instability in Coronary Artery Disease. Circulation. 1997; 96: 4204-10.
16 Koster T, Rosendaal FR, Reitsma PH, van der Velden PA, Briet E, Vandenbroucke JP. Factor VII and fibrinogen levels as risk factors for venous thrombosis. A case-control study of plasma levels and DNA polymorphisms--the Leiden Thrombophilia Study (LETS). Thromb Haemost. 1994; 71: 719-22.
17 Haverkate F, Samama M. Familial dysfibrinogenemia and thrombophilia. Report on a study of the SSC Subcommittee on Fibrinogen. Thromb Haemost. 1995; 73: 151-61.
18 Austin H, Hooper WC, Lally C, Dilley A, Ellingsen D, Wideman C, Wenger NK, Rawlins P, Silva V, Evatt B. Venous thrombosis in relation to fibrinogen and factor VII genes among African-Americans. J Clin Epidemiol. 2000; 53: 997-1001.
19 Kamphuisen PW, Eikenboom JC, Vos HL, Pablo R, Sturk A, Bertina RM, Rosendaal FR. Increased levels of factor VIII and fibrinogen in patients with venous thrombosis are not caused by acute phase reactions. Thromb Haemost. 1999; 81: 680-3.
21 Kerlin B, Cooley BC, Isermann BH, Hernandez I, Sood R, Zogg M, Hendrickson SB, Mosesson MW, Lord S, Weiler H. Cause-effect relation between
hyperfibrinogenemia and vascular disease. Blood. 2004; 103: 1728-34.
22 Machlus KR, Cardenas JC, Church FC, Wolberg AS. Causal relationship between hyperfibrinogenemia, thrombosis, and resistance to thrombolysis in mice. Blood. 2011; 117: 4953-63.
23 Collet JP, Allali Y, Lesty C, Tanguy ML, Silvain J, Ankri A, Blanchet B, Dumaine R, Gianetti J, Payot L, Weisel JW, Montalescot G. Altered fibrin architecture is
associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol. 2006; 26: 2567-73.
24 Undas A, Zalewski J, Krochin M, Siudak Z, Sadowski M, Pregowski J, Dudek D, Janion M, Witkowski A, Zmudka K. Altered plasma fibrin clot properties are
associated with in-stent thrombosis. Arterioscler Thromb Vasc Biol. 2010; 30: 276-82.
25 Undas A, Slowik A, Wolkow P, Szczudlik A, Tracz W. Fibrin clot properties in acute ischemic stroke: relation to neurological deficit. Thromb Res. 2010; 125: 357-61.
26 Undas A, Podolec P, Zawilska K, Pieculewicz M, Jedlinski I, Stepien E, Konarska-Kuszewska E, Weglarz P, Duszynska M, Hanschke E, Przewlocki T, Tracz W. Altered fibrin clot structure/function in patients with cryptogenic ischemic stroke. Stroke. 2009; 40: 1499-501.
27 Undas A, Zawilska K, Ciesla-Dul M, Lehmann-Kopydlowska A, Skubiszak A, Ciepluch K, Tracz W. Altered fibrin clot structure/function in patients with idiopathic venous thromboembolism and in their relatives. Blood. 2009; 114: 4272-8.
28 Martinez MR, Cuker A, Mills AM, Crichlow A, Lightfoot RT, Chernysh IN, Nagaswami C, Weisel JW, Ischiropoulos H. Enhanced lysis and accelerated
establishment of viscoelastic properties of fibrin clots are associated with pulmonary embolism. Am J Physiol Lung Cell Mol Physiol. 2014; 306: L397-404.
31 Lovely RS, Falls LA, Al-Mondhiry HA, Chambers CE, Sexton GJ, Ni H, Farrell DH. Association of gammaA/gamma' fibrinogen levels and coronary artery disease. Thromb Haemost. 2002; 88: 26-31.
32 Mannila MN, Lovely RS, Kazmierczak SC, Eriksson P, Samnegard A, Farrell DH, Hamsten A, Silveira A. Elevated plasma fibrinogen gamma' concentration is associated with myocardial infarction: effects of variation in fibrinogen genes and environmental factors. J Thromb Haemost. 2007; 5: 766-73.
33 Cheung EY, Vos HL, Kruip MJ, den Hertog HM, Jukema JW, de Maat MP. Elevated fibrinogen gamma' ratio is associated with cardiovascular diseases and acute phase reaction but not with clinical outcome. Blood. 2009; 114: 4603-4.
34 Cheung EY, Uitte de Willige S, Vos HL, Leebeek FW, Dippel DW, Bertina RM, de Maat MP. Fibrinogen gamma' in ischemic stroke: a case-control study. Stroke. 2008;
39: 1033-5.
35 van den Herik EG, Cheung EY, de Lau LM, den Hertog HM, Leebeek FW, Dippel DW, Koudstaal PJ, de Maat MP. gamma'/total fibrinogen ratio is associated with short-term outcome in ischaemic stroke. Thromb Haemost. 2011; 105: 430-4.
36 Drouet L, Paolucci F, Pasqualini N, Laprade M, Ripoll L, Mazoyer E, Bal dit Sollier C, Vanhove N. Plasma gamma'/gamma fibrinogen ratio, a marker of arterial
thrombotic activity: a new potential cardiovascular risk factor? Blood Coagul Fibrinolysis. 1999; 10 Suppl 1: S35-9.
37 Uitte de Willige S, de Visser MC, Houwing-Duistermaat JJ, Rosendaal FR, Vos HL, Bertina RM. Genetic variation in the fibrinogen gamma gene increases the risk for deep venous thrombosis by reducing plasma fibrinogen gamma' levels. Blood. 2005;
106: 4176-83.
38 Nowak-Gottl U, Weiler H, Hernandez I, Thedieck S, Seehafer T, Schulte T, Stoll M. Fibrinogen alpha and gamma genes and factor VLeiden in children with
thromboembolism: results from 2 family-based association studies. Blood. 2009; 114: 1947-53.
40 Omarova F, Uitte De Willige S, Ariens RA, Rosing J, Bertina RM, Castoldi E. Inhibition of thrombin-mediated factor V activation contributes to the anticoagulant activity of fibrinogen gamma'. J Thromb Haemost. 2013; 11: 1669-78.
41 Omarova F, Uitte de Willige S, Simioni P, Ariens RA, Bertina RM, Rosing J, Castoldi E. Fibrinogen gamma' increases the sensitivity to activated protein C in normal and factor V Leiden plasma. Blood. 2014; 124: 1531-8.
42 Mosesson MW, Cooley BC, Hernandez I, Diorio JP, Weiler H. Thrombosis risk modification in transgenic mice containing the human fibrinogen thrombin-binding gamma' chain sequence. J Thromb Haemost. 2009; 7: 102-10.
43 Lovely RS, Boshkov LK, Marzec UM, Hanson SR, Farrell DH. Fibrinogen gamma' chain carboxy terminal peptide selectively inhibits the intrinsic coagulation pathway. Br J Haematol. 2007; 139: 494-503.
44 Walton BL, Getz TM, Bergmeier W, Lin FC, Uitte de Willige S, Wolberg AS. The fibrinogen γA/γ’ isoform does not promote acute arterial thrombosis in mice. J Thromb Haemost. 2014; 12: 680-9.
45 Makogonenko E, Tsurupa G, Ingham K, Medved L. Interaction of fibrin(ogen) with fibronectin: further characterization and localization of the fibronectin-binding site. Biochemistry. 2002; 41: 7907-13.
46 Meh DA, Siebenlist KR, Brennan SO, Holyst T, Mosesson MW. The amino acid sequence in fibrin responsible for high affinity thrombin binding. Thromb Haemost. 2001; 85: 470-4.
47 Siebenlist KR, Meh DA, Mosesson MW. Plasma factor XIII binds specifically to fibrinogen molecules containing gamma chains. Biochemistry. 1996; 35: 10448-53.
48 Lucas MA, Fretto LJ, McKee PA. The binding of human plasminogen to fibrin and fibrinogen. J Biol Chem. 1983; 258: 4249-56.
49 Thelwell C, Longstaff C. The regulation by fibrinogen and fibrin of tissue
50 Jerome WG, Handt S, Hantgan RR. Endothelial cells organize fibrin clots into structures that are more resistant to lysis. Microsc Microanal. 2005; 11: 268-77.
51 Maeda N, Seike M, Kume S, Takaku T, Shiga T. Fibrinogen-induced erythrocyte aggregation: erythrocyte-binding site in the fibrinogen molecule. Biochim Biophys Acta. 1987; 904: 81-91.
52 Lominadze D, Dean WL. Involvement of fibrinogen specific binding in erythrocyte aggregation. FEBS Lett. 2002; 517: 41-4.
53 Rampling MW. The binding of fibrinogen and fibrinogen degradation products to the erythrocyte membrane and its relationship to haemorheology. Acta Biol Med Ger. 1981; 40: 373-8.
54 Small M, Lowe GD, Cameron E, Forbes CD. Contribution of the haematocrit to the bleeding time. Haemostasis. 1983; 13: 379-84.
55 Valeri CR, Cassidy G, Pivacek LE, Ragno G, Lieberthal W, Crowley JP, Khuri SF, Loscalzo J. Anemia-induced increase in the bleeding time: implications for treatment of nonsurgical blood loss. Transfusion. 2001; 41: 977-83.
56 Blajchman MA, Bordin JO, Bardossy L, Heddle NM. The contribution of the haematocrit to thrombocytopenic bleeding in experimental animals. Br J Haematol. 1994; 86: 347-50.
57 Ho CH. The hemostatic effect of packed red cell transfusion in patients with anemia. Transfusion. 1998; 38: 1011-4.
58 Sorlie PD, Garcia-Palmieri MR, Costas R, Jr., Havlik RJ. Hematocrit and risk of coronary heart disease: the Puerto Rico Health Program. Am Heart J. 1981; 101: 456-61.
59 Erikssen G, Thaulow E, Sandvik L, Stormorken H, Erikssen J. Haematocrit: a predictor of cardiovascular mortality? J Intern Med. 1993; 234: 493-9.
61 Toss F, Nordstrom A, Nordstrom P. Association between hematocrit in late adolescence and subsequent myocardial infarction in Swedish men. Int J Cardiol. 2013; 168: 3588-93.
62 Tannheimer M, Fusch C, Boning D, Thomas A, Engelhardt M, Schmidt R. Changes of hematocrit and hemoglobin concentration in the cold Himalayan environment in dependence on total body fluid. Sleep Breath. 2010; 14: 193-9.
63 Tefferi A. Polycythemia vera and essential thrombocythemia: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013; 88: 507-16.
64 McMullin MF. The classification and diagnosis of erythrocytosis. Int J Lab Hematol. 2008; 30: 447-59.
65 Landolfi R, Di Gennaro L, Falanga A. Thrombosis in myeloproliferative disorders: pathogenetic facts and speculation. Leukemia. 2008; 22: 2020-8.
66 Marchioli R, Finazzi G, Specchia G, Cacciola R, Cavazzina R, Cilloni D, De Stefano V, Elli E, Iurlo A, Latagliata R, Lunghi F, Lunghi M, Marfisi RM, Musto P, Masciulli A, Musolino C, Cascavilla N, Quarta G, Randi ML, Rapezzi D, Ruggeri M, Rumi E, Scortechini AR, Santini S, Scarano M, Siragusa S, Spadea A, Tieghi A, Angelucci E, Visani G, Vannucchi AM, Barbui T, Group C-PC. Cardiovascular events and
intensity of treatment in polycythemia vera. N Engl J Med. 2013; 368: 22-33.
67 Brown DW, Giles WH, Croft JB. Hematocrit and the risk of coronary heart disease mortality. Am Heart J. 2001; 142: 657-63.
68 Carter C, McGee D, Reed D, Yano K, Stemmermann G. Hematocrit and the risk of coronary heart disease: the Honolulu Heart Program. Am Heart J. 1983; 105: 674-9.
69 Kunnas T, Solakivi T, Huuskonen K, Kalela A, Renko J, Nikkari ST. Hematocrit and the risk of coronary heart disease mortality in the TAMRISK study, a 28-year follow-up. Prev Med. 2009; 49: 45-7.
70 Tsai AW, Cushman M, Rosamond WD, Heckbert SR, Polak JF, Folsom AR.
71 Vaya A, Mira Y, Martinez M, Villa P, Ferrando F, Estelles A, Corella D, Aznar J. Biological risk factors for deep vein trombosis. Clin Hemorheol Microcirc. 2002; 26: 41-53.
72 Braekkan SK, Mathiesen EB, Njolstad I, Wilsgaard T, Hansen JB. Hematocrit and risk of venous thromboembolism in a general population. The Tromso study. Haematologica. 2010; 95: 270-5.
73 Crowl LM, Fogelson AL. Computational model of whole blood exhibiting lateral platelet motion induced by red blood cells. Int J Numer Method Biomed Eng. 2010; 26: 471-87.
74 Atici AG, Kayhan S, Aydin D, Yilmaz YA. Plasma viscosity levels in pulmonary thromboembolism. Clin Hemorheol Microcirc. 2013; 55: 313-20.
75 Lionnet F, Hammoudi N, Stojanovic KS, Avellino V, Grateau G, Girot R, Haymann JP. Hemoglobin sickle cell disease complications: a clinical study of 179 cases. Haematologica. 2012; 97: 1136-41.
76 Booth S, Chohan S, Curran JC, Karrison T, Schmitz A, Utset TO. Whole blood
viscosity and arterial thrombotic events in patients with systemic lupus erythematosus. Arthritis Rheum. 2007; 57: 845-50.
77 Carvalho FA, Connell S, Miltenberger-Miltenyi G, Pereira SV, Tavares A, Ariens RA, Santos NC. Atomic force microscopy-based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano. 2010; 4: 4609-20.
78 De Oliveira S, Vitorino de Almeida V, Calado A, Rosario HS, Saldanha C. Integrin-associated protein (CD47) is a putative mediator for soluble fibrinogen interaction with human red blood cells membrane. Biochim Biophys Acta. 2012; 1818: 481-90.
79 Hermand P, Huet M, Callebaut I, Gane P, Ihanus E, Gahmberg CG, Cartron JP, Bailly P. Binding sites of leukocyte beta 2 integrins (LFA-1, Mac-1) on the human ICAM-4/LW blood group protein. J Biol Chem. 2000; 275: 26002-10.
80 Ihanus E, Uotila L, Toivanen A, Stefanidakis M, Bailly P, Cartron JP, Gahmberg CG. Characterization of ICAM-4 binding to the I domains of the CD11a/CD18 and
81 Hermand P, Gane P, Huet M, Jallu V, Kaplan C, Sonneborn HH, Cartron JP, Bailly P. Red cell ICAM-4 is a novel ligand for platelet-activated alpha IIbbeta 3 integrin. J Biol Chem. 2003; 278: 4892-8.
82 Spring FA, Parsons SF, Ortlepp S, Olsson ML, Sessions R, Brady RL, Anstee DJ. Intercellular adhesion molecule-4 binds alpha(4)beta(1) and alpha(V)-family integrins through novel integrin-binding mechanisms. Blood. 2001; 98: 458-66.
83 Barr JD, Chauhan AK, Schaeffer GV, Hansen JK, Motto DG. Red blood cells mediate the onset of thrombosis in the ferric chloride murine model. Blood. 2013; 121: 3733-41.
84 An X, Schulz VP, Li J, Wu K, Liu J, Xue F, Hu J, Mohandas N, Gallagher PG.
Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood. 2014; 123: 3466-77.
85 Santos MT, Valles J, Marcus AJ, Safier LB, Broekman MJ, Islam N, Ullman HL, Eiroa AM, Aznar J. Enhancement of platelet reactivity and modulation of eicosanoid production by intact erythrocytes. A new approach to platelet activation and
recruitment. J Clin Invest. 1991; 87: 571-80.
86 Silvain J, Pena A, Cayla G, Brieger D, Bellemain-Appaix A, Chastre T, Vignalou JB, Beygui F, Barthelemy O, Collet JP, Montalescot G. Impact of red blood cell
transfusion on platelet activation and aggregation in healthy volunteers: results of the TRANSFUSION study. Eur Heart J. 2010; 31: 2816-21.
87 Silvain J, Abtan J, Kerneis M, Martin R, Finzi J, Vignalou JB, Barthelemy O, O'Connor SA, Luyt CE, Brechot N, Mercadier A, Brugier D, Galier S, Collet JP, Chastre J, Montalescot G. Impact of Red Blood Cell Transfusion on Platelet Aggregation and Inflammatory Response in Anemic Coronary and Non-Coronary Patients The TRANSFUSION-2 study. J Am Coll Cardiol. 2013.
88 Valles J, Santos MT, Aznar J, Martinez M, Moscardo A, Pinon M, Broekman MJ, Marcus AJ. Platelet-erythrocyte interactions enhance alpha(IIb)beta(3) integrin receptor activation and P-selectin expression during platelet recruitment: down-regulation by aspirin ex vivo. Blood. 2002; 99: 3978-84.
90 Peyrou V, Lormeau JC, Herault JP, Gaich C, Pfliegger AM, Herbert JM. Contribution of erythrocytes to thrombin generation in whole blood. Thromb Haemost. 1999; 81: 400-6.
91 Horne MK, 3rd, Cullinane AM, Merryman PK, Hoddeson EK. The effect of red blood cells on thrombin generation. Br J Haematol. 2006; 133: 403-8.
92 Whelihan MF, Zachary V, Orfeo T, Mann KG. Prothrombin activation in blood coagulation: the erythrocyte contribution to thrombin generation. Blood. 2012; 120: 3837-45.
93 Van Der Meijden PE, Van Schilfgaarde M, Van Oerle R, Renne T, ten Cate H, Spronk HM. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J Thromb Haemost. 2012; 10: 1355-62.
94 Jy W, Johansen ME, Bidot C, Jr., Horstman LL, Ahn YS. Red cell-derived
microparticles (RMP) as haemostatic agent. Thromb Haemost. 2013; 110: 751-60.
95 Zecher D, Cumpelik A, Schifferli JA. Erythrocyte-derived microvesicles amplify systemic inflammation by thrombin-dependent activation of complement.
Arterioscler Thromb Vasc Biol. 2014; 34: 313-20.
96 Gersh KC, Nagaswami C, Weisel JW. Fibrin network structure and clot mechanical properties are altered by incorporation of erythrocytes. Thromb Haemost. 2009; 102: 1169-75.
97 Wohner N, Sotonyi P, Machovich R, Szabo L, Tenekedjiev K, Silva MM, Longstaff C, Kolev K. Lytic resistance of fibrin containing red blood cells. Arterioscler Thromb Vasc Biol. 2011; 31: 2306-13.
98 van Gelder JM, Nair CH, Dhall DP. The significance of red cell surface area to the permeability of fibrin network. Biorheology. 1994; 31: 259-75.
99 Liang XM, Han SJ, Reems JA, Gao D, Sniadecki NJ. Platelet retraction force measurements using flexible post force sensors. Lab Chip. 2010; 10: 991-8.
Chapter 2: The fibrinogen γA/γ’ isoform does not promote acute arterial thrombosis in mice2
2.1 Introduction
Fibrinogen is a 340 kDa glycoprotein that circulates in plasma at 2-4 mg/mL, but
during acute inflammation can exceed 7 mg/mL. Fibrinogen is composed of two sets of three
polypeptide chains: Aα, Bβ, and γ. Alternative splicing of the main γA chain leads to the γ’
chain. Molecules containing the γ’ chain circulate as a heterodimer with the γA chain (2Aα,
2Bβ, and γA/ γ’) and comprise 8-15% of total fibrinogen in healthy individuals [1, 2].
Elevated fibrinogen levels are associated with increased risk of arterial thrombosis [3-5], and
we previously showed that when mice are infused with unfractionated human fibrinogen
(~90% γA/γA and 10% γA/γ’) and subjected to FeCl3-mediated carotid artery injury, elevated
plasma fibrinogen shortens the time to vessel occlusion [6]. These findings suggest elevated
fibrinogen is a causative, etiologic agent in arterial thrombosis. However, the specific
contributions of γA/γA and γA/γ’ fibrinogen isoforms to thrombosis in vivo are unknown.
In vitro studies to define the biochemical role of the γ’ chain have shown that clots made with purified γA/γ’ fibrinogen polymerize at a slower rate than clots made with
purified γA/γA fibrinogen [7]. Additionally, the γ’ chain supports high affinity binding to
thrombin exosite II [8, 9], and studies have shown that thrombin binding to the γ’ chain
!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!!
2!This chapter is based on and reproduced in part with permission from:
Walton BL, Getz TM, Bergmeier W, Lin FC, Uitte de Willige S, Wolberg AS.
competitively inhibits mediated platelet activation [10] and reduces
thrombin-mediated FpB cleavage [7], and factor VIII [11] and V [12] activation. These properties
suggest γA/γ’ fibrinogen has anticoagulant activity in vitro. Conversely, the γ’ chain does
not inhibit thrombin-mediated cleavage of FpA [7, 13], and has been reported to support
higher affinity binding of FXIII than the γA chain [14], although more recent studies suggest
only slightly tighter [14], or even similar [15], binding of FXIII to the γA/γ’ isoform
compared to the γA/γA isoform. Additional studies in purified systems report contradictory
effects of the γ’ chain on clot structure and mechanical properties, demonstrating that the γ’
chain induces the formation of alternately smaller [7, 13, 16] or larger [17] pores, and stiffer
[18] or less stiff [17] clots. These conflicting observations make it difficult to predict the role
of γA/γ’ fibrinogen under physiologic conditions in thrombosis in vivo.
The role of the human γ’ chain in thrombosis has previously been tested in two in
vivo studies. Since the murine γ’ chain does not contain the thrombin-binding sequence
found on the human γ’ chain, Mossesson et al. developed a transgenic mouse that replaced
the murine γ’ chain with the human γ’ chain [19]. Following electrolytic injury to the
femoral vein, there was no difference in thrombus volume between mice containing the
human γ’ chain and wild type (WT) controls, although the presence of the human γ’ chain
reduced thrombus volume in mice that were also heterozygous for the factor V Leiden
mutation [19]. However, interpretation of these findings is complicated by the higher total
fibrinogen in WT mice compared to mice expressing the human γ’ chain. In a baboon model
in which an arteriovenous shunt was placed between the femoral artery and vein, an 18
thrombus formation [11]. These studies suggest the γ’ chain reduces fibrin accumulation and
is antithrombotic during venous thrombosis.
Given these findings, it is interesting that retrospective epidemiological studies have
correlated elevated γA/γ’ fibrinogen levels with increased incidence of coronary artery
disease [20], myocardial infarction [21], and stroke [22-24]. In particular, the finding that
some patients have an increased γ’-to-total fibrinogen ratio [22-25] indicates γA/γ’
fibrinogen is not merely a biomarker of increased total fibrinogen, and suggests a specific
role for γA/γ’ in arterial thrombosis. However, these studies do not and cannot demonstrate
causality of γ’ chain-containing fibrinogen in thrombosis. The objective of our study was to
determine the contribution of γA/γA and γA/γ’ fibrinogen to arterial thrombosis.
2.2 Materials and Methods
2.2.1 Proteins and Materials.
Polyclonal rabbit anti-human fibrinogen antibody was from DAKOCytomation
(Carpinteria, CA). Monoclonal anti-fibrin(ogen) antibody (59D8) was a generous gift of Drs.
Marschall Runge (University of North Carolina), Charles Esmon (Oklahoma College of
Medicine), and Rodney Camire (University of Pennsylvania). Mouse anti-human γ’
chain-specific antibody (2.G2.H9) was from Millipore (Temecula, CA). Biotinylated secondary
antibodies were from Vector Laboratories (Burlingame, CA). The AlexaFluor-488 protein
labeling kit and 10% pre-cast Tris-glycine gels were from Invitrogen (Carlsbad, CA).
Human α-thrombin and murine thrombin were from Enzyme Research Laboratories (South
Bend, IN). Lipidated tissue factor (TF, Innovin) was from Siemens (Newark, DE).
were prepared as described [26]. Bovine serum albumin was from Sigma-Aldrich (St. Louis,
MO). Peroxidase substrate was from KPL (Gaithersburg, MD).
2.2.2 Plasma preparation.
Contact-inhibited human normal pooled plasma (hNPP) was prepared from 40
healthy subjects (50% female, 68% nonwhite) as described [27], in a protocol approved by
the UNC Institutional Review Board. γA/γ’ fibrinogen levels in hNPP were measured by
ELISA, as described [28]. Murine normal pooled plasma (mNPP) was prepared by
collecting blood from 49 female C57Bl/6 mice by inferior vena cava (IVC) venipuncture into
3.2% sodium citrate (1:9 ratio sodium citrate:blood). Pooled whole blood was centrifuged
(4000xg, 20 minutes), and platelet-poor plasma was aliquoted and frozen at -80oC.
2.2.3 Isolation of γA/γA and γA/γ’ fibrinogen.
The γA/γA and γA/γ’ fibrinogen variants were separated from human plasminogen-,
von Willebrand Factor-, and fibronectin-depleted human fibrinogen (Enzyme Research
Laboratories Ltd., Swansea, UK), based on the method described previously [7]. After
purification, variants were concentrated using Vivaspin 20 MWCO 100,000 columns (GE
Healthcare, Uppsala, Sweden) and dialyzed into 20 mM N-2-hydroxyethylpiperazine-N′
-2-ethanesulfonic acid (pH 7.4) containing 150 mM NaCl (HBS). Fibrinogen concentrationwas
determined by absorbance at 280 nm using an extinctioncoefficient of 1.51 mL/(mg/cm).
2.2.4 SDS-PAGE and western blotting.
Fibrinogen preparations were assessed by 10% SDS-PAGE and Coomassie Brilliant
Blue staining or western blotting for total fibrinogen or fibrinogen γ’ chain. For western
blots, membranes were blocked with Tris-buffered saline with 1% Tween containing 5%
milk, washed, and probed sequentially with mouse-anti human γ’-specific primary antibody
and AlexaFluor-488 conjugated anti-mouse secondary antibody. Fluorescent signal was
detected on a Typhoon 900 FLA fluorescent scanner.
2.2.5 Clot formation with purified fibrin(ogen).
Purified fibrinogen, thrombin, and CaCl2 (0.5 mg/mL, 5 nM, and 10 mM, final,
respectively) were combined in 96-half-well plates and polymerization was monitored by
turbidity at 405 nm using SpectraMax Plus340 plate reader (Molecular Devices, Sunnyvale,
CA).
2.2.6 Clot formation in plasma.
hNPP or mNPP was spiked with HBS (Control), or γA/γA or γA/γ’ fibrinogen, and
clotting was initiated with TF (1:30,000 dilution of Innovin, final), 10 mM CaCl2, and 4 µM
phospholipid vesicles in 96-well plates. Clot formation was monitored by turbidity at 405
nm.
2.2.7 Intravital microscopy.
Procedures were approved by the UNC Institutional Animal Care and Use Committee.
Briefly, 6-8 week old male C57Bl/6 mice (Charles River Laboratories, Wilmington, MA) were anesthetized and laser injuries were induced with an Ablate! photoablation system equipped with an attenuatable 532 nm pulse laser (Intelligent Imaging Innovations, Denver,
CO). Five minutes before injury, mice were injected via the retro-orbital plexus with AlexaFluor 595-labeled anti-glycoprotein IX antibody (0.3 mg/g body weight; Emfret,
Eibelstadt, Germany), and AlexaFluor 647-labeled murine anti-fibrin antibody (0.2 mg/g body weight), and trace amounts (5% of total fibrinogen) of AlexaFluor 488-labeled γA/γA or γA/γ’ fibrinogen. Five venules maximum were studied per mouse.
2.2.8 FeCl3 thrombosis model.
FeCl3 injury to carotid arteries was performed as described [6]. Briefly, 6-8 week old male C57Bl/6 mice were anesthetized, and human fibrinogen or vehicle (HBS) was
administered through the left saphenous vein cannula on a per-weight basis 5 minutes before
injury. The right common carotid artery was exposed, dried and treated with FeCl3 (10% on 0.5×1.0-mm filter paper) for 2 minutes. We specifically titrated the conditions to perform
these experiments at a threshold at which some mice do not form thrombi, to allow for sensitivity to both increased and decreased procoagulant activity. Blood flow was monitored by Doppler ultrasonic flow probe, and the time to occlusion (TTO) was defined as the time
2.2.9 Measurement of circulating TAT complexes.
TAT levels were measured by ELISA (Enzygnost TAT micro ELISA, Siemens) using
plasma prepared from IVC blood draws from mice subject to FeCl3 carotid artery thrombosis
(blood was drawn ~5 minutes following vessel occlusion). Samples showing hemolysis were
excluded.
2.2.10 Statistical Methods.
Descriptive statistics (mean, median, standard deviation [SD], standard error of the
mean [SEM]) were calculated. Groups were compared using Student’s t-tests
(normally-distributed data determined by Lilliefors test for normality) or Wilcoxon-Mann-Whitney
Rank Sum Tests (non-normally distributed data) in Kaleidagraph v4.1.3. Correlations were
performed using SAS 9.2 (SAS Inc., Cary, NC). P<0.05 was considered statistically
significant.
2.3 Results
2.3.1 γA/γA fibrinogen increases the fibrin polymerization rate to a greater extent than
γA/γ’ fibrinogen.
Purified γA/γA fibrinogen contained all three fibrinogen chains (Aα, Bβ, and γ) at
expected molecular weights (Figures 2.1A-B). No γ’ chain was detected in γA/γA fibrinogen
(Figure 2.1C), whereas purified γA/γ’ fibrinogen showed equal intensities of γA and γ’ bands
(Figures 2.1A-B). We first clotted purified fibrinogens with purified human thrombin and
followed clotting by turbidity. Although fibrinogen γA/γA and γA/γ’ isoforms were not
FXIII does not affect differences in polymerization between γA/γA and γA/γ’ fibrinogen [17]. Indeed, consistent with previous reports [7, 13, 17], purified γA/γA exhibited a faster
Table 2.1. Polymerization of purified fibrinogen isoforms by human and murine thrombin
Human Thrombin Murine Thrombin
Lagtime (seconds)
Change in Turbidity (OD)
Vmax
(mOD/min) Lagtime (seconds)
Change in Turbidity (OD)
To determine the effect of elevated γA/γA and γA/γ’ fibrinogen on plasma clot
formation during in situ thrombin generation, we spiked purified γA/γA, γA/γ’, or HBS
(control) into hNPP. The concentration of fibrinogen in hNPP was 3.1±0.1 mg/mL (100%)
and baseline concentration of γA/γ’ fibrinogen in hNPP was 0.42 mg/mL (13.5% of total
fibrinogen). We increased the total fibrinogen concentration to 3.5 (114%), 3.9 (127%), or
4.4 (143%) mg/mL by spiking in purified γA/γA or γA/γ’, so that the γA/γ’-to-total
fibrinogen ratios ranged from 9.6-40.1% (Table 2.2). These levels span the range of γA/γ’
levels measured in healthy individuals and patients with thrombosis [23-25, 30, 31].
Elevating either γA/γA or γA/γ’ fibrinogen increased final clot turbidity compared to plasma
spiked with HBS (Figure 2.2B, Table 2.2). When total fibrinogen was raised to 114%,
neither γA/γA nor γA/γ’ fibrinogen increased the clot formation rate. However, elevating
total fibrinogen to 127% or 143% with γA/γA or γA/γ’ significantly and dose-dependently
increased the clot formation rate versus baseline (HBS). Notably, at each concentration,
elevating total fibrinogen with γA/γA increased the clot formation rate to a significantly
greater extent than elevating total fibrinogen with γA/γ’ (Figure 2.2C, Table 2.2). Linear
regression analysis showed that the clot formation rate correlated positively with elevated
total fibrinogen (r=0.667, P<0.001) and negatively with the γ’-to-total fibrinogen ratio
(r=-0.0245, P=0.17), although the relationship between γ’-to-total and clot formation rate did not
reach significance. Moreover, the level of γA/γA isoform correlated strongly with the clot
formation rate (r=0.795, P<0.001) whereas the level of γA/γ’ did not.
Spiking purified human γA/γA, γA/γ’, or HBS (Control) into mNPP produced similar
results. For these experiments, the fibrinogen concentration in mNPP was 2.4±0.2 mg/mL
yielding human γ’-to total fibrinogen ratios ranging from 0-41.2%. Consistent with previous
observations [6], the final turbidity of murine plasma clots was lower than that of human
plasma clots, likely reflecting increased fibrin density of murine fibrin networks versus
human networks (unpublished observation). As in human plasma, both γA/γA and γA/γ'
increased the clot formation rate, but γA/γA increased the rate to a greater extent than γA/γ'
at each concentration tested (P<0.02, Figure 2.2F, Table 2.3). These findings suggest that
during in situ thrombin generation, both elevated γA/γA and γA/γ’ fibrinogen promote clot
Figure 2.2. Both γA/γA and γA/γ’ fibrinogen accelerate clotting in human and mouse plasma. A-C) hNPP was spiked with γA/γA or γA/γ’ to increase total fibrinogen to 114%, 127%, or 143% of normal (symbols appear in figure legend), and clot formation was
triggered by addition of TF and CaCl2. D-F) mNPP was spiked with human γA/γA or γA/γ’
to increase total fibrinogen to 135% or 170% of normal (symbols appear in figure legend) and clot formation was triggered by addition of TF and CaCl2. A, D) Polymerization was
monitored by turbidity; for clarity, only a subset of points is shown. B, C, E, F) The


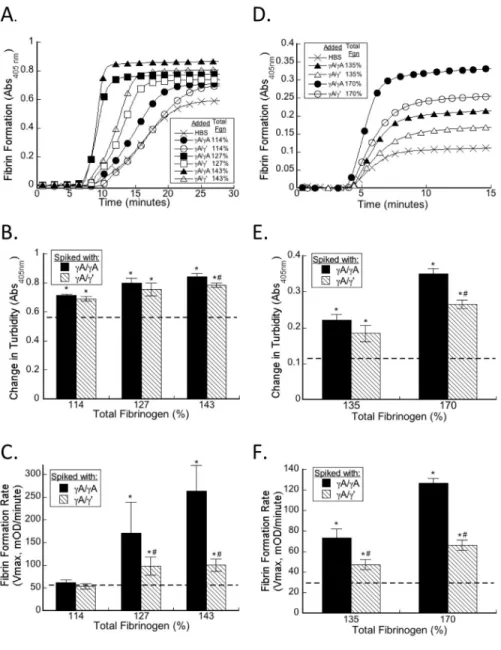
![Table 2.2. Effect of fibrinogen isoforms on human plasma clotting Total Fibrin-ogen (mg/mL [% ]) Fibrin-ogen/ Buffer Infused Human γA/γ’ Final (mg/mL) Human γ’-to-Total Ratio (%) Lagtime (minutes) Change in Turbidity (OD) Vmax (mOD/min) 3.](https://thumb-us.123doks.com/thumbv2/123dok_us/8324758.2207176/60.918.132.815.118.535/effect-fibrinogen-isoforms-clotting-infused-lagtime-change-turbidity.webp)
![Table 2.3. Effect of fibrinogen isoforms on mouse plasma clotting Total Fibrin-ogen (mg/mL [%]) Fibrinogen/ Buffer Infused Human γA/γ’ Final (mg/mL) Human γ’-to-Total Ratio (%) Lagtime (minutes) Change in Turbidity (OD) Vmax (mOD/min) 2.4 (](https://thumb-us.123doks.com/thumbv2/123dok_us/8324758.2207176/61.918.137.811.112.452/fibrinogen-isoforms-clotting-fibrinogen-infused-lagtime-minutes-turbidity.webp)